

Summary
BALB/c mice with pulmonary tuberculosis (TB) develop a T helper cell type 1 that temporarily controls bacterial growth. Bacterial proliferation increases, accompanied by decreasing expression of interferon (IFN)-γ, tumour necrosis factor (TNF)-α and inducible nitric oxide synthase (iNOS). Activation of dendritic cells (DCs) is delayed. Intratracheal administration of only one dose of recombinant adenoviruses encoding granulocyte–macrophage colony-stimulating factor (AdGM-CSF) 1 day before Mycobacterium tuberculosis (Mtb) infection produced a significant decrease of pulmonary bacterial loads, higher activated DCs and increased expression of TNF-α, IFN-γ and iNOS. When AdGM-CSF was given in female mice B6D2F1 (C57BL/6J X DBA/2J) infected with a low Mtb dose to induce chronic infection similar to latent infection and corticosterone was used to induce reactivation, a very low bacilli burden in lungs was detected, and the same effect was observed in healthy mice co-housed with mice infected with mild and highly virulent bacteria in a model of transmissibility. Thus, GM-CSF is a significant cytokine in the immune protection against Mtb and gene therapy with AdGM-CSF increased protective immunity when administered in a single dose 1 day before Mtb infection in a model of progressive disease, and when used to prevent reactivation of latent infection or transmission.
Keywords: gene immunotherapy, latent tuberculosis, lung immunology, transmissibility
Introduction
With more than 1·7 million deaths annually in the world, tuberculosis (TB) is the leading cause of death by a single infectious agent in the history of humanity, and one of the most important causes of mortality in adults infected with human immunodeficiency virus (HIV) [1]. Although efficient chemotherapy is available, TB treatment is long-term and based on several antibiotics, which results in poor compliance, recidivism, toxicity and emergence of multi-drug-resistant (MDR) strains. Mycobacterium tuberculosis (Mtb) can produce progressive disease or latent infection [2]. Indeed, in highly endemic areas infection occurs first in childhood, and in most cases is controlled. Only 10% of these primary infections lead to progressive disease [2,3]. However, some bacilli remain in tissues in a non-replicating dormant or slowly replicating stage for the rest of the life of the individual. This latent TB (LTB) is clinically asymptomatic, and in countries with low or moderate endemicity most active TB cases arise as a result of reactivation of latent bacilli [2,3]. It is estimated that one-third of the world's population carries latent Mtb, and millions of TB reactivation cases are predicted in the coming years [4].
Patients with pulmonary TB are the most important source for Mtb transmission; the risk of infection is determined by the source case infectiousness and the contact closeness. Household contacts, mainly children exposed to adults with TB, have a high risk of infection, and this risk increases with the degree of contact [5,6]. Avoiding house contact infection would be the most appropriate strategy to interrupt transmission and subsequent TB development. Another alternative is preventive chemoprophylaxis based on isoniazid (INH), which is prolonged (6–12 months) [7,8] with low completion rates, reinfection risk [9] and selection of MDR strains [10]. Thus, it is important to develop new and more efficient therapeutic strategies to treat active TB with lower toxicity and simpler administration, as well as to develop new therapies to prevent LTB reactivation and protect healthy close contacts against Mtb transmission.
Granulocyte–macrophage colony-stimulating factor (GM-CSF) is a pleiotropic cytokine able to induce effects on survival, proliferation and differentiation priming of myeloid and non-myeloid precursor cells [11]. Furthermore, GM-CSF exerts a specific physiological role regulating the surfactant replacement priming for the efficient function of alveolar macrophages (AMs) [11]. GM-CSF regulates the inflammatory response in pulmonary infections by activation of the Jak kinase–signal transducer and activator of transcription (STAT) factor pathway, inducing the expression of interferon-regulating factor 5 (IRF5). High expression of IRF5 results in classical macrophage activation (M1) or inflammatory macrophage induction [12], and absolute or relative GM-CSF deficiencies produce severe AM dysfunction with a phenotype of pulmonary alveolar proteinosis and abnormalities in host defence [13]. Exogenous GM-CSF administration through aerosol induces an increased number of AMs [14]. Therefore, GM-CSF has a specific role in lung physiology and would mediate the control of intracellular infections such as TB, as it is able to induce the production of interleukin (IL)-12, tumour necrosis factor (TNF)-α and interferon (IFN)-γ [11,12]. Furthermore, in-vivo pulmonary over-expression of GM-CSF by local administration of recombinant adenoviruses encoding this cytokine (AdGM-CSF) induces early differentiation and activation of dendritic cells (DCs) with potent immunostimulatory function [15,16]. This is important, considering that in experimental murine pulmonary TB there is a delay in DC recruitment and maturation in both lungs and mediastinal lymph nodes, which apparently contributes to early Mtb immune evasion [17,18]. Thus, GM-CSF should be a significant participant in the immune response against Mtb and could be a target for immunotherapy. The aim of the present study was to determine in a murine model of progressive pulmonary TB the effect of the intratracheal (i.t.) administration of AdGM-CSF 1 day before infection, as well as its effect in preventing reactivation in a mouse model of LTB and avoiding infection of healthy mice co-housed with tuberculous animals in a transmissibility model.
Materials and methods
Kinetics of GM-CSF gene expression and cytokine location during progressive pulmonary TB in BALB/c mice
We used the murine model of i.t. infection described previously [19,20]. Briefly, virulent Mtb strain H37Rv was cultured in Proskauer and Beck medium. After 1 month of culture, mycobacteria were harvested and adjusted to 2·5 × 105 cells in 100 μl of phosphate-buffered saline (PBS), aliquoted and maintained at −70°C until used. Pathogen-free male BALB/c mice, 6–8 weeks old, were anaesthetized (sevoflurane; Abbott Laboratories, Abbott Park, IL, USA) and 100 μl of isotonic sterile endotoxin-free saline solution with 2·5 × 105 viable bacilli were inoculated i.t. using a stainless steel cannula. Animals were then maintained in cages fitted with micro-isolators in a P-3 biosecurity level facility. Following infection, mice were killed by exsanguination under anaesthesia at days 1, 3, 7, 14, 21, 28, 60 and 120 post-infection; lungs were collected immediately to perform quantitative reverse transcription–polymerase chain reaction (RT–PCR) and immunohistochemistry (IHC).
mRNA extraction and reverse transcription (RT)
Five lungs per group per day of killing were placed into 2 ml of RPMI-1640 medium (Invitrogen Life Technologies, Carlsbad, CA, USA) containing 0·5 mg/ml of collagenase type 2 (Worthington, Lakewood, NJ, USA) and incubated for 1 h at 37°C; the lung was then macerated and passed through a sterile 70-μm cell sieve (BD Biosciences, Bedford, MA, USA). The cell suspension was centrifuged at 250 g for 1 min at 4°C and washed with RPMI-1640 medium. The supernatant was removed and red cells were lysed with 1 ml of lysis buffer [0·34 M ammonium chloride, 0·12 mM ethylenediamine tetraacetic acid (EDTA) and 1 mM potassium carbonate], and finally the cells were washed and centrifuged under the same conditions. Five × 106 cells were counted, and 350 μl of buffer RLT was added (Qiagen, Hilden Germany) with β-mercaptoethanol. RNA was isolated using the RNeasy minikit (Qiagen, Hilden, Germany), passing the sample through a column, centrifuged at 10 000 g for 1 min at 4°C. The RNA bound to the column was washed with 700 μl of two other buffers of the mini RNeasy kit (Qiagen, Hilden, Germany) and finally eluted with 50 μl of RNase-free water. The RNA was treated with one unit of DNase (Invitrogen Life Technologies) per microgram of RNA. The quality and quantity of RNA were also evaluated by spectrophotometry (260 nm and A260/280 ratio, NanoDrop-1000; Thermo Fisher Scientific, Waltham, MA, USA) and agarose gels. The cDNA was synthesized using Omniscript RT kit (Qiagen, Hilden, Germany), oligo dT (Promega Corporation, Madison, WI, USA) and 100 ng of RNA. The expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was confirmed by conventional PCR and the cDNAs were amplified with Taq DNA polymerase Hot Start (Qiagen, Germantown, MD, USA).
Real-time PCR
Real-time PCR was developed using the computer real-time PCR system 7500 (Applied Biosystems, Bedford, MA, USA). We used 100 ng of cDNA, 12·5 μl of the mix Quantitect SYBR Green PCR (Qiagen, USA): QuantiTect SYBR Green PCR buffer containing Tris-Cl, KCl, (NH4)2SO4, 5 mM MgCl2, pH 8·7, the mixture of dNTPs (dATP, dCTP, dGTP, dTTP/dUTP), SYBR green I and ROX, plus primer sense and primer anti-sense (50 pmol of each). The formation of a single PCR product and the expected amplicon size were confirmed previously by electrophoresis of the conventional PCR product. The standard curves of PCR products, quantified and diluted, and negative controls were included in each real-time PCR run. The specific primers were designed using Primer Express software (Applied Biosystems) for the following targets: GAPDH: 5′-GGCGCTCACCAAAACATCA-3′, 5′-CCGGAATGCCATTCCTGTTA-3′ [232 base pairs (bp) expected amplicon size]; inducible NO synthase (iNOS): 5′-AGCGAGGAGCAGGTGGAAG-3′, 5′-CATTTCGCTGTCTCCCCAA-3′ (206 bp expected amplicon size); TNF-α: 5′-TGTGGCTTCGACCTCTACCTC-3′, 5′-GCCGAGAAAGGCTGCTTG-3′ (205 bp expected amplicon size); IFN-γ: 5′-GGTGACATGAAAATCCTGCAG-3′, 5′-CCTCAAACTTGGCAATACTCATGA-3′ (180 bp expected amplicon size); GM-CSF: 5′-GCCATCAAAGAAGCCCTGAA-3′, 5′-GCGGGTCTGCACACATGTTA-3′ (114 bp expected amplicon size); and IL-12: 5′-GGATGGAAGAGTCCCCCAAA-3′, 5′-GCTCTGCGGGCATTTAACAT-3′ (125 bp expected amplicon size).
Conditions used were: initial denaturation at 95°C for 15 min, followed by 40 cycles at 95°C for 20 s, 60°C or 58°C for 20 s and 72°C for 34 s. The number of copies of each cytokine mRNA were related to a million copies of GAPDH mRNA. Data were reported as mean ± standard deviation (s.d.) of five different mice for each of two independent experiments.
IHC for F4/80 and GM-CSF
The same paraffin-embedded tissues were used for IHC; 5-μm sections were obtained on slides loaded with poly L-lysine (Biocare Medical, Lake Concord, CA, USA). For dewaxing, the slides were placed at 60–70°C for 20 min, then incubated for 5 min into xylene. The slides were changed five times into the medium in the following sequence: (i) xylene-alcohol (1:1), (ii) absolute alcohol, (iii) alcohol 96% and (iv) distilled H2O. Once hydrated, endogenous peroxidase was blocked with methanol–10% H2O2. The washings were performed with HEPES-buffered saline (HBS)-Tween 20 (10 mM HEPES, 150 mM NaCl, 2 mM CaCl2, 0·05% Tween 20). The areas of tissue were delineated and then blocked with 100 μl of HBS with 2% background sniper (Biocare Medical) and incubated for 30 min in a humid chamber. The slides were then incubated with monoclonal antibody anti-F4/80 conjugated with biotin (eBioscience, San Diego, CA, USA) for 12 h at room temperature. Subsequently, slides were washed and 100 μl of antibody–horseradish peroxidase (AB/HRP) complex (Vectastain ABC System, Burlingame, CA, USA) was added and incubated for 30 min to be revealed with 100 μl of diaminobenzidine/H2O2 (0·004 g diaminobenzidine + 10 ml HBS + 4 μl H2O2). Slides were washed and contrasted with haematoxylin. Staining for GM-CSF took a primary antibody monoclonal anti-GM-CSF (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and incubated subsequently with biotin-conjugated secondary antibody (Invitrogen) for 1 h, washed, and the processing was performed based on the same procedure as above. To obtain the percentage of F4/80-positive AMs or GM-CSF-positive cells, an image analyser was used (Q Win Leica, Milton Keynes, UK). We analysed 50 different random fields with an increase of ×100 in interstitial, pneumonic, peribronchial and perivascular areas as well as all the granulomas found per slide. Measurements were taken by investigators who were blinded to the treatment groups, and the data are reported as the mean values ± s.d. from five different mice at each time-point in each of two different experiments.
Determination of the most effective dose of AdGM-CSF to induce GM-CSF transcription and cytokine production in healthy BALB/c mice
The construction, expression, biological effect and titration of AdGM-CSF and its vector control Addl70-3 have been reported previously [15]. Groups of five healthy male 8-week-old BALB/c mice were anaesthetized with sevoflurane vapour (Abbott Laboratories, Mexico City, Mexico) in a sealed acrylic cage. Anaesthetized mouse was fixed on cardboard and recombinant adenoviruses were administered through a stainless steel cannula (Thomas Scientific, Swedesboro, NJ, USA) connected to an insulin syringe. The cannula was introduced first into the mouth and then directed into the trachea, where the selected dose of recombinant adenoviruses suspended in 100 μl of isotonic sterile endotoxin-free saline solution was injected. Three different doses were tested: 1 × 107, 5 × 107 and 1 × 108 plaque-forming units (pfu). Groups of five mice per each dose were euthanized by exsanguination under terminal anaesthesia after 1 and 7 days post-administration; one lung lobe, right or left, was collected immediately and frozen in liquid nitrogen for total RNA isolation and determination of GM-CSF gene expression by quantitative RT–PCR (qRT-PCR), as described above, while the other lung was perfused with absolute ethanol for fixation and embedded in paraffin for histological evaluation and GM-CSF detection by IHC.
Studies on the effect of AdGM-CSF administration in the experimental model of progressive pulmonary TB
We used the previously described model of progressive TB to study the effect of IT AdGM-CSF [19,20]. Male BALB/c 6–8-week-old mice were treated with 1 × 108 pfu of AdGM-CSF or Addl70-3 i.t., as described above, then 1 day later animals were anaesthetized with sevoflurane, and 100 μl of isotonic sterile endotoxin-free saline solution with 2·5 × 105 viable bacilli was inoculated i.t. Following infection, mice were killed by exsanguination under anaesthesia at days 1, 3, 7, 14, 21, 28, 60 and 120 post-infection to obtain the lungs and perform bacteriological, histomorphometric and molecular biology studies. Five mice per group were euthanized at every time-point selected for the various analyses.
For quantification of bacilli loads by colony-forming units (CFUs), lungs were collected at the selected times, as mentioned, and homogenized using a polytron (Kinematica, Lucerne, Switzerland) homogenizer. The suspensions were then diluted with 0·05% Tween-80 to a final 1-ml volume. Three consecutive logarithmic dilutions were made from this homogenate. Ten μl of each dilution were plated in duplicate on BactoMiddlebrook 7H10 agar (Difco, Detroit, MI, USA) enriched with oleic acid, albumin, dextrose and catalase. Plates were then incubated at 37°C and 5% CO2 for 21 days to quantify the CFUs.
For histological study, the right or left lungs from five different mice per group were perfused i.t. with absolute ethanol. Parasaggital sections were dehydrated and embedded in paraffin (Oxford Labware, St Louis, MO, USA), sectioned and stained with haematoxylin and eosin (H&E). The granuloma area (measured in square microns) and percentage of lung surface affected by pneumonia were determined using an automated image analyser (Q Win Leica), as described previously [20]. Measurements were performed blind, and the data are reported as the mean values ± s.d. from five different mice at each time-point in each of two different experiments. For IHC detection of GM-CSF, the procedure was as described above.
For quantification of activated DCs by cytofluorometry, five lungs per group per day of killing were placed into 2 ml of RPMI-1640 medium (Invitrogen Life Technologies) containing 0·5 mg/ml of collagenase type 2 (Worthington) and incubated for 1 h at 37°C; the lung was then macerated and passed through a 70-μm sterile strainer (BD Biosciences). The cell suspension was centrifuged at 250 g for 1 min at 4°C and washed with RPMI-1640 medium. The supernatant was removed and red cells were lysed with 1 ml of lysis buffer (0·34 M ammonium chloride, 0·12 mM EDTA and 1 mM potassium carbonate). Cells were washed and centrifuged under the same conditions; 1 × 106 viable cells were counted (by trypan blue exclusion) and stained for activated DCs determination [major histocompatibility complex class II (MHC II)+CD11c+CD86+]. The antibodies used were: fluorescein isothiocyanate-labelled anti-I-A/I-E (MHC II-FITC; BD Pharmingen), allophycocyanin-labelled anti-CD11c (CD11c-APC; BD Pharmingen), phycoerythrin-labelled anti-CD86 (CD86-PE; BD Pharmingen). In each cell suspension for each day of sacrifice, staining controls were included to check specificity. Individual stains were made with each of the antibodies used as positive controls and their respective isotype controls. Finally, we included negative controls (unstained) and a dual control mark (MHC II+CD11c+). The double-positive control was the negative parameter for determining the zone of histogram considered positive for CD86; 1 × 105 events were acquired for each sample using fluorescence activated cell sorter (FACS)Calibur cytofluorometer and CellQuest software (BD Biosciences). The data collected were analysed using FlowJo software version 6·1. Data were reported as mean ± s.d. of five different mice for each of two independent experiments.
For cytokines and iNOS gene expression determined by qRT–PCR, five lungs, right or left, from two different experiments were removed and used for isolating RNA from the different groups, following the protocol as published previously [21].
Experimental model of chronic infection similar to LTB
The murine model of LTB has been described previously [22]. Two groups of five 8-week-old female mice B6D2F1 (C57BL/6J X DBA/2J) were infected i.t. with 4000 viable bacteria H37Rv strain suspended in 100 μl of isotonic sterile endotoxin-free saline solution. After 7 months of infection, groups of five mice were treated with AdGM-CSF (1 × 108 pfu) or Addl70-3 (1 × 108 pfu) administered by i.t. instillation as described above. One month later, corticosterone was then administered in drinking water (3 mg/l) for 1 month. Mice were killed under terminal anaesthesia and the lungs were obtained and used to determine bacilli burdens and pneumonia by CFU quantification and automated histomorphometry, as described above.
Mtb transmissibility experimental model in BALB/c mice
The transmissibility experimental model has been described previously [23]. Five BALB/c mice infected (2·5 × 105 bacilli) with Mtb H37Rv or highly virulent Beijing strain 9001000 were co-housed in the same micro-isolator from the first day of infection with five healthy non-infected BALB/c mice (contacts), which received AdGM-CSF (1 × 108 pfu IT) by the i.t. route. Control groups received Addl70-3 (1 × 108 pfu IT) or INH by intragastric cannulation (0·2 mg/day). Delayed-type hypersensitivity (DTH) to mycobacterial antigens was performed in the footpads after 2 months of co-housing, following the reported method [20]. Animals were killed after 2 months of co-housing and their lungs were collected and homogenized for CFU determination. The suspensions were diluted with 0·05% Tween-80 to a final volume of 1 ml. One part of the lung suspension was diluted into three parts of PBS and 100 μl of each dilution were plated in triplicate on BactoMiddlebrook 7H10 agar enriched with oleic acid, albumin, dextrose and catalase. Plates were then incubated at 37°C and 5% of CO2 for 21–45 days to quantify the CFU.
Statistical analysis
Results are expressed as mean ± s.d. Student's two-tailed t-test was used for comparing experimental groups, with a P < 0·05 value considered significant.
Ethical approval
Animal studies were approved by the Institutional Ethics Committee of the National Institute of Medical Sciences and Nutrition ‘Salvador Zubirán’ in accordance to the guidelines of the Mexican national regulations on Animal Care and Experimentation NOM 062-ZOO-1999.
Results
Kinetics of endogenous GM-CSF gene expression during progressive pulmonary TB
When BALB/c mice are infected by the i.t. route with a high dose of the reference Mtb strain H37Rv, an early phase of temporal bacilli growth control is produced and dominated by high expression of TNF-α and IFN-γ with granuloma formation. After 3 weeks of infection, a progressive disease phase develops, characterized by high pulmonary bacilli burdens, tissue damage (progressive pneumonia), lower production of TNF-α and IFN-γ with high expression of T helper type 2 (Th2) cytokines, such as IL-4 and IL-13 [19,20]. In order to investigate the potential role of GM-CSF in this model, gene expression kinetics was determined by qRT–PCR. After Mtb infection a low but progressive increase of GM-CSF gene expression was seen peaking at day 7, followed by a progressive decrease to day 120, when the lowest level was detected (Fig. 1a). IHC and automated morphometry showed that during the course of infection the principal GM-CSF cellular source was the bronchial and bronchiolar epithelium; from days 1 to 14 after infection 10–20% of airway epithelial cells showed strong GM-CSF immunostaining, rising to maximal percentage at day 21 (90%) but with lower staining per cell, followed by progressive decrease until day 120, when 30% of these cells were immunostained (Fig. 1b). Macrophages also showed GM-CSF immunostaining in the alveolar–capillary interstitium at day 14 and during late infection at days 60 and 120, when 10–14% showed positive staining. Granulomas at days 60 and 120 also showed immunostained macrophages (Fig. 1b–d).
Figure 1.
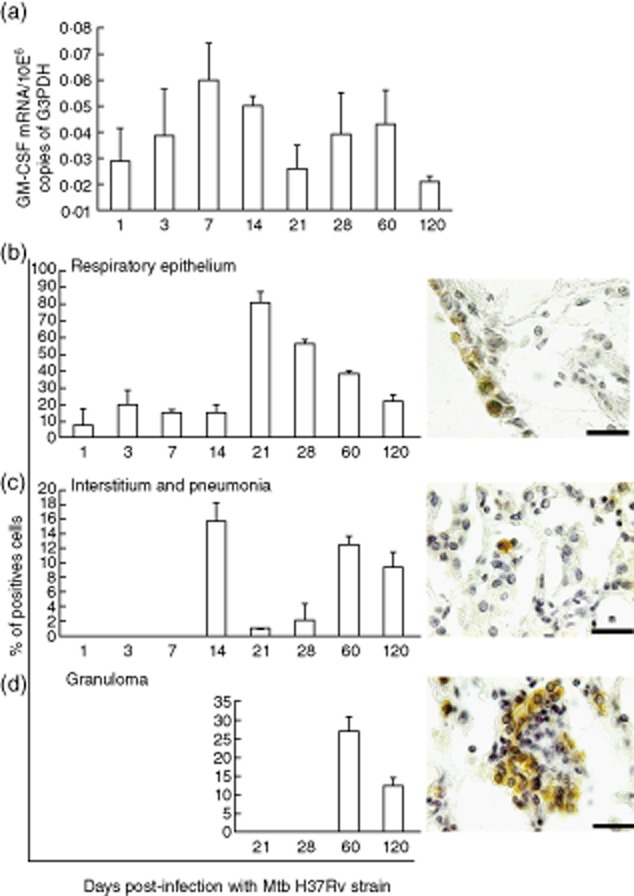
Kinetics of granulocyte–macrophage colony-stimulating factor (GM-CSF) during progressive pulmonary tuberculosis (TB). (a) Five lungs from the same number of Mycobacterium tuberculosis (Mtb)-infected mice at each indicated time-point were used to isolate total RNA and determine the gene expression of GM-CSF by quantitative reverse transcription–polymerase chain reaction (RT–PCR). (b–d) GM-CSF protein expression was detected by immunohistochemistry and the percentage of positive cells was determined in the indicated lung compartments by automated morphometry; representative histological figures of each compartment are to the right of the morphometry graphs (scale bar represents 20 μm). All values are means ± standard deviation (s.d.) from two independent experiments with five mice per group (408 × 552 mm; 150 × 150 DPI).
Determination of the most suitable AdGM-CSF dose to induce transgenic GM-CSF expression in the lungs of healthy mice
Three different doses of AdGM-CSF administered by the i.t. route in healthy BALB/c mice were used to define the most suitable dose to induce GM-CSF gene expression. GM-CSF expression was dose-dependent, with the 1 × 108 pfu dose inducing the highest transgene expression (Fig. 2a). The control group received the empty adenovirus vector (Addl70-3). The cellular source of GM-CSF was determined by IHC; at day 1 post-treatment it was detectable and the bronchiolar epithelium showed strong immunostaining. No detectable basal expression was seen in the alveolar epithelium or AMs. Addl70-3 did not induce GM-CSF production detectable by IHC in healthy mice (Fig. 2b,c).
Figure 2.
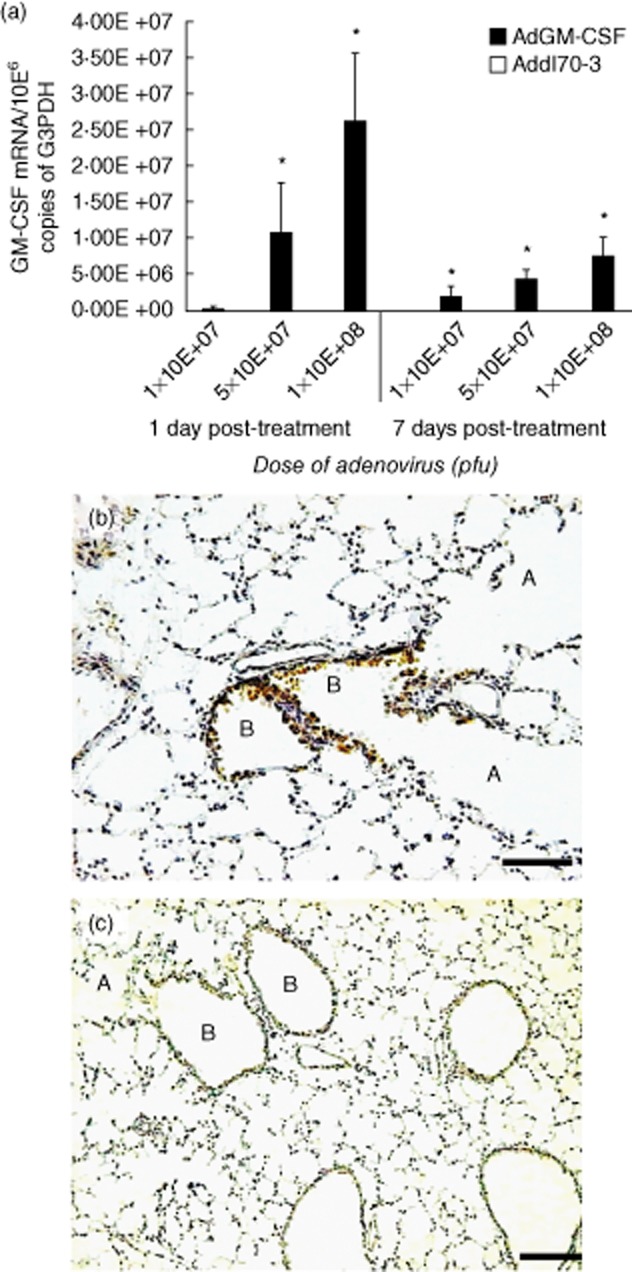
Granulocyte–macrophage colony-stimulating factor (GM-CSF) expression after intratracheal (i.t.) administration of different doses of adenoviruses encoding GM-CSF (AdGM-CSF). (a) Groups of healthy mice were treated with the indicated dose of AdGM-CSF (black bars) or the control adenoviruses Addl70-3 (white bars, undetectable levels), and euthanized after 1 and 7 days; the lungs were used to determine the expression of GM-CSF reverse transcription–polymerase chain reaction (RT–PCR), values are means ± standard deviation (s.d.) from two independent experiments with five mice per group; *P < 0·05. (b) GM-CSF protein expression was detected by immunohistochemistry; the mouse lung after 1 day of 1 × 108 plaque-forming units (pfu) i.t. administration shows strong immunostaining in the airways epithelium in bronchioles (B) and negative immunostaining in alveolar walls (A). (c) In contrast, the mouse lung treated with the same dose of control adenovirus Addl70-3 does not show immunostaining (scale bar represents 60 μm) (207 × 386 mm; 300 × 300 DPI).
Effect of transgenic GM-CSF expression by AdGM-CSF delivered 1 day before infection in the murine model of progressive pulmonary TB
Considering the low percentage of GM-CSF immunostained cells during early infection, two independent experiments were performed to assess the effect of i.t. administration of AdGM-CSF or vector control Addl70-3 1 day before i.t. infection with Mtb H37Rv. The AdGM-CSF administration effect was determined by changes in lung bacterial loads, the histomorphometry of tissue damage (percentage of pneumonic area) and number and size of granulomas. A single dose of AdGM-CSF (1 × 108 pfu) 1 day before infection induced a significant decrease of bacterial loads from day 14 post-infection until the end of the experiment (day 120) (Fig. 3a). With regard to the histological changes, animals that received AdGM-CSF exhibited a significantly lower pneumonic area and more and bigger granulomas than mice treated with Addl70-3 (Fig. 3b–h).
Figure 3.
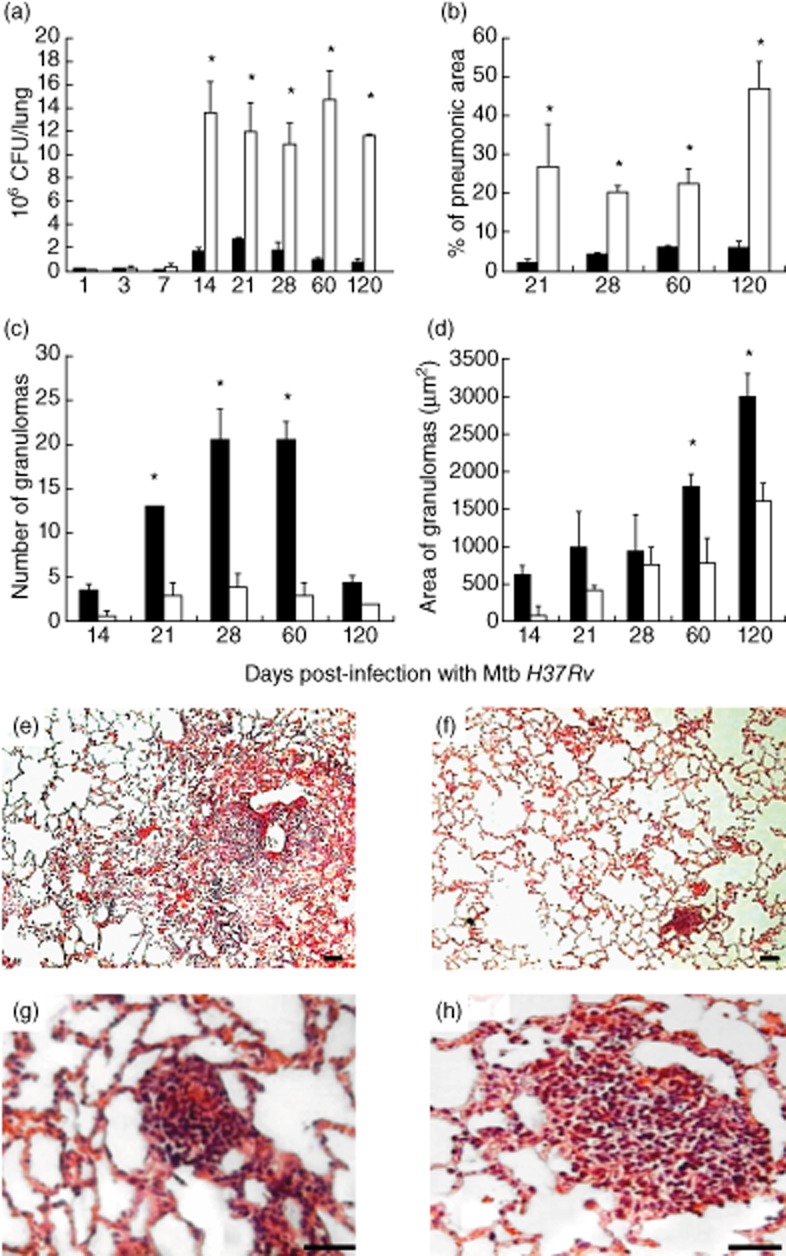
Effect of a single administration of adenoviruses encoding granulocyte–macrophage colony-stimulating factor (AdGM-CSF) 1 day before infection in the murine model of progressive pulmonary tuberculosis (TB). (a) Groups of mice were treated with AdGM-CSF (black bars) or control virus Addl70-3 (white bars) 1 day before intratracheal (i.t.) infection with a high dose of Mycobacterium tuberculosis (Mtb) H37Rv; five mice were killed at each day indicated and the lungs were used to determine bacterial loads by colony-forming units (CFU). A significant CFU decrease was produced by AdGM-CSF administration. (b) The morphometry study showed a significantly lower percentage of lung surface affected by pneumonia in animals treated with AdGM-CSF. (c) The number of granulomas at days 21, 28 and 60 was significantly higher in the AdGM-CSF-treated group, as well as the granuloma size at days 60 and 120 (d). Representative histopathology of the lung from treated mice after 120 days of infection with Mtb, (e) the mouse lung treated with Addl70-3 shows extensive areas of pneumonia. (f) In contrast, mouse treated with AdGM-CSF shows scarce inflammation. (g) Small granulomas are seen in mice treated with Addl70-3. (h) In comparison, bigger granulomas are formed in mouse treated with AdGM-CSF; scale bars represent 60 μm (e) and (f), and 20 μm (g) and (h). All values of bacilli loads and histomorphometry are means ± standard deviation (s.d.) of two independent experiments with five mice per group; *P < 0·05 (466 × 771 mm; 150 × 150 DPI).
In this model of murine pulmonary TB there is a delay in DC activation [17]. Considering that GM-CSF is a significant factor in promoting DC activation, this effect could be a mechanism which explains the observed protective effect of AdGM-CSF. To this end, flow cytometry was used to quantify activated DCs (MHC II+CD11c+CD86+) in lung cell suspensions from each selected day after infection. Mice treated with AdGM-CSF showed a significantly higher number of lung-activated DCs on day 7 post-infection compared with the group treated with Addl70-3, in which there was an increase of these cells until day 21 post-infection (Fig. 4a). Both groups showed a progressive decrease of activated DC during late disease.
Figure 4.

Effect of a single administration of adenoviruses encoding granulocyte–macrophage colony-stimulating factor (AdGM-CSF) on the production of activated dendritic cells and macrophages in the murine model of progressive pulmonary tuberculosis (TB). (a) Groups of mice were treated with AdGM-CSF (black symbols) or Addl70-3 (white symbols) 1 day before infection with Mycobacterium tuberculosis (Mtb) H37Rv; lungs were used to obtain cell suspensions and the number of activated dendritic cells was determined by flow cytometry using antibodies against MHC II-fluorescein isothiocyanate (FITC), CD11c-allophycocyanin (APC) and CD86-phycoerythrin (PE). For each lung sample 1 × 105 events were acquired and the population of cells positive for FITC (FL1) and APC (FL4) (i.e. MHC II+ CD11c+ cells) were selected for obtaining the histogram for the number of positive events for PE (FL2) (i.e. MHC II+ CD11c+ CD86+ cells). The treatment with AdGM-CSF induced more rapid and higher numbers of activated dendritic cells. (b) Lung sections from treated animals with adenoviruses were used to detect the marker of activated macrophages F4/80 by immunohistochemistry, and the percentage of immunostained cells were determined in the indicated lung compartments and lesions. Results are expressed as means ± standard deviation (s.d.) from five mice per time-point and per group in two independent experiments. Representative immunohistochemistry detection of F4/80+ cells in the lungs of animals treated with AdGM-CSF or Add170-3 at the indicated time-point (left corner) in pneumonic areas (c) or granulomas (d). There are more F4/80+ cells in animals treated with AdGM-CSF; scale bar represents 20 μm (518 × 553 mm; 200 × 200 DPI).
GM-CSF induces the local differentiation and activation of AMs [14], and this could be another participating mechanism in the observed efficient protective activity of AdGM-CSF. High expression of F4/80 has been associated with the presence of inflammatory macrophages, over-expression of MHC II, CD80, CD11b and increased production of NO and IL-12 [24,25]. Similarly, F4/80 has been related to the early formation of granulomas induced by bacillus Calmette–Guérin (BCG) [26]. Thus, we used the F4/80 marker to monitor macrophages determined by IHC in different histological compartments (perivascular, peribronchial and alveolar–capillary interstitium areas) and lesions (pneumonia and granulomas). In comparison with the control group, mice treated with AdGM-CSF showed significant earlier and higher recruitment of activated AM F4/80+ in perivascular, peribronchial and interstitial areas at day 1. This significantly higher recruitment of AM F4/80+ remained in perivascular and peribronchial areas at day 3 (Fig. 4b). At day 14 post-infection, significantly higher AM F4/80+ was detected only in the interstitial area in the group treated with AdGM-CSF compared with the control group. At day 28, in the peribronchial and interstitial areas a significantly higher number of F4/80-positive cells was seen in the AdGM-CSF treated group. At late infection, day 60, in the peribronchial and pneumonic areas the number of F4/80-positive AMs was significantly higher in the AdGM-CSF-treated group (Fig. 4b,c), while in granulomas this group showed a significantly earlier and higher recruitment of AMs at days 21 and 28 (Fig. 4b,d).
In human and murine TB, protective immunity is mediated by Th1 responses (IFN-γ, IL-12) and activated macrophages which produce TNF-α and NO by activity of the enzyme-iNOS [27]. GM-CSF promotes the production of Th1 cytokines [11,12]. Thus, higher induction of the Th1 response is another mechanism which could be involved in the AdGM-CSF protective activity. Murine lungs from each treated group were homogenized for total RNA extraction and subsequent retrotranscription and real-time PCR (qRT–PCR) to quantify the transcription of GM-CSF, IFN-γ, IL-12, TNF-α and iNOS. In comparison with the control group, mice treated with AdGM-CSF showed higher transcription of GM-CSF, which began at the same time as IL-12 expression (Fig. 5a). Similarly, an earlier and significantly higher transcription of IFN-γ, TNF-α and iNOS was seen in the AdGM-CSF-treated group, showing a peak at days 21 and 90 post-infection (Fig. 5a). The cellular production of GM-CSF protein was evaluated by IHC. In the group treated with AdGM-CSF, the bronchial and bronchiolar epithelium showed strong GM-CSF immunostaining at various examined time-points, while by comparison with the control group the immunostaining was much weaker at these time-points (Fig. 5b). Animals treated with AdGM-CSF showed GM-CSF immunostaining at days 1 and 3 post-infection in pneumocytes type II, while AMs were negative at the same time-points and showed the strongest positivity later, suggesting that these cells were the most important GM-CSF cellular source. In contrast, the lungs from the control group showed GM-CSF positivity until 14 day post-infection in type II pneumocytes and AMs (Fig. 5c). In granulomas, mice treated with AdGM-CSF showed immunostained macrophages with heterogeneous distribution, predominantly in the granuloma periphery, until day 28 post-infection. A similar distribution was seen in the control group until day 60 post-infection (Fig. 5d).
Figure 5.

Effect of a single administration of adenoviruses encoding granulocyte–macrophage colony-stimulating factor (AdGM-CSF) on the cytokines and inducible nitric oxide synthase (iNOS) gene transcription in the murine model of progressive pulmonary tuberculosis (TB). (a) Groups of mice infected by the intratracheal (i.t.) route with a high dose of Mycobacterium tuberculosis (Mtb) to induce progressive TB were treated 1 day before with AdGM-CSF (black symbols) or Addl70-3 (white symbols), and their lungs were used to isolate total RNA and quantify the expression of the indicated cytokines by real-time reverse transcription–polymerase chain reaction (RT–PCR). Animals treated with AdGM-CSF showed a higher expression of cytokines and iNOS. Results are expressed as means ± standard deviation (s.d.) of two independent experiments with five mice per group; *P < 0·05. Representative immunohistochemistry of GM-CSF cellular detection in treated animals with AdGM-CSF or Add170-3 at different time-points in bronchial epithelium (b), alveolar–capillary interstitium (c) and granulomas (d); more immunostained cells are observed in animals treated with AdGM-CSF, the strongest being labelled the airways epithelium; scale bar represents 20 μm (439 × 475 mm; 200 × 200 DPI).
Effect of transgenic GM-CSF expression on reactivation of chronic LTB infection
When female B6D2F1 mice are infected by the i.t. route with a low dose of Mtb strain H37Rv, a chronic form of TB, similar to latent infection, is produced [22]. It is characterized by a low and stable lung bacillary load (fewer than 500 CFU) without weight loss, tissue damage, spontaneous reactivation or death. If a 3-mg/l concentration of corticosterone is administered by drinking water during late infection (7 months), supraphysiological plasma levels are reached, and the disease reactivation is then manifested by an increment of bacilli growth and progressive pneumonia. This chronic/latent infection model was used to determine if transgenic expression of GM-CSF by AdGM-CSF delivered at 7 months post-infection could prevent reactivation. One month after AdGM-CSF delivery, reactivation was induced with cortisone administered for 1 month in drinking water. Pulmonary Mtb burdens and histopathology were evaluated at 9 months post-infection. Mice treated with AdGM-CSF showed significantly lesser bacterial loads and exhibited lower pneumonic areas than mice that received the control vector Addl70-3 (Fig. 6a,b).
Figure 6.
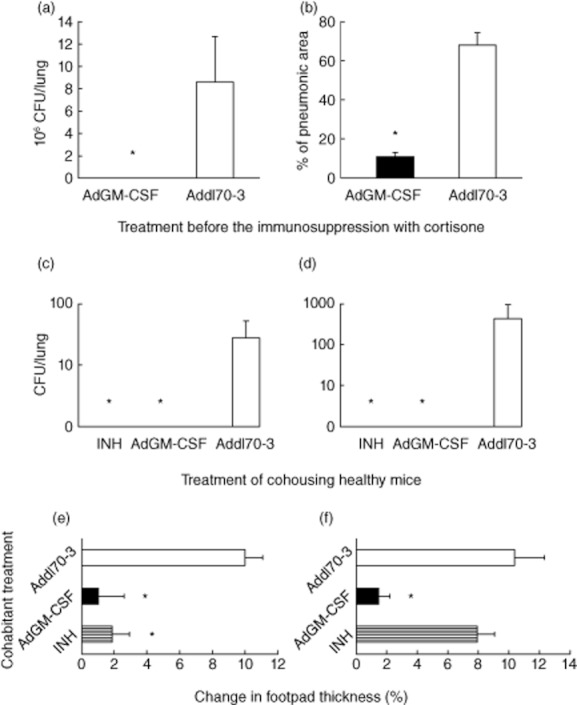
Adenovirus encoding granulocyte–macrophage colony-stimulating factor (AdGM-CSF) treatment prevents reactivation of chronic tuberculosis (TB) similar to latent infection following immunosuppression and prevention of transmission in co-housed healthy mice. (a) Mice were infected with low-dose Mycobacterium tuberculosis (Mtb) H37Rv to induce chronic infection similar to latent infection, and after 7 months animals were treated with AdGM-CSF (black bars) or Addl70-3 (white bars); after 1 month mice were treated with cortisone for 1 month to induce disease reactivation. Animals treated with AdGM-CSF showed lower pulmonary bacilli loads and (b) lesser tissue damage (pneumonia) than mice treated with Addl70-3. (c) With regard to evaluation of the effect of AdGM-CSF treatment in the prevention of transmission, groups of mice were infected with mild virulent Mtb strain H37Rv or (d) with the highly virulent Beijing strain 9001000 and co-housed with healthy mice treated with isoniazid (INH; hatched bar), AdGM-CSF (black bar) or Addl70-3(white bar) for 2 months; the lungs were used for bacilli burden determination by quantification of colony-forming units (CFUs). In comparison with animals treated with Addl70-3, the lungs of animals treated with INH or AdGM-CSF do not show bacilli growth. (e) Significantly lower delayed-type hypersensitivity (DTH) was measured in animals treated with INH or AdGM-CSF than mice treated with Addl70-3 that were co-housed with mice infected with Mtb strain H37Rv. (f) In contrast, there was no DTH difference in animals treated with INH or Addl70-3 when co-housed with mice infected with highly virulent Beijing strain, while animals treated with AdGM-CSF showed significantly lower DTH. All values are means ± standard deviation (s.d.) from two independent experiments with five mice per group;*P < 0·05 (561 × 632 mm; 200 × 200 DPI).
Effect of transgenic GM-CSF expression on the transmission of Mtb from infected to healthy mice
Our transmissibility model is based on long co-housing between infected and healthy mice [23]. This model was used to determine whether or not AdGM-CSF treatment was able to prevent infection of healthy mice co-housed with animals infected with moderate (H37Rv) or highly virulent (Beijing 900–1000) strains. The negative control group was represented by mice treated with Addl70-3, while the positive control animals received antibiotic INH treatment. Cultures of lung homogenates from healthy mice (contacts) treated with AdGM-CSF or INH did not show Mtb growth after 2 months of co-housing with infected mice with H37Rv (Fig. 6c) or Beijing (Fig. 6d) strains. In contrast, contact mice treated with Addl70-3 showed bacillary loads after 2 months of co-housing, being highest in the group that co-housed with mice infected with the highly virulent strain Beijing 900–1000. As an independent verification of the status of Mtb transmission, cutaneous DTH response to mycobacterial antigens was also performed in healthy contact mice after 2 months of co-housing with infected mice. Co-housed contact mice with H37Rv-infected animals treated with INH or AdGM-CSF showed a significantly lower DTH response than contact mice treated with Addl70-3 (Fig. 6e). Co-housed contact mice with animals infected with the highly virulent Beijing strain and treated with AdGM-CSF showed significantly less DTH than contact mice treated with INH or Add170-3, and no difference was found between these control groups (Fig. 6f).
Discussion
GM-CSF was first identified in mouse lung following lipopolysaccharide injection by its ability to stimulate proliferation of bone marrow cells and generate colonies of granulocytes and macrophages [28]. Interestingly, mice with homozygous deletion of the GM-CSF gene develop normally without significant alteration of haematopoiesis, but they develop lung abnormalities such as extensive accumulation of pulmonary surfactant phospholipids and increased susceptibility to opportunistic bacterial and fungal infections [13]. Thus, GM-CSF has significant physiological and immunological regulatory activity in the lung, such as increasing myeloperoxidase activity in neutrophils [29], stimulating differentiation and activation of macrophages and Toll-like receptor 4 (TLR-4) expression and T cell activation [30]. Indeed, most of the GM-CSF-mediated effects on T cells are believed to be exerted indirectly through antigen-presenting cells (APCs). Regarding its participation in mycobacterial infection, it was reported that mice lacking GM-CSF died rapidly, showing severe necrosis when exposed to an aerosol-delivered Mtb infection because of their inability to mount a Th1 response [31]. Our results confirm and extend these observations, by showing progressive GM-CSF expression during early infection peaking at day 7 and maintaining high levels until day 28, which coincide with the transitory bacilli growth control in this model mediated by a predominant Th1 response and proinflammatory cytokine expression [19,20].
GM-CSF can be produced by a wide variety of cells, including macrophages, fibroblasts, endothelial cells, T cells, mesothelial and epithelial cells, among others [32]. In these cells, bacterial antigens and inflammatory cytokines such as IL-1, IL-6 and TNF-α are potent inducers [33,34]. Thus, these factors should contribute to the observed higher expression of this growth factor, considering that all these cytokines are produced preferentially during the first month after infection in this model [20]. During late stages of the disease there was a progressive decrease of GM-CSF expression, showing its lowest level at day 120 post-infection. The expression of GM-CSF can be inhibited by IL-10 and IL-4 [35] or glucocorticoids [36]. These anti-inflammatory factors might be related to GM-CSF decrease during advanced disease, when there is a significant decline in immune protection in this model co-existing with extensive tissue damage and high bacilli burdens [19,20].
The significant participation of GM-CSF in Mtb infection was supported by our current results obtained after the administration of only one dose of recombinant adenoviruses expressing this cytokine, 1 day before infection with Mtb. This pretreatment resulted in efficient GM-CSF transgenic expression in the airways epithelium that induced fourfold more activated DCs and significantly higher expression of the immune protective factors TNF-α, IL-12, IFN-γ and iNOS, which led to more and bigger granulomas, more AMs F4/80+, lesser pneumonia and a prolonged decrease in bacilli loads. Thus, although GM-CSF is produced preferentially in our model during early infection it appears not to be enough, as is indicated by the late emergence of activated DCs, and overproduction by AdGM-CSF administration boosts the Th1 cells and DC activation, producing a significant improvement in the immune protective response.
Cytokines involved in the activation of Th1 lymphocytes can be used clinically as a form of immunotherapy to increase anti-mycobacterial activity through activation of DCs, AMs and T lymphocytes. In this regard, the use of recombinant cytokines such as IFN-γ [37] or aerosolized IFN-α [38], in conjunction with antibiotics, produced clinical improvement in patients with TB. Using the same murine TB model, it was showed that gene therapy based on adenoviruses encoding IFN-γ controlled disease progression successfully in mice infected with drug-sensible and drug-resistant strains [21]. Furthermore, vaccines based on recombinant viral vectors encoding mycobacterial antigens [39] or vaccines based on recombinant BCG [40,41] have shown effectiveness in experimental models of TB, and have also demonstrated that the respiratory mucosal route of administration is the best way to induce an efficient protective immune response against respiratory infections. This is greatly augmented when GM-CSF is used as adjuvant [42], reaching improved protection against disseminated infection associated with expansion and activation of APCs [40,41]. Moreover, it has been shown recently that recombinant BCG expressing GM-CSF is highly efficient in preventing TB in a mouse model [41], inducing a similar cytokine response that we observed when AdGM-CSF was administered 1 day before infection. In fact, administration of GM-CSF, or inducing its expression, has been useful in the treatment against other pulmonary infections such as aspergillosis [43], Chlamydia trachomatis [44] and pneumococcal pneumonia [45]. Moreover, the delivery of recombinant human GM-CSF by inhalation was shown to be beneficial in the treatment of alveolar proteinosis and it was well tolerated in healthy subjects [46]. However, there are no available data about their prophylactic use to augment anti-micobacterial host defence for mainly immunodeficient patients. In line with this, one disadvantage is that recombinant cytokines are extremely expensive and have a short in-vivo half-life. Thus, as shown in our current study, the use of only one dose of adenoviruses encoding GM-CSF could have a potential benefit during early Mtb infection.
Both adenoviral vectors AdGM-CSF and Addl70-3 have been well characterized; they are expressed temporarily in the lung (for 12–14 days) [47] and essentially infect the respiratory epithelium resulting in detectable GM-CSF in the lung until the third week post-administration, without evidence of serum increments, when administered in healthy mice. This transient expression is important, considering that transgenic mice that over-express GM-CSF showed macrophage accumulation, blindness and severe damage to various tissues [48]; these mice also exhibited a significant increment of many cytokines and inflammatory mediators, and failed to focus T cells and macrophages into sites of Mtb infection [31], suggesting that high and permanent expression of GM-CSF leads to defects in cytokine and chemokine regulation. Therefore, excess of GM-CSF does not induce an overly Th1 response, and very fine control of this cytokine is needed to fight infections. With regard to Addl70-3, they did not induce detectable levels of GM-CSF and did not cause significant viral-mediated inflammation [47].
Our experimental results suggest that the immunostimulatory effect of GM-CSF promotes not only protection against primary TB, but also prevention of LTB reactivation. Compared with mice treated with Addl70-3, a single dose of AdGM-CSF was able to prevent reactivation after immunosuppression induced by the administration of corticosterone in mice, with chronic infection similar to latent infection. These preclinical results offer an alternative for future immunotherapeutic trials in high-risk individuals preventing reactivation of LTB, such as patients with rheumatological diseases treated with anti-TNF-α therapy [49], or HIV/AIDS patients who can develop TB after primary infection or after LTB reactivation. However, this therapeutic approach in HIV infection should be taken with some caution, considering that some studies have shown an increase of viral replication in HIV-infected patients due to higher immune cell activation induced by GM-CSF [50], while others report beneficial therapeutic effects induced by this cytokine [51].
Another clinical scenario for the potential application of gene therapy codifying cytokines such as GM-CSF is to prevent infection in the healthy contacts of TB patients. The World Health Organization (WHO) recommends the screening of household contacts from a TB source case to identify infected individuals, principally children under 6 years of age, in order to supply prompt treatment to ill individuals and provide preventive chemotherapy to subjects who do not develop the disease [1]. The only current alternative to BCG vaccination for preventing TB is chemoprophylaxis, particularly with INH administered for at least 6 months. However, a systematic review of TB prophylaxis in HIV-infected adults showed that INH reduced the incidence of TB in those subjects with positive DTH, but was ineffective in those with negative DTH, suggesting that chemoprophylaxis does not prevent primary infection [7]. Our current results in the BALB/c mouse model of transmissibility confirm this suggestion, particularly in animals co-housed with mice infected with the highly virulent Beijing strain, as contact mice that were treated with INH chemoprophylaxis did not show any statistically significant difference in DTH compared with the Addl70-3 control group, confirming that INH did not prevent primary infection, although it inhibited the growth and viability of Mtb efficiently. In contrast, administration of AdGM-CSF was more efficient in preventing transmission, as suggested by the lower DTH without detectable CFU exhibited by treated contact mice. These results are in agreement with clinical trials which have shown that administration of INH as chemoprophylaxis in children living in a TB-endemic zone did not reduce the risk of infection, illness or death due to TB [52]. However, INH has shown effectiveness in preventing active TB in individuals who have had contact with TB patients [53]. Considering the increased prevalence of MDR and XDR strains, our results, using the murine transmissibility model, suggest that gene-based immunotherapeutics using adenoviruses encoding GM-CSF could be useful to prevent infection in close contacts of TB patients. In addition, chemoprophylaxis with INH takes 6–9 months and is potentially hepatotoxic; substitution by only one administration of AdGM-CSF without toxic effects could have significant implications in the control of the transmissibility of this disease.
In conclusion, GM-CSF is a significant cytokine in the immune protection against Mtb. Gene therapy based on adenoviruses encoding GM-CSF increased protective immunity when administered in a single dose 1 day before Mtb infection in BALB/c mice, after recombinant adenoviruses infected the airways epithelium and macrophages increased production of GM-CSF, which induced rapid and efficient activation of DCs that enhanced the production of IFN-γ, TNF-α and iNOS, permitting the efficient control of bacilli growth. The same treatment was effective in preventing LTB reactivation and transmission. Whether administration of AdGM-CSF during late progressive disease could also be beneficial in drug-susceptible and drug-resistant disease is currently under investigation in our laboratory.
Acknowledgments
We thank I. Estrada-García, Y. Serafín-López, L. Sánchez-Torres, M. Perez-Tapia, J. Luna and L. Flores-Romo for valuable suggestions and help. A.F.C. was responsible for design, performance and data interpretation of the study, and wrote the primary writing of the manuscript. D.A.M.E. performed and analysed experiments. Z.X. kindly provided and characterized AdGM-CSF and Addl70-3. R.H.P conceived, designed and provided funding of the study and reviewed the final manuscript. The authors thank Cristina Parada for her technical assistance. The study was supported by Consejo Nacional de Ciencia y Tecnología (CONACyT; contract 84456). A.F.C. is a recipient of post-graduate fellowship by CONACyT.
Disclosure
The authors have no conflicting financial interests.
References
- 1.Geneva, Switzerland: World Health Organization; 2009. Global tuberculosis control: epidemiology, strategy, financing. WHO report 2009. [Google Scholar]
- 2.Parrish NM, Dick JD, Bishai WR. Mechanisms of latency in Mycobacterium tuberculosis. Trends Microbiol. 1998;6:107–112. doi: 10.1016/s0966-842x(98)01216-5. [DOI] [PubMed] [Google Scholar]
- 3.Tufariello J, Chan J, Flynn J. Latent tuberculosis: mechanisms of host and bacillus that contribute to persistent infection. Lancet Infect Dis. 2003;3:578–590. doi: 10.1016/s1473-3099(03)00741-2. [DOI] [PubMed] [Google Scholar]
- 4.Kaufmann SH. How can immunology contribute to the control of tuberculosis? Nat Rev Immunol. 2001;1:20–30. doi: 10.1038/35095558. [DOI] [PubMed] [Google Scholar]
- 5.Nakaoka H, Lawson L, Squire SB. Risk for tuberculosis among children. Emerg Infect Dis. 2006;12:1383–1388. doi: 10.3201/eid1209.051606. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khalilzadeh S, Masjedi H, Hosseini M, Safavi A, Masjedi MR. Transmission of Mycobacterium tuberculosis to households of tuberculosis patients: a comprehensive contact tracing study. Arch Iran Med. 2006;9:208–212. [PubMed] [Google Scholar]
- 7.Akolo C, Adetifa I, Shepperd S, Volmink J. Treatment of latent tuberculosis infection in HIV infected persons. Cochrane Database Syst Rev. 2010;(1) doi: 10.1002/14651858.CD000171.pub3. CD000171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaplan JE, Benson C, Holmes KH, Brooks JT, Pau A, Masur H Centers for Disease Control and Prevention (CDC); National Institutes of Health; HIV Medicine Association of the Infectious Diseases Society of America. Guidelines for prevention and treatment of opportunistic infections in HIV-infected adults and adolescents: recommendations from CDC, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. MMWR Recomm Rep. 2009;58:1–207. [PubMed] [Google Scholar]
- 9.Quigley MA, Mwinga A, Hosp M. Long-term effect of preventive therapy for tuberculosis in a cohort of HIV-infected Zambian adults. AIDS. 2001;15:215–222. doi: 10.1097/00002030-200101260-00011. et al. [DOI] [PubMed] [Google Scholar]
- 10.Balcells ME, Thomas SL, Godfrey-Faussett P, Grant AD. Isoniazid preventive therapy and risk for resistant tuberculosis. Emerg Infect Dis. 2006;12:744–751. doi: 10.3201/eid1205.050681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. 2008;8:533–544. doi: 10.1038/nri2356. [DOI] [PubMed] [Google Scholar]
- 12.Krausgruber T, Blazek K, Smallie T. IRF5 promotes inflammatory macrophage polarization and TH1–TH17 responses. Nat Immunol. 2011;12:231–238. doi: 10.1038/ni.1990. et al. [DOI] [PubMed] [Google Scholar]
- 13.Stanley E, Lieschke GJ, Grail D. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci USA. 1994;91:5592–5596. doi: 10.1073/pnas.91.12.5592. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rose RM, Kobzik L, Dushay K. The effect of aerosolized recombinant human granulocyte macrophage colony-stimulating factor on lung leukocytes in nonhuman primates. Am Rev Respir Dis. 1992;146:1279–1286. doi: 10.1164/ajrccm/146.5_Pt_1.1279. et al. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Snider DP, Hewlett BR. Transgenic expression of granulocyte–macrophage colony-stimulating factor induces the differentiation and activation of a novel dendritic cell population in the lung. Blood. 2000;95:2337–2345. et al. [PubMed] [Google Scholar]
- 16.Miller G, Pillarisetty VG, Shah AB, Lahrs S, Xing Z, DeMatteo RP. Endogenous granulocyte–macrophage colony-stimulating factor overexpression in vivo results in the long-term recruitment of a distinct dendritic cell population with enhanced immunostimulatory function. J Immunol. 2002;169:2875–2885. doi: 10.4049/jimmunol.169.6.2875. [DOI] [PubMed] [Google Scholar]
- 17.García-Romo GS, Pedroza-González A, Aguilar-León D. Airways infection with virulent Mycobacterium tuberculosis delays the influx of dendritic cells and the expression of costimulatory molecules in mediastinal lymph nodes. Immunology. 2004;112:661–668. doi: 10.1046/j.1365-2567.2004.01904.x. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balboa L, Romero MM, Yokobori N. Mycobacterium tuberculosis impairs dendritic cell response by altering CD1b, DC-SIGN and MR profile. Immunol Cell Biol. 2010;88:716–726. doi: 10.1038/icb.2010.22. et al. [DOI] [PubMed] [Google Scholar]
- 19.Hernández-Pando R, Orozco H, Sampieri A. Correlation between the kinetics of Th1/Th2 cells and pathology in a murine model of experimental pulmonary tuberculosis. Immunology. 1996;89:26–33. et al. [PMC free article] [PubMed] [Google Scholar]
- 20.Hernández-Pando R, Orozco EH, Arriaga AK, Sampieri A, Larriva-Sahd J, Madrid MV. Analysis of the local kinetics and localization of interleukin-1α, tumor necrosis factor-α and transforming growth factor-β, during the course of experimental pulmonary tuberculosis. Immunology. 1997;90:607–617. doi: 10.1046/j.1365-2567.1997.00193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mata-Espinosa DA, Mendoza-Rodríguez V, Aguilar-León D, Rosales R, López-Casillas F, Hernández-Pando R. Therapeutic effect of recombinant adenovirus encoding interferon-gamma in a murine model of progressive pulmonary tuberculosis. Mol Ther. 2008;16:1065–1072. doi: 10.1038/mt.2008.69. [DOI] [PubMed] [Google Scholar]
- 22.Arriaga AK, Orozco EH, Aguilar LD, Rook GA, Hernández-Pando R. Immunological and pathological comparative analysis between experimental latent tuberculous infection and progressive pulmonary tuberculosis. Clin Exp Immunol. 2002;128:229–237. doi: 10.1046/j.1365-2249.2002.01832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marquina-Castillo B, García-García L, Ponce-de-León A. Virulence, immunopathology and transmissibility of selected strains of Mycobacterium tuberculosis in a murine model. Immunology. 2009;128:123–133. doi: 10.1111/j.1365-2567.2008.03004.x. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lifshitz L, Tabak G, Gassmann M, Mittelman M, Neumann D. Macrophages as novel target cells for erythropoietin. Haematologica. 2010;95:1823–1831. doi: 10.3324/haematol.2010.025015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghosn EE, Cassado AA, Govoni GR. Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proc Natl Acad Sci USA. 2010;107:2568–2573. doi: 10.1073/pnas.0915000107. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Egen JG, Rothfuchs AG, Feng CG, Winter N, Sher A, Germain RN. Macrophage and T cell dynamics during the development and disintegration of mycobacterial granulomas. Immunity. 2008;28:271–284. doi: 10.1016/j.immuni.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cooper AM, Flynn JL. The protective immune response to Mycobacterium tuberculosis. Curr Opin Immunol. 1995;7:512–516. doi: 10.1016/0952-7915(95)80096-4. [DOI] [PubMed] [Google Scholar]
- 28.Burgess AW, Camakaris J, Metcalf D. Purification and properties of colony-stimulating factor from mouse lung-conditioned medium. J Biol Chem. 1977;252:1998–2003. [PubMed] [Google Scholar]
- 29.Frossard JL, Saluja AK, Mach N. In vivo evidence for the role of GM-CSF as a mediator in acute pancreatitis-associated lung injury. Am J Physiol Lung Cell Mol Physiol. 2002;283:L541–548. doi: 10.1152/ajplung.00413.2001. et al. [DOI] [PubMed] [Google Scholar]
- 30.Wada H, Noguchi Y, Marino MW, Dunn AR, Old LJ. T cell functions in granulocyte/macrophage colony-stimulating factor deficient mice. Proc Natl Acad Sci USA. 1997;94:12557–12561. doi: 10.1073/pnas.94.23.12557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonzalez-Juarrero M, Hattle JM, Izzo A. Disruption of granulocyte macrophage-colony stimulating factor production in the lungs severely affects the ability of mice to control Mycobacterium tuberculosis infection. J Leukoc Biol. 2005;77:914–922. doi: 10.1189/jlb.1204723. et al. [DOI] [PubMed] [Google Scholar]
- 32.Griffin JD, Cannistra SA, Sullivan R, Demetri GD, Ernst TJ, Kanakura Y. The biology of GM-CSF: regulation of production and interaction with its receptor. Int J Cell Cloning. 1990;8(Suppl. 1):35–44. doi: 10.1002/stem.5530080705. [DOI] [PubMed] [Google Scholar]
- 33.Huleihel M, Douvdevani A, Segal S, Apte RN. Different regulatory levels are involved in the generation of hemopoietic cytokines (CSFs and IL-6) in fibroblasts stimulated by inflammatory products. Cytokine. 1993;5:47–56. doi: 10.1016/1043-4666(93)90023-x. [DOI] [PubMed] [Google Scholar]
- 34.Shi Y, Liu C, Roberts A. Garnulocyte–macrophage colony-stimulating factor and T cell responses; what we do and don't know. Cell Res. 2006;16:126–133. doi: 10.1038/sj.cr.7310017. et al. [DOI] [PubMed] [Google Scholar]
- 35.Sagawa K, Mochizuki M, Sugita S, Nagai K, Sudo T, Itoh K. Suppression by IL-10 and IL-4 of cytokine production induced by two-way autologous mixed lymphocyte reaction. Cytokine. 1996;8:501–506. doi: 10.1006/cyto.1996.0068. [DOI] [PubMed] [Google Scholar]
- 36.Adcock IM, Caramori G. Cross-talk between pro-inflammatory transcription factors and glucocorticoids. Immunol Cell Biol. 2001;79:376–384. doi: 10.1046/j.1440-1711.2001.01025.x. [DOI] [PubMed] [Google Scholar]
- 37.Condos R, Rom WH, Schluger NW. Treatment of multidrug resistant pulmonary TB with interferon-gamma via aerosol. Lancet. 1997;349:1513–1515. doi: 10.1016/S0140-6736(96)12273-X. [DOI] [PubMed] [Google Scholar]
- 38.Giosué S, Casarini M, Alemanno L. Effects of aerosolized interferon-alpha in patients with pulmonary TB. Am J Respir Crit Care Med. 1998;158:1156–1162. doi: 10.1164/ajrccm.158.4.9803065. et al. [DOI] [PubMed] [Google Scholar]
- 39.Roediger EK, Kugathasan K, Zhang X, Lichty BD, Xing Z. Heterologous boosting of recombinant adenoviral prime immunization with a novel vesicular stomatitis virus-vectored tuberculosis vaccine. Mol Ther. 2008;16:1161–1169. doi: 10.1038/mt.2008.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ryan AA, Wozniak TM, Shklovskaya E. Improved protection against disseminated tuberculosis by Mycobacterium bovis bacillus Calmette–Guérin secreting murine GM-CSF is associated with expansion and activation of APCs. J Immunol. 2007;179:18–24. doi: 10.4049/jimmunol.179.12.8418. et al. [DOI] [PubMed] [Google Scholar]
- 41.Nambiar JK, Ryan AA, Kong CU, Britton WJ, Triccas JA. Modulation of pulmonary DC function by vaccine-encoded GM-CSF enhances protective immunity against Mycobacterium tuberculosis infection. Eur J Immunol. 2010;40:153–161. doi: 10.1002/eji.200939665. [DOI] [PubMed] [Google Scholar]
- 42.Zhang X, Divangahi M, Ngai P. Intramuscular immunization with a monogenic plasmid DNA tuberculosis vaccine: enhanced immunogenicity by electroporation and co-expression of GM-CSF transgene. Vaccine. 2007;25:1342–1352. doi: 10.1016/j.vaccine.2006.09.089. et al. [DOI] [PubMed] [Google Scholar]
- 43.Quezada G, Koshkina NV, Zweidler-McKay P, Zhou Z, Kontoyiannis DP, Kleinerman ES. Intranasal granulocyte–macrophage colony-stimulating factor reduces the Aspergillus burden in an immunosuppressed murine model of pulmonary aspergillosis. Antimicrob Agents Chemother. 2008;52:716–718. doi: 10.1128/AAC.00760-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu H, Xing Z, Brunham RC. GM-CSF transgene-based adjuvant allows the establishment of protective mucosal immunity following vaccination with inactivated Chlamydia trachomatis. J Immunol. 2002;169:6324–6331. doi: 10.4049/jimmunol.169.11.6324. [DOI] [PubMed] [Google Scholar]
- 45.Steinwede K, Tempelhof O, Bolte K. Local delivery of GM-CSF protects mice from lethal pneumococcal pneumonia. J Immunol. 2011;187:5346–5356. doi: 10.4049/jimmunol.1101413. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tazawa R, Trapnell BC, Inoue Y. Inhaled granulocyte/macrophage-colony stimulating factor as therapy for pulmonary alveolar proteinosis. Am J Respir Crit Care Med. 2010;181:1345–1354. doi: 10.1164/rccm.200906-0978OC. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lei XF, Ohkawara Y, Stämpfli MR. Compartmentalized transgene expression of granulocyte–macrophage colony-stimulating factor (GM-CSF) in mouse lung enhances allergic airways inflammation. Clin Exp Immunol. 1998;113:157–165. doi: 10.1046/j.1365-2249.1998.00652.x. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lang RA, Metcalf D, Cuthbertson RA. Transgenic mice expressing a hemopoietic growth factor gene (GM-CSF) develop accumulations of macrophages, blindness, and a fatal syndrome of tissue damage. Cell. 1987;51:675–686. doi: 10.1016/0092-8674(87)90136-x. et al. [DOI] [PubMed] [Google Scholar]
- 49.Keane J, Gershon S, Wise RP. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med. 2001;345:1098–1104. doi: 10.1056/NEJMoa011110. et al. [DOI] [PubMed] [Google Scholar]
- 50.McClure J, van't Wout AB, Tran T. Granulocyte–monocyte colony-stimulating factor upregulates HIV-1 replication in monocyte-derived macrophages cultured at low density. J Acquir Immune Defic Syndr. 2007;44:254–261. doi: 10.1097/QAI.0b013e318030f5c5. et al. [DOI] [PubMed] [Google Scholar]
- 51.Kedzierska K, Mak J, Mijch A. Granulocyte-macrophage colony-stimulating factor augments phagocytosis of Mycobacterium avium complex by human immunodeficiency virus type 1-infected monocytes/macrophages in vitro and in vivo. J Infect Dis. 2000;181:390–394. doi: 10.1086/315191. et al. [DOI] [PubMed] [Google Scholar]
- 52.Madhi SA, Nachman S, Violari A. Primary isoniazid prophylaxis against tuberculosis in HIV-exposed children. N Engl J Med. 2011;365:21–31. doi: 10.1056/NEJMoa1011214. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smieja MJ, Marchetti CA, Cook DJ, Smaill FM. Isoniazid for preventing tuberculosis in non-HIV infected persons. Cochrane Database Syst Rev. 2000;(2) doi: 10.1002/14651858.CD001363. CD001363. [DOI] [PMC free article] [PubMed] [Google Scholar]