

Abstract
Adefovir is an adenosine analogue approved by the Food and Drug Administration for the treatment of chronic hepatitis B. Mutations occurring in the hepatitis B virus (HBV) reverse transcriptase (rt) domains are shown to confer resistance to antiviral drugs. The role of the rtI233V mutation and adefovir resistance remains contradictory. In this study, it was attempted to evaluate the impact of putative rtI233V substitution on adefovir action by homology modeling and docking studies. The HBVrt nucleotide sequence containing rtI233V mutation was obtained from the treatment-naive chronic hepatitis B subject. The three dimensional model of HBV polymerase/rt was constructed using the HIV-1rt template (PDB code: 1RTD A) and the model was evaluated by the Ramachandran plot. Autodock was employed to dock the HBV polymerase/rt and adefovir. The modelled structure showed the amino acid rtI233 to be located away from the drug interactory site. The substitution of isoleucine to valine did not appear to affect the catalytic sites of the protein. In addition, it does not alter the conformation of bent structure formed by residues 235 to 240 that stabilizes the binding of dNTPs. Therefore, it was predicted that rtI233V substitution may not independently affect the antiviral action of adefovir and incoming dNTP binding.
Keywords: Hepatitis B virus, Adefovir, Mutation, Homology model, Docking, Drug resistance
Background
Hepatitis B virus (HBV) related liver disease is a global health problem [1]. Though there are several options for the treatment of chronic hepatitis B infection, management of HBV still remains a major challenge [2, 3]. Antiviral resistance is considered to be one of the most important factors associated with HBV treatment failure [4]. Antiviral resistance is primarily mediated by mutations in the antiviral target sites thereby altering the drug interactory mechanism. Identification and characterization of these resistant mutations is important for appropriate tailoring of therapy and the design of newer drugs to challenge the resistant strains [5]. Adefovir, a nucleotide analogue of adenosine is one of the therapeutic options for chronic hepatitis B infection. The nucleotide analogues lack the 3-hydroxyl group and the incorporation of these analogues prevents the formation of phosphodiester linkage that is essential for DNA elongation. It inhibits the enzymatic action of HBV reverse transcriptase (HBVrt) and thus acts as a chain terminator of DNA synthesis [6]. The primary adefovir-resistant mutations significantly associated with treatment failure are rtN236T and rtA181T/V [7, 8]. The role of rtI233V mutation and adefovir response remains contradictory. Some studies have shown rtI233V mutation to be associated with adefovir resistance [9, 10]. In another study, it was not shown to affect adefovir response [11].
Computational methods like molecular modeling and docking studies have helped researchers understand the structural features of protein, drug-protein interaction and the effect of resistance mutations and drug interaction [12–14]. Knowledge of HBV reverse transcriptase (HBVrt) structure would thus be valuable for understanding the molecular basis of drug resistance. We have previously reported the putative rtI233V mutation in 4 treatment-naive subjects [15]. It has been documented that rtI233V mutation occurs in approximately 2% of treatment-naive chronic hepatitis B virus carriers [16]. The three-dimensional (3D) polymerase model of HBV has shown to assist in understanding the interactions between HBV polymerase and the antiviral agents [17–19]. We attempted to study the impact of this putative rtI233V substitution and adefovir binding by molecular modeling and docking studies.
Methodology
Study subject:
Blood sample was collected from treatment-naive chronic hepatitis B subject after obtaining written informed consent to participate in the study. The subject was recruited as a part of the investigation to characterize HBV antiviral resistance mutations. The study was approved by the institutional review oard of Christian Medical College, Vellore.
HBV polymerase/rt gene PCR and sequencing:
HBV polymerase gene covering the entire rt region was amplified and sequenced as described previously [15]. Obtained bidirectional sequences were analyzed using BioEdit v7.0.9 and the consensus was generated. The nucleotide sequence has been deposited in GenBank with the accession number GU799007.
Homology model of hepatitis B virus polymerase/rt:
Homology model of HBVrt was built in MODELLER 9v8 using the crystal structure of HIV-1rt template [Protein Data Bank (PDB) code: 1RTD chain A]. The nucleotide sequences were translated into the amino acid sequences using BioEdit v7.0.9. The amino acid sequence containing rtI233V mutation was substituted with valine for the construction of wild type model for comparison. The translated target sequences were aligned with HIV-1rt template using ClustalW (http://www.ebi.ac.uk/Tools/msa/clustalw2/). The target-template alignments were used to build the three-dimensional model of target protein. At least five models were generated and the model with lowest Discrete Optimization Protein Energy (DOPE) is selected. The structure validation was performed in PROCHECK using the Structure Analysis and Verification Server (http://nihserver.mbi.ucla.edu/SAVES/).
Molecular docking studies:
To the modelled protein, the two magnesium (Mg2+) ions and the template primer DNA duplex [d (GCXCCGGCGCTC)- d(GAGCGCCGG)] were located based on the co-ordinates of PDB: 1RTD chain A of HIV-1rt. The ‘X’ in the DNA duplex was substituted to the complementary base of the rt inhibitor adefovir (adenosine analogue, X=G). The docking studies of HBV polymerase/rt wild type and mutant models with adefovir were performed using Autodock (v1.5.2). All the possible torsion angles in the ligand molecules were set to rotate freely and polar hydrogen molecules were added. Kollman united atom partial charges were assigned for the receptor. Grid box was generated at the centre of the protein with the grid box size of 46, 40 and 40 Å for x, y and z respectively.
Lamarckian genetic algorithm was used for docking analysis with the population level of 200 and number of evals set to long. All other parameters were set to default and the best docking complex was identified using root mean square deviation (RMSD) cluster analysis. Based on the binding free energy best binding pose was identified. PyMOL molecular visualization tool was used to analyze the interactions between entecavir and the target protein [20].
Results and Discussion
Initially for model building, protein BLAST of the query (target) sequence showed close identity to HIV-2 rt (PDB: 1MU2 B) and Moloney murine leukemia virus (MULV) rt (PDB: 1NND A). The query coverage (39% and 58% respectively) and identity score (E value: 0.009 and 0.022 respectively) for these templates were good when compared to previously described template of HIV-1rt [PDB: 1RTD A (query coverage 22% and E value 0.75)]. However, the three aspartate amino acids that form the catalytic sites in HIV-1 rt is well conserved in HBVrt amino acid positions 85 (A domain), 203 and 204 (C domain). Likewise, most of the amino acids interacting with the template primer and the incoming dNTP substrates are conserved in both HIV- 1rt and HBVrt [16]. Moreover, the nucleos(t)ide analogues lamivudine, adefovir and tenofovir used for chronic HBV treatment were initially developed for HIV infection and their drug interactory mechanisms are very well documented [20, 21]. Therefore, modeling and docking studies of HBV using HIV-1rt template would be a suitable model for the prediction of drug resistance as demonstrated previously [16–18].
Homology model of HBV polymerase/rt:
As described for HIVrt the modelled HBV polymerase has fingers, palm and thumb subdomains. According to the nomenclature of Stuyver et al. [21] the fingers subdomain covers the HBVrt codons 1 to 55 and 121 to 171, palm region extends between 56 to 92 and 172 to 265 and thumb subdomain occupies position 266 to 344. The two magnesium (Mg2+) ions, thymidine triphoshate and the DNA template were located using the coordinates of PDB: 1RTD A of HIV-1rt.
Structure validation:
The model was evaluated by PROCHECK and the stereochemical quality of the structure was good with the overall G factor of -0.22. The Ramachandran plot shows the phi (φ)-psi (ψ) torsion angles for all residues except glycine and proline in the structure [22]. The distribution of φ, ψ angles showed 82% residues in the most favourable core region 13.8% of residues in allowed region and 3.4% residues in the generous region. Overall 99.1% of the residues were within the allowed region.
Effect of rtI233V mutation and adefovir action: Molecular modeling and docking analysis:
The modelled structure showed the amino acid position rtI233 to be located away from the drug interactory site. The substitution of isoleucine to valine did not show to affect the catalytic sites of aspartate residues at HBVrt positions 83, 205 and 206 respectively. However, as observed in the wild type (rtI233) model rtD83, rtD205 and rtN33 did not participate in the H-bond interaction with the ligand molecule and instead rtK32 formed H-bonds in the mutant model (Figure 1). The wild type model exhibited the best docking energy of -5.97 Kcal/mol and the rtI233V mutation decreased the docking score to less than 1 Kcal/mol (-5.19 Kcal/mol; Table 1 (see supplementary material). Therefore rtI233V mutation does not show any significant changes in the binding of adefovir.
Figure 1.
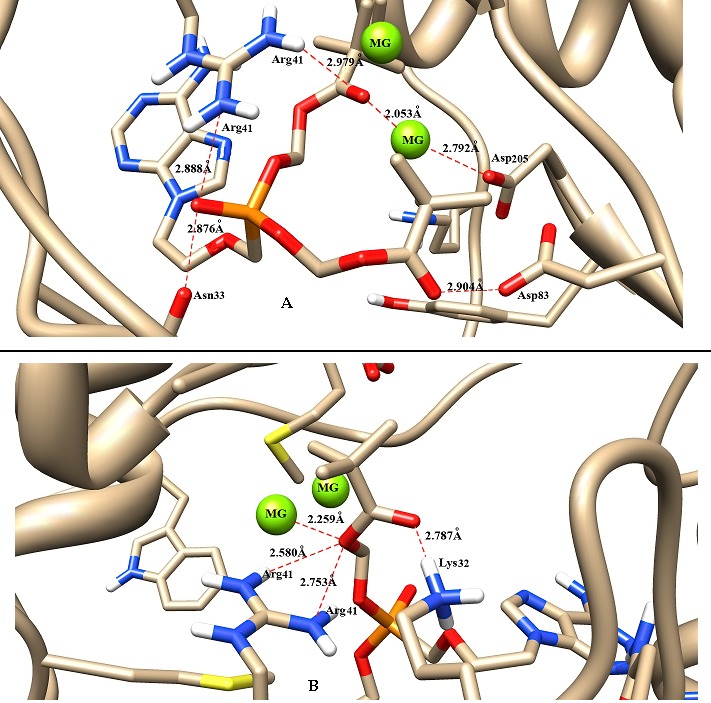
Impact of HBV rtI233V mutation and adefovir binding. Binding conformation of A) wild type (rtI233) and B) mutant (rtI233V) protein-ligand complex derived from Autodock. The ligand (adefovir) and interacting residues are shown in stick format and red dotted lines represent H-bond. The image was prepared using Chimera 1.6.2 software.
It has been proposed that residues 235 to 240 form a bent structure and stabilizes the binding of incoming dNTPs [23]. The wild type isoleucine (rtI233) is just located three amino acids away from the crucial adefovir resistance amino acid position asparagine (rtN236), which in-part forms the bent structure. It was further attempted to study whether rtI233V substitution would alter the relative positions of neighbouring residues and alter the conformation. In wild type model the relative distance of the bent structure formed by the HBVrt amino acids L235, N236, P237, N238, K239 and T240 is 7.8 angstrom (Å). Substitution of valine reduced its relative distance to 7.7 Å. The overall conformation of the bent structure is maintained and the 0.1 Å difference in relative distance may not impose a spatial constraint to dNTP binding (Figure 2). Therefore, it was predicted that rtI233V substitution in the reverse transcriptase domain may not affect the antiviral action of adefovir and dNTP binding. Furthermore, two subjects with pre-existing rtI233V mutation at baseline (treatment-naive) responded to lamivudine and entecavir subsequently in our center (unpublished data). This again shows that rtI233V mutation does not alter the antiviral efficacy to any of these drugs.
Figure 2.
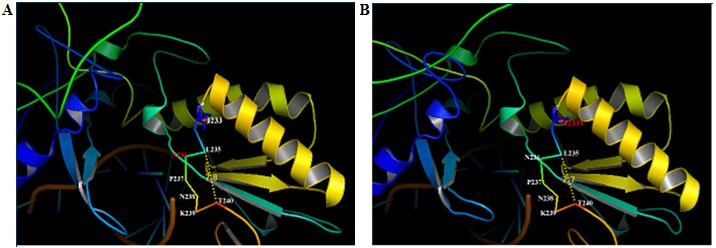
A) Homology model of HBV polymerase/rt wild type rtI233 was compared with; B) rtI233V mutation. The relative distance between the residues 235 to 240 crucial for dNTP binding that form the bent structure is shown as yellow dots. Substitution of valine for isoleucine (rtI233V) reduced its relative distance by only 0.1 Angstrom.
Conclusions
The rtI233V amino acid substitution did not show to alter the catalytic sites and adefovir binding. In addition, the conformation of bent structure formed by residues 235 to 240 that stabilizes the binding of dNTPs is maintained. Our prediction model enabled to identify the impact of rtI233V mutation which has been debated in the recent years. Adding evidence to the findings of Curtis et al. [10] we show that rtI233V mutation cannot affect the antiviral efficacy of adefovir.
Conflicts of interest
None declared
Supplementary material
Acknowledgments
The sequences were generated as part of HBV antiviral resistance mutations characterization study supported by Indian Council for Medical research (ICMR Grant No.5/8/7/7/2008-ECD-I).
Footnotes
Citation:Ismail et al, Bioinformation 9(3): 121-125 (2013)
References
- 1. http://www.who.int/mediacentre/factsheets/fs204/en/
- 2.Hoofnagle JH, et al. Hepatology. 2007;45:1056. doi: 10.1002/hep.21627. [DOI] [PubMed] [Google Scholar]
- 3.Mohamed R, et al. J Gastroenterol Hepatol. 2004;19:958. doi: 10.1111/j.1440-1746.2004.03420.x. [DOI] [PubMed] [Google Scholar]
- 4.Locarnini S, et al. J Hepatol. 2006;44:422. doi: 10.1016/j.jhep.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 5.Lok AS, et al. Hepatology. 2007;46:254. [Google Scholar]
- 6.Thomas H, et al. J Hepatol. 2003;39:S93. doi: 10.1016/s0168-8278(03)00207-1. [DOI] [PubMed] [Google Scholar]
- 7.Villeneuve JP, et al. J Hepatol. 2003;39:1085. doi: 10.1016/j.jhep.2003.09.022. [DOI] [PubMed] [Google Scholar]
- 8.Borroto-Esoda K, et al. J Hepatol. 2007;47:492. doi: 10.1016/j.jhep.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 9.Schildgen O, et al. N Engl J Med. 2006;354:1807. doi: 10.1056/NEJMoa051214. [DOI] [PubMed] [Google Scholar]
- 10.Schildgen O, et al. J Clin Microbiol. 2010;48:631. doi: 10.1128/JCM.01073-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Curtis M, et al. J Infect Dis. 2007;196:1483. doi: 10.1086/522521. [DOI] [PubMed] [Google Scholar]
- 12.Wang W, et al. Proc Natl Acad Sci U S A. 2001;98:14937. doi: 10.1073/pnas.251265598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Das K, et al. Proc Natl Acad Sci U S A. 2008;105:1466. doi: 10.1073/pnas.0711209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tse H, et al. Exp Biol Med (Maywood) 2011;236:915. doi: 10.1258/ebm.2011.010264. [DOI] [PubMed] [Google Scholar]
- 15.Ismail AM, et al. Intervirology. 2012;55:36. doi: 10.1159/000323521. [DOI] [PubMed] [Google Scholar]
- 16.Chang TT, Lai CL. N Engl J Med. 2006;355:322. doi: 10.1056/NEJMc066267. [DOI] [PubMed] [Google Scholar]
- 17.Das K, et al. J Virol. 2001;75:4771. doi: 10.1128/JVI.75.10.4771-4779.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langley DR, et al. J Virol. 2007;81:3992. doi: 10.1128/JVI.02395-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yadav V, Chu CK. Bioorg Med Chem Lett. 2004;14:4313. doi: 10.1016/j.bmcl.2004.05.075. [DOI] [PubMed] [Google Scholar]
- 20.Seeliger D, et al. J Comput Aided Mol Des. 2010;24:417. doi: 10.1007/s10822-010-9352-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stuyver LJ, et al. Hepatology. 2001;33:751. doi: 10.1053/jhep.2001.22166. [DOI] [PubMed] [Google Scholar]
- 22.Morris AL, et al. Proteins. 1992;12:345. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]
- 23.Warner N, et al. Antimicrob Agents Chemother. 2007;51:2285. doi: 10.1128/AAC.01499-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.