

Summary
Previous studies have indicated phenotypical differences in glutamic acid decarboxylase 65 autoantibodies (GADA) found in type 1 diabetes (T1D) patients, individuals at risk of developing T1D and stiff-person syndrome (SPS) patients. In a Phase II trial using aluminium-formulated GAD65 (GAD-alum) as an immunomodulator in T1D, several patients responded with high GADA titres after treatment, raising concerns as to whether GAD-alum could induce GADA with SPS-associated phenotypes. This study aimed to analyse GADA levels, immunoglobulin (Ig)G1–4 subclass frequencies, b78- and b96·11-defined epitope distribution and GAD65 enzyme activity in sera from four cohorts with very high GADA titres: T1D patients (n = 7), GAD-alum-treated T1D patients (n = 9), T1D high-risk individuals (n = 6) and SPS patients (n = 12). SPS patients showed significantly higher GADA levels and inhibited the in-vitro GAD65 enzyme activity more strongly compared to the other groups. A higher binding frequency to the b78-defined epitope was found in the SPS group compared to T1D and GAD-alum individuals, whereas no differences were detected for the b96·11-defined epitope. GADA IgG1–4 subclass levels did not differ between the groups, but SPS patients had higher IgG2 and lower IgG4 distribution more frequently. In conclusion, the in-vitro GADA phenotypes from SPS patients differed from the T1D- and high-risk groups, and GAD-alum treatment did not induce SPS-associated phenotypes. However, occasional overlap between the groups exists, and caution is indicated when drawing conclusions to health or disease status.
Keywords: GAD65 immunotheraphy, GADA, stiff-person syndrome, type 1 diabetes
Introduction
Glutamic acid decarboxylase (GAD) is a pyroxidal 5′-phosphate (PLP)-dependent enzyme responsible for synthesis of the main inhibitory neurotransmitter γ-aminobutyric acid (GABA) from glutamate. The two GAD isoforms, GAD65 and GAD67, have 65% identical amino acid sequences, with 74% homology in the C-terminal and 25% homology in the N-terminal regions [1,2]. In humans, GAD65 is expressed both in pancreatic β cells and in the synaptic vesicles of neurones, while GAD67 is restricted to the neural cytoplasm [3]. The function of GAD65 in β cells still remains uncertain.
Stiff-person syndrome (SPS) is a rare autoimmune neurological disorder estimated to affect one per million in the general population [4,5], where clinical examination shows progressive muscle stiffness and spasms [6]. Symptoms arise due to deficient GABA levels which have been attributed to the inhibition of GAD65 enzyme activity, as GADA-positive serum from SPS patients has been shown to inhibit the GAD65 catalysed decarboxylation of glutamate to GABA [7,8]. Approximately 60% of SPS patients have high GADA levels in sera [8], and autoantibodies are also present in the cerebrospinal fluid (CSF) [9–11].
Type 1 diabetes (T1D) results from a selective autoimmune destruction of the pancreatic insulin-producing β cells, where GAD65 acts as one of the major autoantigens [12]. Approximately 70–80% of newly diagnosed T1D patients have detectable GADA in serum [13], and the presence of persistent GADA together with other T1D-associated autoantibodies is a strong predictor for progression to disease in healthy individuals [14–16]. Both T1D and SPS are characterized by GAD65-specific cellular and humoral immune responses [17]. Whereas the majority of GADA in T1D are directed to GAD65 [18], SPS patients show high levels of GADA specific for both isoforms [19,20]. The shared immunological aetiology is reflected in the co-existence of both diseases in as many as 30% of SPS patients who also develop T1D [1,9]; however, only one in 10 000 individuals diagnosed with T1D is affected by SPS [21].
There are differences in the GADA phenotypes present in these two diseases, as expressed in titres, recognized epitopes, immunoglobulin (Ig)G subclass distribution and ability to inhibit GAD65 enzyme activity. It has been reported that SPS patients display significantly higher GADA titres compared to T1D individuals [22]. GAD65-specific monoclonal antibodies and their recombinant Fab (rFab) have been used previously to map GADA epitopes associated with T1D and SPS. The GADA epitope defined by monoclonal antibody b96·11 is located in the middle region of GAD65, and appears to be associated with progression to T1D [23–25]. In contrast, SPS patients recognize a GADA epitope defined by monoclonal antibody b78, which is located in the C-terminal region [7]. While sera from SPS patients characteristically inhibit GAD65 enzyme activity, an event associated with the presence of b78-defined GADA [26], this phenomenon is observed for only a minority of GADA-positive T1D patients [8]. Furthermore, previous studies of GADA IgG subclass distribution have shown that IgG1 is the dominant subclass in newly diagnosed T1D patients and individuals at risk of developing T1D as well as in SPS patients [11,17,27,28]. However, SPS patients present a broader range of subclasses other than IgG1 more frequently [11,17], whereas T1D patients show higher levels of IgG3 [17]. In contrast, individuals with a susceptibility to T1D, who display a higher frequency of GADA IgG2 [28] and/or IgG4 [27], remain non-diabetic for longer than those with a broader subclass response lacking the emergence of IgG4.
During a previous clinical Phase II trial, using aluminium formulated GAD65 (GAD-alum) as an immunomodulator for T1D [29], treated patients displayed up to a 57-fold increase in GADA titres. These findings raised concerns as to whether induction of GADA titres by treatment could be accompanied by the development of a GADA phenotype similar to that observed in SPS patients. Thus, the aim of the present study was to determine phenotypical differences in GADA titres, the ability to inhibit GAD65 enzyme activity as well as GADA epitope and IgG subclass distribution in four groups of high GADA titres, T1D patients, T1D patients treated with GAD-alum, individuals at high risk for T1D and SPS patients.
Material and methods
Study populations
Four groups of high GADA-positive individuals were included in the present study; for a detailed description of patient characteristics, see Table 1.
Table 1.
Patient characteristics of SPS and T1D patients, GAD-alum treated T1D patients and healthy high-risk T1D individuals.
| Patient | Age at sampling | Sex | Age T1D | Age SPS |
|---|---|---|---|---|
| SPS 1 | 53 | M | 15 | 51 |
| SPS 2 | 48 | F | 25 | n.a. |
| SPS 3 | 45 | M | 24 | 42 |
| SPS 4 | 48 | F | 32 | 47 |
| SPS 5 | 65 | F | 60 | 63 |
| SPS 6 | 71 | F | No | 69 |
| SPS 7 | 37 | F | No | 34 |
| SPS 8 | 33 | F | 28 | 31 |
| SPS 9 | 61 | F | No | 60 |
| SPS 10 | 71 | F | 31 | 68 |
| SPS 11 | 64 | M | 29 | 49 |
| SPS 12 | 56 | F | No | 47 |
| T1D 1 | 10 | F | 10 | No |
| T1D 2 | 17 | M | 17 | No |
| T1D 3 | 10 | F | 10 | No |
| T1D 4 | 5 | M | 5 | No |
| T1D 5 | 12 | F | 12 | No |
| T1D 6 | 4 | F | 4 | No |
| T1D 7 | 17 | M | 13 | No |
| High-risk 1 | 5 | F | No | No |
| High-risk 2 | 8 | M | 13 | No |
| High-risk 3 | 8 | F | No | No |
| High-risk 4 | 8 | F | 11 | No |
| High-risk 5 | 8 | M | No | No |
| High-risk 6 | 8 | M | 13 | No |
| GAD-alum 1 | 18 | F | 17 | No |
| GAD-alum 2 | 15 | F | 15 | No |
| GAD-alum 3 | 17 | F | 16 | No |
| GAD-alum 4 | 12 | M | 11 | No |
| GAD-alum 5 | 15 | F | 14 | No |
| GAD-alum 6 | 18 | M | 18 | No |
| GAD-alum 7 | 17 | F | 17 | No |
| GAD-alum 8 | 11 | F | 10 | No |
| GAD-alum 9 | 15 | F | 15 | No |
SPS: stiff-person syndrome; T1D: type 1 diabetes; GAD-alum: aluminium-formulated glutamic acid decarboxylase; M: male; F: female; n.a.: not applicable.
T1D patients
Samples from the T1D group (n = 7) were obtained from patients participating in a Swedish nationwide prospective cohort study, Better Diabetes Diagnosis (BDD), involving newly diagnosed T1D patients aged ≤ 18 years recruited from 40 paediatric clinics [30]. For the current study, samples with the highest GADA titres (> 95th percentile of GADA-positive patients) were selected from BDD patients recruited at the Linköping University Hospital paediatric clinic (n = 198).
T1D high-risk individuals
The high-risk group (n = 6) was selected from the ABIS (All Babies in Southeast of Sweden) cohort, where 17 055 children born from 1997 to 1999 have been followed prospectively with regular biological sampling [31]. From this cohort, children testing positive for several T1D-associated autoantibodies at ≥ two time-points (n = 23) have been classified as having a high risk for developing the disease [32]. In this study we included six of the children with the highest GADA levels, three of whom developed manifest T1D after sample collection.
SPS patients
Serum from the SPS group (n = 12) were chosen based exclusively on sample availability; all SPS patients were GADA-positive. Serum samples from 10 patients were kindly donated by Mohammed Hawa and David Leslie at the Queen Mary University of London, UK, while two samples were collected from patients recruited from the Östergötland county council, Sweden. Eight of 12 SPS individuals were also diagnosed with T1D.
T1D patients treated with GAD-alum
Samples from the GAD-alum group (n = 9) were selected from a previous clinical Phase II trial described elsewhere [29]. The treatment increased GADA levels significantly compared to patients receiving placebo, with the highest levels detected 3 months after initiation of treatment. At this time-point approximately one-third (n = 11) of patients receiving GAD-alum displayed a GADA fold-change of 10–35 times, while the remaining two-thirds of the patients (n = 24) displayed a GADA fold-change of less than 10 times compared to baseline. The maximum increase of GADA from baseline observed during the trial was a fold-change of 57 times, detected in one patient at 3 months. For the present study, serum samples from the 3-month visit were selected based on the highest quartile of GADA levels within the treated group.
Determination of GADA titres
Serum GADA titres were determined using a radio-binding assay employing 35S-labelled recombinant human GAD65, as described previously [33]. The assay is validated through the Diabetes Autoantibody Standardization Program (DASP) workshop, and in 2010 the assay had 100% specificity and 80% sensitivity.
GAD65 enzyme activity assay
Recombinant human GAD65 enzyme activity was measured in duplicate in the presence of patient serum by a 14CO2-trapping method based on the enzymatic conversion of glutamate to GABA, as described previously [33]. Mean results were expressed as a percentage of the maximum GAD65 enzyme activity.
Epitope-specific radioligand binding assay (ES-RBA)
Monoclonal antibodies b96·11 and b78 were derived from a patient with autoimmune polyendocrine syndrome – type 2 [34], and recognize conformational epitopes formed by the three-dimensional structure of amino acid residues 308–365 and 451–585, respectively. Both monoclonal antibodies (mAbs) recognized GAD65 in its native conformation and do not bind GAD67. The capacity of their recombinant Fab (rFab) to inhibit GAD65 binding by human serum GADA was tested in a competitive ES-RBA, as described previously [25]. The two rFab were added to separate wells at a concentration sufficient to compete binding of the originating intact mAb to GAD65 by at least 80%. Non-competitive GAD65 binding was established by no addition of rFab. The cut-off for specific competition was determined as > 15% by using a negative control rFab CG7C7 specific to insulin, at 2 μg/ml. Each sample was measured in triplicate, and the mean value was calculated. A control serum was included on each plate to correct for interplate variations. Binding of GADA to GAD65 in the presence of rFab was expressed as follows: ratio = GADA counts per minute (cpm) in the presence of rFab (competed)/GADA cpm in the absence of rFab (non-competed). A higher binding to GAD65 in the presence of an rFab indicates a lower proportion of GADA binding to the respective epitope. Cases where the rFab-competed sample resulted in higher cpm than the non-competed sample were regarded to have a 100% binding capacity (i.e. no GADA with the epitope specificity in question).
GADA IgG subclass assay
The GADA IgG1, 2, 3 and 4 subclasses were measured using a modification of the conventional GADA assay, as described previously [33]. The cut-off value for each subclass was determined using a GADA-negative control, which was run in duplicate in each assay. Results were expressed as cpm, and the positivity of each sample was calculated by subtraction of the mean cpm value plus three times the standard deviation (s.d.) obtained for the negative control. Due to sample limitations in the SPS group, GADA subclass distribution analysis were performed for 10 of 12 patients.
Statistics
As data sets were determined to be significantly different from a Gaussian distribution using the Shapiro–Wilk test, non-parametric tests corrected for ties were used. Unpaired analysis was performed using the Kruskal–Wallis test followed by the Mann–Whitney U-test and correlations were calculated using Spearman's rank correlation coefficient test. Differences within the groups were analysed by Friedman's test followed by Wilcoxon's signed-rank test; P-values < 0·05 were considered statistically significant. The statistical analyses were performed using IBM spss statistics version 19 (SPSS, Inc., Chicago, IL, USA).
Ethics
Informed consent from the participants or their guardians was obtained as part of previous clinical and epidemiological studies according to the Helsinki Declaration.
Results
Higher GADA levels in SPS patients
The SPS group displayed higher GADA levels (median: 424 300 U/ml, range: 2019–4 992 000) compared to the T1 (median: 21 140 U/ml, range: 12 040–48 000; P = 0·003), GAD-alum (median: 14 770 U/ml, range: 9145–158 300; P = 0·002) and high-risk (median: 13 678 U/ml, range: 1020–70 350; P = 0·003) groups (Fig. 1). On average, SPS patients had a 20-fold higher GADA titre compared to the T1D group. While the co-existence of T1D in SPS patients did not affect GADA levels, and GADA levels of these individuals were distributed evenly within the SPS group, the four SPS patients without T1D showed GADA titres above the median level for the whole group (Fig. 1).
Figure 1.
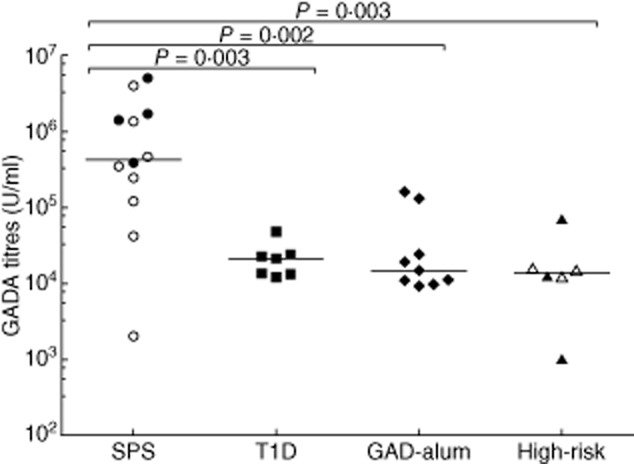
Serum glutamic acid decarboxylase antibody (GADA) titres (U/ml) in stiff-person syndrome (SPS) (circles, n = 12), type 1 diabetes mellitus (T1D) (squares, n = 7), glutamic acid decarboxylase (GAD)-alum (rhombuses, n = 9) and high-risk (triangles, n = 6) groups. Empty circles in the SPS group (n = 8) represent individuals with co-existent T1D, whereas empty triangles in the high-risk group (n = 3) represent individuals who developed T1D after sampling. Significant differences are indicated as P-values and horizontal lines represent the median.
T1D, GAD-alum and high-risk individuals inhibit GAD65 enzyme activity to a lower extent compared to SPS patients
The in-vitro GAD65 enzyme activity was significantly lower in SPS patients (median: 45%; range: 34–67%) compared to the T1D (median: 66%; range: 42–81%; P = 0·010), GAD-alum (median: 93%; range: 54–100%; P < 0·001) and high-risk (median: 75%; range: 38–88%; P = 0·032) groups (Fig. 2). Sera from GAD-alum-treated patients inhibited the activity to a lesser extent than sera from the T1D group (P = 0·042). Co-existence of T1D and SPS did not seem to affect the enzymatic inhibition differently. In addition, one T1D patient and one high-risk individual inhibited GAD65 enzyme activity to the same extent as the median inhibition observed for the SPS group.
Figure 2.
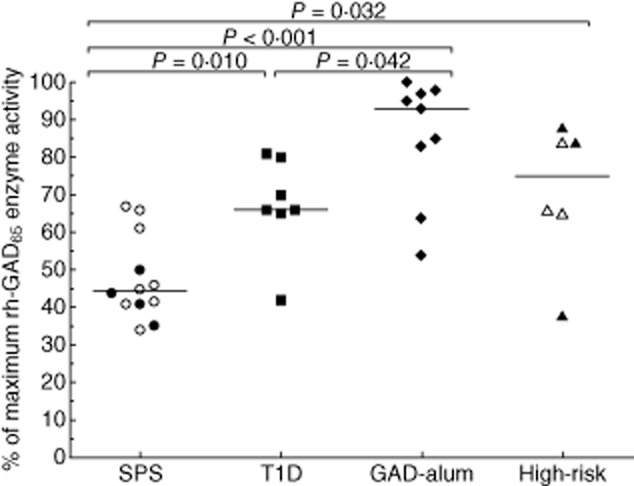
Recombinant human glutamic acid decarboxylase (GAD)65 in-vitro enzyme activity in the presence of sera from stiff-person syndrome (SPS) (circles, n = 12), type 1 diabetes mellitus (T1D) (squares, n = 7), GAD-alum (rhombuses, n = 9) and high-risk (triangles, n = 6) groups. Open circles in the SPS group (n = 8) represent individuals with co-existent T1D, whereas open triangles in the high-risk group (n = 3) represent individuals who developed T1D after sampling. Results are expressed as a percentage of maximum GAD65 enzyme activity. Significant differences are indicated as P-values and horizontal lines represent the median.
Correlation analysis revealed a relationship between high GADA titres and low GAD65 enzyme activity in the GAD-alum (r = −0·883; P = 0·002) and high-risk groups (r = −0·812; P = 0·050), and a trend in T1D patients (r = −0·721; P = 0·068) (Fig. 3a–d). However, this association was not observed for the SPS patients. No other association was observed between GAD65 enzyme inhibition, GADA titres, IgG subclass distribution or epitope pattern.
Figure 3.
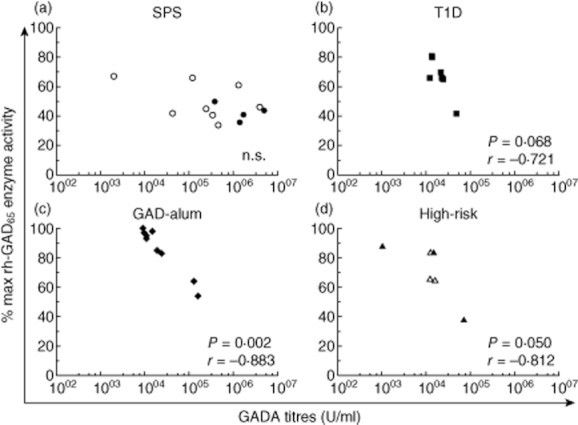
Correlation between glutamic acid decarboxylase antibody (GADA) titres and glutamic acid decarboxylase (GAD)65 enzyme activity in stiff-person syndrome (SPS) (a) and type 1 diabetes mellitus (T1D) patients (b), GAD-alum treated T1D patients (c) and high-risk T1D individuals (d). Open circles in the SPS group (n = 8) represent individuals with co-existent T1D, whereas open triangles in the high-risk group (n = 3) represent individuals who developed T1D after sampling. Significant differences or trends are indicated as P-values, and the correlation coefficient as r.
Higher frequency of GADA with the b78 phenotype in SPS patients
For the majority of SPS patient sera (11 of 12, 92%), binding to GAD65 was reduced significantly in the presence of rFab b78, while only two of seven T1D (28%), three of nine GAD-alum (33%) and two of six high-risk individuals (33%) were affected (Fig. 4a). The majority of individuals in all groups showed significant reduction in binding to GAD65 in the presence of rFab b96, with no significant differences between the groups (Fig. 4b).
Figure 4.
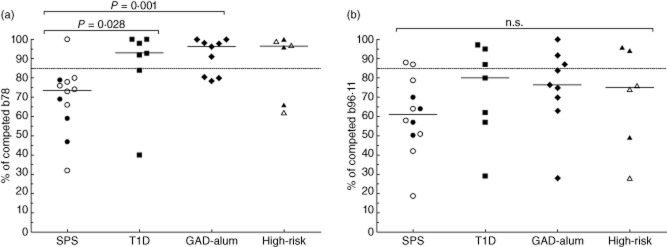
(a,b) Binding to GAD65 in the presence of rFab b78 (a) and rFab b96·11 (b) presented as a ratio of competed/non-competed in stiff-person syndrome (SPS) (circles, n = 12), type 1 diabetes mellitus (T1D) (squares, n = 7), glutamic acid decarboxylase (GAD)-alum (rhombuses, n = 9) and high-risk (triangles, n = 6) groups. Open circles in the SPS group (n = 8) represent individuals with co-existent T1D, whereas open triangles in the high-risk group (n = 3) represent individuals who developed T1D after sampling. A higher binding to GAD65 in the presence of rFab indicates a lower proportion of glutamic acid decarboxylase antibody (GADA) binding to the respective epitope. Samples with a calculated value below the 85% cut-off limit, represented as a dotted line, were regarded as positive for binding to the respective epitope. Significant differences are indicated as P-values and horizontal lines represent the median.
No significant differences in GADA IgG1–4 subclass distribution
Analysis of the GADA IgG1–4 subclass distribution (in cpm) revealed no significant differences between the groups (Fig. 5). We further assessed the relative contribution (%) of each subclass to the entire GADA titre for each individual; still no differences were found between the groups (data not shown). However, while the levels of each subclass did not differ between the groups, a difference in subclass hierarchy within the groups was found. The most frequent subclass in all groups was IgG1. While IgG2 was the lowest prevalent in T1D, GAD-alum and high-risk individuals, similar distribution of the other classes was found in the T1D and GAD-alum groups (IgG3>IgG4>IgG2), but not in the high-risk group (IgG4>IgG3>IgG2). In contrast, SPS patients showed lower proportions of IgG4 (IgG3> IgG2>IgG4) more frequently.
Figure 5.
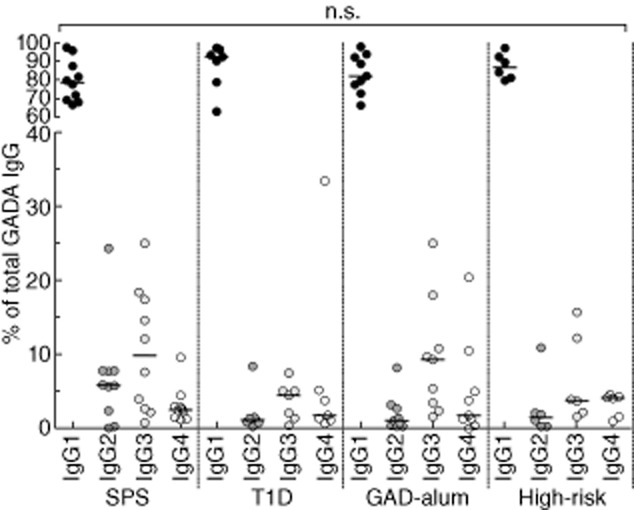
Serum glutamic acid decarboxylase antibody (GADA) IgG 1–4 subclass distribution in stiff-person syndrome (SPS) (n = 10), type 1 diabetes mellitus (T1D) (n = 7), glutamic acid decarboxylase (GAD)-alum (n = 9) and high-risk (n = 6) groups. Results are expressed as the relative contribution of each subclass (% of total GADA) and positivity of each sample was calculated by subtraction of the mean cpm value plus three times the standard deviation (s.d.) obtained for the negative control. Horizontal lines represent the median.
Discussion
In this study we assessed whether GADA phenotype characteristics observed in different groups of individuals with very high GADA titres correlated with disease status. Only high GADA titre groups were included when comparing GADA phenotypes to SPS patients, in contrast to most previous studies which have selected individuals based solely on GADA positivity. This evaluation is also of clinical relevance, as GAD-alum immunization triggers a significant increase in GADA titres, raising concerns about the possible induction of SPS-like GADA phenotypes. While our data support previous findings of disease-specific GADA phenotypes on a group basis, we found phenotypical overlaps among individuals from the different groups. A previous report including high-titre patient groups suggested that GADA phenotypical patterns may be associated with the high GADA titres found usually in SPS patients, rather than the disease per se [35]. Even though sera from GAD-alum, T1D and high-risk groups for this study were selected based on their very high GADA titres, levels in SPS patients were still significantly higher than the other groups. It has been reported previously that GADA titres in SPS patients are 50–500-fold greater than those found in general T1D populations [21,22]. The 20-fold difference in GADA levels between the SPS and T1D group in our study is considerably lower, as our T1D cohort was selected on the basis of extremely high GADA titres. Even though GADA titres in the SPS group overall exceeded that of the other groups, some SPS patients had levels similar to those found in the other cohorts. Further, two individuals in the GAD-alum group had titres similar to the SPS group median level.
A similar pattern was observed when analysing the inhibition of GAD65 enzyme activity. Thus, while sera from SPS patients inhibited the in-vitro GAD65 enzyme activity significantly more compared to the other groups, the inhibition in three SPS patients was close to the median inhibition observed for T1D patients, and an overlap for certain individuals was observed within each group. The lower inhibition of enzyme activity observed by sera from the GAD-alum-treated group compared to that of T1D individuals further supports the safety of GAD-alum treatment in T1D patients. An inverse correlation was found between GADA titres and GAD65 enzyme activity in the GAD-alum and high-risk group, but not for SPS individuals. Previous studies have not been able to establish a correlation between GADA titres in serum or CSF with disease severity in SPS patients [7,36], which might explain the lack of correlation between GADA titres and enzyme inhibition in our SPS group. Due to ethical and practical reasons it was unfortunately not possible to include CSF sampling as a part of this study.
The analysis of GADA epitopes showed that the b78-defined epitope, described previously as a marker for SPS [7], was indeed recognized significantly better by SPS patients. Our results, including selected high-titre GAD-alum-treated patients, are in line with our previous report including the whole study cohort, where we found no change in recognition of the b78-defined epitope and only a transient increase in binding to the b96·11-defined epitope [37]. Here we add new data, showing that even a 57-fold increase in GADA titres did not induce an SPS-associated phenotype. It is noteworthy that all patients participating in the GAD-alum Phase II trial have been followed at 4 [38], 5 and 6 years after the trial was initiated (unpublished data) and no neurological or other clinical adverse events have been reported. No induction of SPS-associated GADA phenotypes were detected during the Phase II GAD-alum trial [33,37], and after several years none of the participants in the trial has developed neurological complications. To be able to assess the persistence of the GADA phenotypes observed during the study, additional future sampling is needed.
Previous studies have shown that the b96·11-defined epitope is recognized commonly by GADA in T1D individuals [7] and in individuals progressing to T1D [39]. The majority of samples from all groups recognized the b96·11-defined epitope with no significant differences between the groups. This may be due to the fact that the majority of SPS patients were also diagnosed with T1D, and half the high-risk individuals developed T1D after sampling.
Analysis of GADA IgG1–4 subclass distribution in absolute values (cpm) or relative contribution (%) revealed no differences between the groups. As described previously [11,17,27], IgG1 was the most frequent subclass in all groups, and the similar IgG subclass hierarchy observed for the T1D and GAD-alum groups is in line with a previous study showing higher IgG3 frequencies in T1D patients [17]. The subclass hierarchy observed for the high-risk individuals is also in agreement with previous findings showing relatively higher IgG4 frequencies [27], and the two individuals with highest IgG4 levels have not yet developed T1D. In contrast, SPS patients displayed a higher prevalence of the IgG3 and IgG2 subclasses and low IgG4 more frequently. Indeed, it has been reported that SPS patients show a broader subclass distribution [17] more frequently, including a higher frequency of the IgG4 subclass. However, another study could detect only IgG1 and IgG2, but no IgG3 and IgG4 in sera and CSF from SPS patients [11]. It has also been proposed that increased frequency of IgG2, IgG3 and IgG4 may reflect the high antibody titres normally found in SPS individuals [35], highlighting the difficulty in establishing a consistent subclass hierarchy for these patients. Due to the lack of samples at the time of T1D diagnosis for the SPS patients with co-existing diseases, it was impossible to assess GADA phenotypes during this period.
In conclusion, in this study we show that in-vitro phenotypes of GADA from SPS patients differed from high GADA titre-positive T1D patients and T1D high-risk individuals, and that GAD65 injections did not induce SPS-associated phenotypes in T1D patients responding with very high GADA titres to GAD-alum treatment. However, despite the low number of patients in each group, overlaps between these groups exist, suggesting caution when drawing conclusions from in-vitro analyses regarding the association of GADA phenotypes to health or disease status.
Acknowledgments
We thank all participating patients for sample donations. We also acknowledge Mohammed Hawa and David Leslie (Queen Mary University of London, UK), as well as Kim Leerbeck (Motala Hospital, Sweden) and Olof Danielsson (Linköping University Hospital, Sweden) for their kind donations of SPS sera. The authors thank Ingela Johansson and Gosia Smolinska-Konefal for excellent technical assistance. This research was supported by grants from The Swedish Child Diabetes Foundation (Barndiabetesfonden), Östgöta Brandstodsbolag, Research Council of Souteast Sweden (FORSS) and an unrestricted grant from Diamyd Medical.
Disclosure
None.
References
- 1.Ali F, Rowley M, Jayakrishnan B, Teuber S, Gershwin ME, Mackay IR. Stiff-person syndrome (SPS) and anti-GAD-related CNS degenerations: protean additions to the autoimmune central neuropathies. J Autoimmun. 2011;37:79–87. doi: 10.1016/j.jaut.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 2.Karlsen AE, Hagopian WA, Grubin CE. Cloning and primary structure of a human islet isoform of glutamic acid decarboxylase from chromosome 10. Proc Natl Acad Sci USA. 1991;88:8337–8341. doi: 10.1073/pnas.88.19.8337. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim J, Richter W, Aanstoot HJ. Differential expression of GAD65 and GAD67 in human, rat, and mouse pancreatic islets. Diabetes. 1993;42:1799–1808. doi: 10.2337/diab.42.12.1799. et al. [DOI] [PubMed] [Google Scholar]
- 4.Murinson BB. Stiff-person syndrome. Neurologist. 2004;10:131–137. doi: 10.1097/01.nrl.0000126587.37087.1a. [DOI] [PubMed] [Google Scholar]
- 5.Solimena M, Folli F, Denis-Donini S. Autoantibodies to glutamic acid decarboxylase in a patient with stiff-man syndrome, epilepsy, and type I diabetes mellitus. N Engl J Med. 1988;318:1012–1020. doi: 10.1056/NEJM198804213181602. et al. [DOI] [PubMed] [Google Scholar]
- 6.Hadavi S, Noyce AJ, Leslie RD, Giovannoni G. Stiff person syndrome. Pract Neurol. 2011;11:272–282. doi: 10.1136/practneurol-2011-000071. [DOI] [PubMed] [Google Scholar]
- 7.Raju R, Foote J, Banga JP. Analysis of GAD65 autoantibodies in stiff-person syndrome patients. J Immunol. 2005;175:7755–7762. doi: 10.4049/jimmunol.175.11.7755. et al. [DOI] [PubMed] [Google Scholar]
- 8.Dinkel K, Meinck HM, Jury KM, Karges W, Richter W. Inhibition of gamma-aminobutyric acid synthesis by glutamic acid decarboxylase autoantibodies in stiff-man syndrome. Ann Neurol. 1998;44:194–201. doi: 10.1002/ana.410440209. [DOI] [PubMed] [Google Scholar]
- 9.Solimena M, Folli F, Aparisi R, Pozza G, De Camilli P. Autoantibodies to GABA-ergic neurons and pancreatic beta cells in stiff-man syndrome. N Engl J Med. 1990;322:1555–1560. doi: 10.1056/NEJM199005313222202. [DOI] [PubMed] [Google Scholar]
- 10.Hanninen A, Soilu-Hanninen M, Hampe CS. Characterization of CD4+ T cells specific for glutamic acid decarboxylase (GAD65) and proinsulin in a patient with stiff-person syndrome but without type 1 diabetes. Diabetes Metab Res Rev. 2010;26:271–279. doi: 10.1002/dmrr.1083. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skorstad G, Hestvik AL, Torjesen P. GAD65 IgG autoantibodies in stiff person syndrome: clonality, avidity and persistence. Eur J Neurol. 2008;15:973–980. doi: 10.1111/j.1468-1331.2008.02221.x. et al. [DOI] [PubMed] [Google Scholar]
- 12.Baekkeskov S, Aanstoot HJ, Christgau S. Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature. 1990;347:151–156. doi: 10.1038/347151a0. et al. [DOI] [PubMed] [Google Scholar]
- 13.Notkins AL, Lernmark A. Autoimmune type 1 diabetes: resolved and unresolved issues. J Clin Invest. 2001;108:1247–1252. doi: 10.1172/JCI14257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parikka V, Nanto-Salonen K, Saarinen M. Early seroconversion and rapidly increasing autoantibody concentrations predict prepubertal manifestation of type 1 diabetes in children at genetic risk. Diabetologia. 2012;55:1926–1936. doi: 10.1007/s00125-012-2523-3. et al. [DOI] [PubMed] [Google Scholar]
- 15.Ziegler AG, Bonifacio E. Age-related islet autoantibody incidence in offspring of patients with type 1 diabetes. Diabetologia. 2012;55:1937–1943. doi: 10.1007/s00125-012-2472-x. [DOI] [PubMed] [Google Scholar]
- 16.Orban T, Sosenko JM, Cuthbertson D. Pancreatic islet autoantibodies as predictors of type 1 diabetes in the Diabetes Prevention Trial–Type 1. Diabetes Care. 2009;32:2269–2274. doi: 10.2337/dc09-0934. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lohmann T, Hawa M, Leslie RD, Lane R, Picard J, Londei M. Immune reactivity to glutamic acid decarboxylase 65 in stiffman syndrome and type 1 diabetes mellitus. Lancet. 2000;356:31–35. doi: 10.1016/S0140-6736(00)02431-4. [DOI] [PubMed] [Google Scholar]
- 18.Jayakrishnan B, Hoke DE, Langendorf CG, Buckle AM, Rowley MJ. An analysis of the cross-reactivity of autoantibodies to GAD65 and GAD67 in diabetes. PLoS ONE. 2011;6:e18411. doi: 10.1371/journal.pone.0018411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pittock SJ, Yoshikawa H, Ahlskog JE. Glutamic acid decarboxylase autoimmunity with brainstem, extrapyramidal, and spinal cord dysfunction. Mayo Clin Proc. 2006;81:1207–1214. doi: 10.4065/81.9.1207. et al. [DOI] [PubMed] [Google Scholar]
- 20.Saiz A, Blanco Y, Sabater L. Spectrum of neurological syndromes associated with glutamic acid decarboxylase antibodies: diagnostic clues for this association. Brain. 2008;131:2553–2563. doi: 10.1093/brain/awn183. et al. [DOI] [PubMed] [Google Scholar]
- 21.Levy LM, Dalakas MC, Floeter MK. The stiff-person syndrome: an autoimmune disorder affecting neurotransmission of gamma-aminobutyric acid. Ann Intern Med. 1999;131:522–530. doi: 10.7326/0003-4819-131-7-199910050-00008. [DOI] [PubMed] [Google Scholar]
- 22.Daw K, Ujihara N, Atkinson M, Powers AC. Glutamic acid decarboxylase autoantibodies in stiff-man syndrome and insulin-dependent diabetes mellitus exhibit similarities and differences in epitope recognition. J Immunol. 1996;156:818–825. [PubMed] [Google Scholar]
- 23.Padoa CJ, Banga JP, Madec AM. Recombinant Fabs of human monoclonal antibodies specific to the middle epitope of GAD65 inhibit type 1 diabetes-specific GAD65Abs. Diabetes. 2003;52:2689–2695. doi: 10.2337/diabetes.52.11.2689. et al. [DOI] [PubMed] [Google Scholar]
- 24.Gilliam LK, Binder KA, Banga JP. Multiplicity of the antibody response to GAD65 in Type I diabetes. Clin Exp Immunol. 2004;138:337–341. doi: 10.1111/j.1365-2249.2004.02610.x. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schlosser M, Banga JP, Madec AM. Dynamic changes of GAD65 autoantibody epitope specificities in individuals at risk of developing type 1 diabetes. Diabetologia. 2005;48:922–930. doi: 10.1007/s00125-005-1719-1. et al. [DOI] [PubMed] [Google Scholar]
- 26.Manto MU, Hampe CS, Rogemond V, Honnorat J. Respective implications of glutamate decarboxylase antibodies in stiff person syndrome and cerebellar ataxia. Orphanet J Rare Dis. 2011;6 doi: 10.1186/1750-1172-6-3. : doi: 10.1186/1750-1172-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ronkainen MS, Hoppu S, Korhonen S. Early epitope- and isotype-specific humoral immune responses to GAD65 in young children with genetic susceptibility to type 1 diabetes. Eur J Endocrinol. 2006;155:633–642. doi: 10.1530/eje.1.02271. et al. [DOI] [PubMed] [Google Scholar]
- 28.Couper JJ, Harrison LC, Aldis JJ, Colman PG, Honeyman MC, Ferrante A. IgG subclass antibodies to glutamic acid decarboxylase and risk for progression to clinical insulin-dependent diabetes. Hum Immunol. 1998;59:493–499. doi: 10.1016/s0198-8859(98)00040-8. [DOI] [PubMed] [Google Scholar]
- 29.Ludvigsson J, Faresjo M, Hjorth M. GAD treatment and insulin secretion in recent-onset type 1 diabetes. N Engl J Med. 2008;359:1909–1920. doi: 10.1056/NEJMoa0804328. et al. [DOI] [PubMed] [Google Scholar]
- 30.Delli AJ, Lindblad B, Carlsson A. Type 1 diabetes patients born to immigrants to Sweden increase their native diabetes risk and differ from Swedish patients in HLA types and islet autoantibodies. Pediatr Diabetes. 2010;11:513–520. doi: 10.1111/j.1399-5448.2010.00637.x. et al. [DOI] [PubMed] [Google Scholar]
- 31.Ludvigsson J, Ludvigsson M, Sepa A. Screening for prediabetes in the general child population: maternal attitude to participation. Pediatr Diabetes. 2001;2:170–174. doi: 10.1034/j.1399-5448.2001.20405.x. [DOI] [PubMed] [Google Scholar]
- 32.Gullstrand C, Wahlberg J, Ilonen J, Vaarala O, Ludvigsson J. Progression to type 1 diabetes and autoantibody positivity in relation to HLA-risk genotypes in children participating in the ABIS study. Pediatr Diabetes. 2008;9:182–190. doi: 10.1111/j.1399-5448.2008.00369.x. [DOI] [PubMed] [Google Scholar]
- 33.Cheramy M, Skoglund C, Johansson I, Ludvigsson J, Hampe CS, Casas R. GAD-alum treatment in patients with type 1 diabetes and the subsequent effect on GADA IgG subclass distribution, GAD65 enzyme activity and humoral response. Clin Immunol. 2010;137:31–40. doi: 10.1016/j.clim.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 34.Tremble J, Morgenthaler NG, Vlug A. Human B cells secreting immunoglobulin G to glutamic acid decarboxylase-65 from a nondiabetic patient with multiple autoantibodies and Graves' disease: a comparison with those present in type 1 diabetes. J Clin Endocrinol Metab. 1997;82:2664–2670. doi: 10.1210/jcem.82.8.4171. et al. [DOI] [PubMed] [Google Scholar]
- 35.Piquer S, Belloni C, Lampasona V. Humoral autoimmune responses to glutamic acid decarboxylase have similar target epitopes and subclass that show titer-dependent disease association. Clin Immunol. 2005;117:31–35. doi: 10.1016/j.clim.2005.06.009. et al. [DOI] [PubMed] [Google Scholar]
- 36.Rakocevic G, Raju R, Dalakas MC. Anti-glutamic acid decarboxylase antibodies in the serum and cerebrospinal fluid of patients with stiff-person syndrome: correlation with clinical severity. Arch Neurol. 2004;61:902–904. doi: 10.1001/archneur.61.6.902. [DOI] [PubMed] [Google Scholar]
- 37.Skoglund C, Cheramy M, Casas R, Ludvigsson J, Hampe CS. GAD autoantibody epitope pattern after GAD-alum treatment in children and adolescents with type 1 diabetes. Pediatr Diabetes. 2012;13:244–250. doi: 10.1111/j.1399-5448.2011.00802.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ludvigsson J, Hjorth M, Cheramy M. Extended evaluation of the safety and efficacy of GAD treatment of children and adolescents with recent-onset type 1 diabetes: a randomised controlled trial. Diabetologia. 2011;54:634–640. doi: 10.1007/s00125-010-1988-1. et al. [DOI] [PubMed] [Google Scholar]
- 39.Maruyama T, Koyama A, Hampe CS. Latent autoimmune diabetes in an adult. Ann NY Acad Sci. 2008;1150:267–269. doi: 10.1196/annals.1447.022. [DOI] [PubMed] [Google Scholar]