

Abstract
Community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA) has recently emerged as a nosocomial pathogen to the community which commonly causes skin and soft-tissue infections (SSTIs). This strain (MW2) has now become resistant to the most of the beta-lactam antibiotics; therefore it is the urgent need to identify the novel drug targets. Recently fructose 1,6 biphosphate aldolase-II (FBA) has been identified as potential drug target in CA-MRSA. The FBA catalyses the retro-ketolic cleavage of fructose-1,6-bisphosphate (FBP) to yield dihydroxyacetone phosphate (DHAP) and glyceraldehyde-3-phosphate (G3P) in glycolytic pathway. In the present research work the 3D structure of FBA was predicted using the homology modeling method followed by validation. The molecular dynamics simulation (MDS) of the predicted model was carried out using the 2000 ps time scale and 1000000 steps. The MDS results suggest that the modeled structure is stable. The predicted model of FBA was used for virtual screening against the NCI diversity subset-II ligand databases which contain 1364 compounds. Based on the docking energy scores, it was found that top four ligands i.e. ZINC01690699, ZINC13154304, ZINC29590257 and ZINC29590259 were having lower energy scores which reveal higher binding affinity towards the active site of FBA. These ligands might act as potent inhibitors for the FBA so that the menace of antimicrobial resistance in CA-MRSA can be conquered. However, pharmacological studies are required to confirm the inhibitory activity of these ligands against the FBA in CA-MRSA.
Keywords: Fructose biphosphate aldolase, CA-MRSA, Modeling, Molecular dynamics simulation, Docking, Virtual screening
Background
Methicillin-resistant Staphylococcus aureus (MRSA) is a common pathogen in healthcare facilities which frequently causes infections to the patients. Treatment of Staphylococcal infections is complicated by antibiotic resistant strains, most notably those with resistance to penicillin and γ-lactamase resistant γ-lactam antibiotics [1]. In the past decade, MRSA has emerged as a challenging pathogen in the community settings besides healthcare settings. In addition to the widespread problem of MRSA in hospitals, community-acquired methicillin-resistant S. aureus (CA-MRSA) has emerged as a nosocomial pathogen to the community [2]. The most common type of CA-MRSA infections are Skin and soft-tissue infections (SSTIs) which accounts for approximately 90% of cases, of which 90% are abscesses and/or cellulitis with purulent drainage [3]. CA-MRSA strains also appear to be especially virulent with the capacity to cause severe infections, such as necrotizing fasciitis, necrotizing pneumonia, bone and joint infections accompanied by septic thromboembolic disease, purpura fulminans with or without Waterhouse-Friderichsen syndrome, orbital cellulitis and endophthalmitis, infections of the central nervous system, bacteremia and endocarditis [4]. In comparison to infections caused by healthcare-associated MRSA (HA-MRSA) strains, CA-MRSA infections have been associated with lethal infections and worse clinical outcomes giving rise to the clinical impression that CA-MRSA strains are more virulent than other strains [5]. CA-MRSA has had intense impact on traditional therapy of suspected staphylococcal infection. Most beta-lactam antibiotics are no longer effective for a variety of common staphylococcal infections and skin and soft-tissue infections in particular. This underlines the high urgency to find novel treatment alternatives for drug resistant S. aureus [6].
Availability of the complete genome sequence of CA-MRSA (strain MW2) has paved the new way to identify the novel drug targets which have been unexplored yet. Recently Fructose 1,6 biphosphate aldolase-II (FBA) has been identified as potential drug target in CA-MRSA through metabolic pathways analysis [7]. FBA has also been reported as potential therapeutic drug target in Mycobacterium tuberculosis and Candida albicans [8, 9]. Fructose 1,6-biphosphate aldolase enzyme (EC: 4.1.2.13) catalyses the retro-ketolic cleavage of fructose-1,6-bisphosphate (FBP) to yield dihydroxyacetone phosphate (DHAP) and glyceraldehyde-3-phosphate (G3P) in glycolytic pathway. It also plays important role in gluconeogenesis and the Calvin cycle, in which it catalyses the reversible aldol condensation of DHAP with G3P [10, 11]. The present research work was undertaken to determine the 3D structure of FBA using molecular modeling method, to perform the molecular dynamics simulation of modelled structure, and to perform the virtual screening for the target using a ligand database.
Methodology
Molecular modeling:
The amino acid sequence of target protein fructose 1, 6- biphosphate aldolase (FBA) was retrieved from the UniProtKB database (Uniprot accession no: P67478) (www.uniprot.org). The length of target protein was 286 amino acid residues. In order to find the most similar template(s) for the target protein, database searching was performed against the PDB database using the BLASTp program [12]. Based on high sequence identity (76%) a template (pdb id: 3Q94) was selected, subsequently multiple sequence alignment was carried out between the target and template sequences using the clustalW2 program (http://www.ebi.ac.uk/Tools/msa/clustalw2/). The target-template aligned sequences were submitted in PIR/NBRF file format to the HHpred server (http://toolkit.tuebingen.mpg.de/hhpred). HHpred is an interactive server for protein homology detection and structure prediction [13]. The 3D structural models of FBA were predicted by the MODELLER 9v9 software from those target-template alignments (http://www.salilab.org/modeller). To evaluate and identify any anomalies in the predicted model of FBA, it was submitted to the Structural Analysis and Verification Server (http://nihserver.mbi.ucla.edu/SAVES/). SAVES is a metaserver for analyzing and validating protein structures which integrates five modules i.e. Procheck, What_check, Errat, Verify_3D and Prove. To improve the quality of predicted model of FBA, energy minimization was performed with the GROMOS 96 forcefield [14] implementation of DeepView v4.04 (spdb viewer) tool. This force field permits to evaluate the energy of the modeled structure as well as overhaul distorted geometries through energy minimization. All computations during energy minimization were done in vacuo, without reaction field. The predicted 3D structure of FBA was visualized by PyMOL v1.3 Viewer (http:// www.pymol.org/).
Molecular dynamics simulation:
The molecular dynamics simulation of modelled 3D structure of the Fructose 1,6 biphosphate aldolase was performed using the Gromacs ver 4.53 program [15] to track the motion of individual atoms. Two methods energy minimization and molecular dynamics were employed to optimize structure and simulate the natural motion of atoms respectively. Before starting simulation, Gromacs environment was set up and input files necessary for the simulation were prepared, and the structure was solvated in water, minimized and equlibrated. In order to prepare the topology from the pdb file, ‘pdb2gmx’ tool was used. The solvent water was added around the protein to generate a simulation box using the ‘genbox’ program. The dimensions of the box were set to as 0.9 nm from the protein molecules periphery. The energy minimization was performed in 1000 steps using the steepest descent minimization algorithm. GROMOS 96 force field [14] was chosen for the calculation of potential energy of the structure. A standard cutoff of the 1.0 nm, both for the neighbourlist generation and the Coulomb & Lennard-Jones interactions was employed. The system was neutralized by adding a Cl¯ ion in the model. To avoid the unnecessary distortion of the protein, an equilibrium run of water around the protein was performed using the 100 pico second (ps) time scale and 50000 steps (iterations). Finally molecular dynamics simulation was performed using the 2000 ps time scale and 1000000 steps at 300 °K temperature and 1 atm. pressure. The simulation results were analyzed using the ‘grace’ program.
Virtual screening:
In order to perform the virtual screening for fructose 1, 6- biphosphate aldolase (FBA) in CA-MRSA, the active site was predicted in the modelled structure using the metaPocket 2.0 server (http://projects.biotec.tu-dresden.de/pocket/). The metaPocket server [16, 17] is a meta server to identify ligand binding sites on protein surface based on a consensus method, in which the predicted binding sites from eight methods: LIGSITEcs, PASS, QSiteFinder, SURFNET, Fpocket, GHECOM, ConCavity and POCASA are combined together to improve the prediction success rate. In the 3D structure of 1, 6-biphosphate aldolase, 3 potential ligand binding sites were predicted and ranked according to their z-score. Out of all 3 predicted active sites, second active site was chosen for the screening of a set of ligand databases. This active site was predicted by LIGSITEcs (LCS) and SURFNET (SFN) methods and average was taken from these two methods. Using the protein-ligand docking method, virtual screening was performed for the fructose 1, 6- biphosphate aldolase against the NCI diversity subset-II molecules retrieved from the ZINC databases. ZINC is a free database of commercially-available compounds for virtual screening. ZINC contains over 21 million purchasable compounds in ready-to-dock, 3D formats. ZINC database is provided by the Shoichet Laboratory at the University of California, San Francisco (UCSF) (http://zinc.docking.org/) [18]. The virtual screening was carried out using the Autodockvina package (http://vina.scripps.edu/). Before performing the screening process, a set of 1,364 compounds (NCI Diversity subset-II) available in mol2 file format were converted into pdbqt file format using a small python script prepare_ligand4.py. The receptor molecule (target) was also converted into pdbqt format using prepare_receptor4.py script available in Autodock Tools package. After performing the virtual screening, a python script in the MGL tools package was used to analyze the best docked ligands for the fructose 1, 6- biphosphate aldolase (FBA) based on their energy score. Top ranked ligand-receptor complexes were further analyzed to study the protein-ligand interactions using the LigPlot+ v.1.4.3 program. LigPlot+ is a successor to the original LIGPLOT program for automatic generation of 2D ligand-protein interaction diagrams ( http://www.ebi.ac.uk/thornton-srv/software/LigPlus /) [19]. Using this program the hydrogen and hydrophobic interactions between the ligand and amino acid residues within the active site of the FBA were analyzed.
Discussion
Homology modeling and validation:
The availability of 3D structure of the target is foremost step to proceed for the structure-based drug designing. Since the 3D structure of fructose 1, 6-biphosphate aldolase (FBA) in CAMRSA is not yet available in the Protein Data Bank (PDB), therefore unknown structure of FBA was predicted by the homology modeling method using the HHPred server [13]. This server uses Modeller 9v9 program for 3D structure prediction. To identify the best template(s) for the target protein, database searching was carried out using the BLASTp program. A template (PDB ID: 3Q94_A) having 76% sequence identity, 1.2e- 100 E-value and alignment score(S) of 711.74, was selected for generating the 3D model of the FBA. The sequence identities and E-values are important parameters which are considered during the template(s) selection. The templates which are having high sequence identity, high alignment score(S), and low E-value are often selected for the reliable model generation of the target protein. The expect(E) value is a parameter that measures the significance of similarity between sequences during the database searching. This value decreases exponentially with the Score(S) that is assigned to a match between two sequences. The multiple sequence alignment was performed for the target-template sequences using the clustalW2 program and aligned target-template sequences were submitted to the HHPred server. Then 3D model of the FBA was created (Figure 1A). In order to evaluate the stereochemical properties as well as to find any anomaly in the predicted model, it was uploaded to the SAVES server. Procheck [20] results show that on the Ramachandran plot [21] , 94.7% amino acid residues fall in core (most favored) regions, 4.9% in allowed, 0.4% in generously allowed, and no any residue falls in disallowed regions (Figure 1B).
Figure 1.
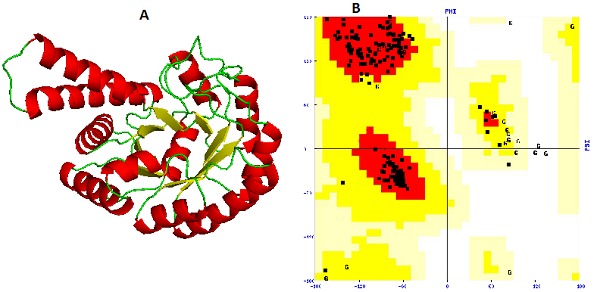
(A) The 3D model of fructose biphosphate aldolase; (B) The Ramachandran plot of modeled structure validated by PROCHECK program.
A good quality model would be expected to have more than 90% amino acid residues in the most favoured (core) regions and the results suggest that the predicted model is of good quality. But total 3 bad contacts were present in the modelled structure which could have lead to instability to the structure. In order to minimize the bad contacts in the model, it was subjected to the energy minimization using the GROMOS 96 forcefield [14] implementation of DeepView v4.04 (spdb viewer) tool [22]. After the minimization, the modelled structure of FBA was further subjected to the SAVES server and it was found that there was no any bad contact present in the structure. Verify_3D [23] result shows that 88.85% of residues had an average 3D-1D score >0.2 and the model was passed. According to Errat module [24], the overall quality factor of the predicted model was found to be 94.60 which reveal good quality of the predicted 3D structure of FBA in CA-MRSA.
MDS:
The protein dynamics of modelled 3D structure of fructose 1, 6- biphosphate aldolase was studied using the Gromacs v4.53 tool. Before performing molecular dynamics simulation (MDS), the protein was solvated in a water box followed by equilibration using the Newton's laws of motion. Using steepest descent minimization method, the energy of the structure was minimized in 1000 steps at 100 ps time scale to obtain the preferred conformation, which was converged in 465 steps. The average potential energy of the structure was calculated using the GROMOS 96 force field which was found to 5.345e+05 KJ/Mole. The starting structure was having potential energy of -4.646+e05 KJ/Mole and the minimization were converging at - 4.670e+05 KJ/Mole (Figure 2A) which shows the stability of the protein structure. Since our model has a net charge of +1.00, therefore one chloride ion was added to neutralize the net charge. Before starting the actual MDS, position-restrained molecular dynamics was performed. In this process the atom positions of the protein molecule were restrained (partially freezed) while solvent was allowed to move. This was done to soak the water molecule in to the protein molecule. The relaxation time of 100 ps was given to water molecule.
Figure 2.
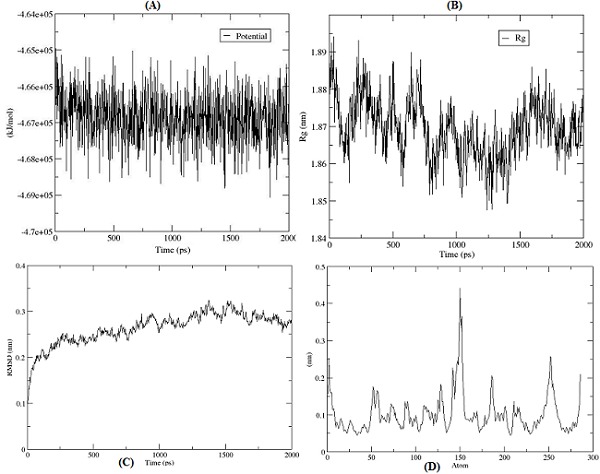
(A) Potential energy of the structure; (B) RMSD of protein from backbone; (C) Radius of gyration; (D) RMSF of amino acid residues of protein.
The molecular dynamics simulation of the modelled structure was performed using the 2000 ps time scale and 1000000 steps (iterations) at 300 °K temperature and 1 atm pressure. Thetrajectory of the MDS was analyzed using the ‘Grace’ program. The trajectory of radius of gyration (Rg) of modelled structure was analyzed which gives a measure of the mass of the atom(s) relative to the center of mass of the molecule. This quantity gives a measure of the compactness of the structure. The average of Rg of the protein was found to be 1.87 nm which reveals that the modeled structure is compact (Figure 2B). One of the most important criteria to analyze the stability of the protein structure is to measure the root mean square deviations (RMSD). The deviations from the original starting structure have been measured over the course of simulation. RMSD increases pretty rapidly in the beginning of the simulation, but stabilizes around 0.275 nm (Figure 2C). The trajectory of root mean square fluctuation (RMSF) of individual amino acid residues of protein was also analyzed, which computes the fluctuations of atomic positions in the structure. Except very few amino acid residues, the RMSF (i.e. standard deviation) of almost all amino acid residues was found between the 0.05 nm to 0.21 nm, which indicates that the structure is stable (Figure 2D).
Virtual screening:
Active site identification in the target protein is the starting point for virtual screening. Using the metaPocket 2.0 server, ligand binding site (pocket) was located in the 3D structure of the fructose biphosphate aldolase (FBA) of CA-MRSA. Three pockets were located in the target, and based on further analysis of amino acid residues involved in the active site, 2nd pocket was chosen for the docking. This pocket was having 13 residues in which Asn26, Gln237, Asp85, His86, His181 and His209 have been reported to be conserved in the active site of FBA [25]. The active site in the 3D structure of FBA on X, Y & Z coordinates were located as -22.686Å, 55.957Å and 36.002Å respectively. Before performing the virtual screening for the FBA as a drug target, the receptor was prepared using a Python script in the MGL tools package. The grid size for the receptor for docking was given as 30 Å, 30Å and 30Å on X, Y & Z coordinates respectively, which makes sure that the search space is large enough for the ligand to rotate in. Using the Autodock vina package, 1364 molecules from the NCI diversity subset II were screened by the protein-ligand docking method. The Autodock vina algorithm searches the ligands in different orientations in the active site of receptor. Two components searching and scoring are involved in most of the docking algorithms. The vina scoring function amalgamates knowledgebased potentials and empirical scoring functions, which extracts empirical information from both the conformational preferences of the receptor-ligand complexes and the experimental affinity measurements [26]. After performing the virtual screening using the vina package, the docking results were analyzed from the log files using a Python script in the ADT (Auto Dock Tool). Based on the energy score, top 10 ligands from the NCI diversity subset II molecules were selected for further analysis Table 1 (see supplementary material). Using the LigPlot+ program, schematic diagrams of protein-ligand interactions for top four receptor-ligand docked complexes were generated in 2D space. This diagram represents the hydrogen and hydrophobic interactions between ligand and active site residues of the FBA (Figure 3). The molecule ZINC01690699 was interacting with Lys65 and Glu68 residue through hydrogen bond at a distance of 3.01 Å and 2.97 Å respectively while Tyr61, Met66, Asn27, Tyr56, Leu261, Tyr260, Pro257, Glu29, Leu28 & Gly69 residues were involved in hydrophobic interactions.
Figure 3.
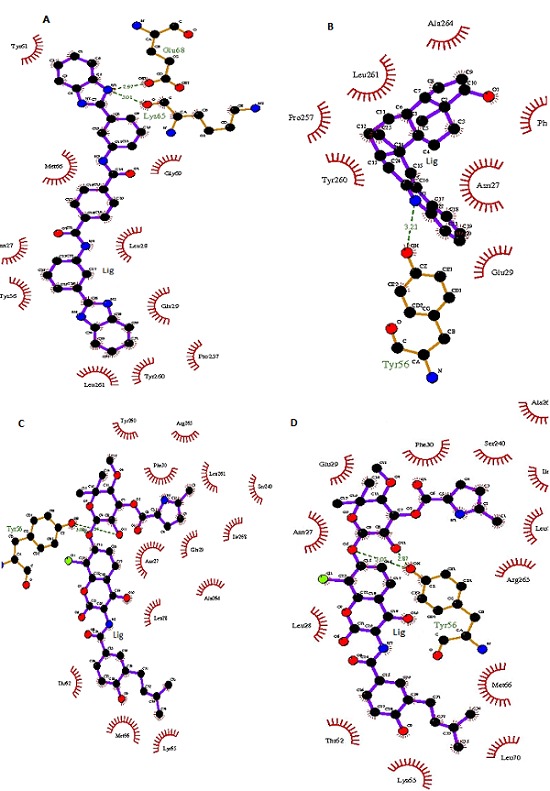
Schematic representation of FBA-Ligand interactions in CA-MRSA A) ZINC01690699; B) ZINC13154304; C) ZINC29590257; D) ZINC29590259.
The molecule ZINC13154304 was interacting with Tyr56 residue through hydrogen bond at a distance of 3.21 Å, and Ala264, Leu261, Pro257, Tyr260, Glu29, Asn27 & Phe30 residues were involved in hydrophobic interactions. The molecule ZINC29590257 was interacting with Tyr56 residue through hydrogen bond at a distance of 3.08 Å, and Tyr260, Arg265, Phe30, Leu261, Ser240, Ile268, Glu29, Asn27, Ala264, Leu28, Lys65, Met66 & Thr62 residues were involved in hydrophobic interactions. The fourth molecule ZINC29590259 was interacting with Tyr56 residue through hydrogen bond at a distance of 2.87 & 3.05 Å respectively, and Phe30, Ser240, Ala264, Ile268, Leu261, Arg265, Met66, Leu70, Lys65, Thr62, Leu28, Asn27 & Glu29 residues were involved in hydrophobic interactions. Interestingly it was found that Tyr56 amino acid residue was involved in hydrogen bond interactions within the active site of FBA in all top ranked ligands except in ZINC01690699. This Tyr56 residue might play crucial role in ligand interaction with the active site of FBA receptor in CAMRSA. Top four ligands i.e. ZINC01690699, ZINC13154304, ZINC29590257 and ZINC29590259 are having lower docking energy scores which show higher binding affinity towards the FBA. These ligands might act as potent inhibitors for the FBA in CA-MRSA.
Conclusion
The three dimensional structure of fructose biphosphate aldolase (FBA) in CA-MRSA was predicted and the quality of the model was validated which confirms the good quality model. The molecular dynamics study was carried out at 2000 ps and results suggest that the modeled structure is stable. The predicted model of FBA was used for virtual screening against the NCI diversity subset-II ligand databases which contain 1364 compounds. Based on the docking energy scores, it was found that top four ligands i.e. ZINC01690699, ZINC13154304, ZINC29590257 and ZINC29590259 are having lower energy scores which reveal higher binding affinity towards the active site of FBA. These ligands might prove to be potent inhibitors for the FBA so that the menace of antimicrobial resistance in CA-MRSA can be conquered. However, pharmacological studies are required to confirm the inhibitory activity of these ligands against the FBA in CA-MRSA.
Supplementary material
Acknowledgments
The authors are grateful to Sam Higginbottom Institute of Agriculture, Technology & Sciences (Deemed University), Allahabad for providing facilities and support to complete the present research work. We are thankful to Dr. Roman Laskowski for providing the academic license of LigPlot+ program.
Footnotes
Citation:Yadav et al, Bioinformation 9(3): 158-164 (2013)
References
- 1.Barber M, Rozwadowska-Dowzenko M. Lancet. 1948;2:641. doi: 10.1016/s0140-6736(48)92166-7. [DOI] [PubMed] [Google Scholar]
- 2.CDC. JAMA. 1999;282:1123. [PubMed] [Google Scholar]
- 3.Moran GJ, et al. N Engl J Med. 2006;355:666. doi: 10.1056/NEJMoa055356. [DOI] [PubMed] [Google Scholar]
- 4.Chambers HF, DeLeo FR. Nat Rev Microbiol. 2009;7:629. doi: 10.1038/nrmicro2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davis SL, et al. J Clin Microbiol. 2007;45:1705. doi: 10.1128/JCM.02311-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Otto M. Med Res Rev. 2010;30:1. doi: 10.1002/med.20160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yadav PK, et al. Bioinformation. 2012;8:664. doi: 10.6026/97320630008664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pegan SD, et al. J Mol Biol. 2009;386:1038. doi: 10.1016/j.jmb.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodaki A, et al. Eukaryot Cell. 2006;5:1371. doi: 10.1128/EC.00115-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fonvielle M, et al. Chemistry. 2008;14:8521. doi: 10.1002/chem.200800857. [DOI] [PubMed] [Google Scholar]
- 11.Labbé G, et al. Protein Expr Purif. 2011;80:224. doi: 10.1016/j.pep.2011.06.020. [DOI] [PubMed] [Google Scholar]
- 12.Altschul SF, et al. Nucleic Acids Res. 1997;25:3389. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soding J, et al. Nucleic Acids Res. 2005;33:W244. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Gunsteren WF, et al. Biomolecular simulation: the GROMOS96 manual and user guide. 1996;1:1042. [Google Scholar]
- 15.Berendsen HJC, et al. Comput Phys Commun. 1995;91:43. [Google Scholar]
- 16.Huang B. Omics. 2009;13:325. doi: 10.1089/omi.2009.0045. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Z, et al. Bioinformatics. 2011;27:2083. doi: 10.1093/bioinformatics/btr331. [DOI] [PubMed] [Google Scholar]
- 18.Irwin JJ, Shoichet BK. J Chem Inf Model. 2005;45:177. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laskowski RA, Swindells MB. J Chem Inf Model. 2011;51:2778. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 20.Laskowski RA, et al. J Appl Cryst. 1993;26:283. [Google Scholar]
- 21.Ramachandran GN, Sasiskharan V. Adv Protein Chem. 1968;23:283. doi: 10.1016/s0065-3233(08)60402-7. [DOI] [PubMed] [Google Scholar]
- 22.Guex N, Peitsch MC. Electrophoresis. 1997;18:2714. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 23.Bowie JU, et al. Science. 1991;253:164. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- 24.Colovos C, Yeates TO. Protein Sci. 1993;2:1511. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cooper SJ, et al. Structure. 1996;4:1303. doi: 10.1016/s0969-2126(96)00138-4. [DOI] [PubMed] [Google Scholar]
- 26.Trott O, Olson AJ. J Comput Chem. 2010;31:455. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.