

Abstract
Although the potent anti-tumor activity of nitric oxide (NO) supports its promise as an anti-neoplastic agent, effective and selective delivery and action on tumor and not normal cells remains a limiting factor. Nanoparticle-based delivery of NO has been considered as one approach to overcome these limitations. Therefore, we determined the utility of NO delivery using silica nanoparticles and evaluated their anti-tumor efficacy against human ovarian tumor and nontumor cells. The NO-releasing nanoparticles exhibited enhanced growth inhibition of ovarian tumor cells when compared to both control nanoparticles and a previously reported small molecule NO donor, PYRRO/NO. In addition, the NO-releasing nanoparticles showed greater inhibition of the anchorage-independent growth of tumor-derived and Ras-transformed ovarian cells. Confocal microscopy analysis revealed that fluorescently-labeled NO-releasing nanoparticles entered the cytosol of the cell and localized to late endosomes and lysosomes. Furthermore, we observed a nanoparticle size dependency on efficacy against normal versus transformed ovarian cells. Our study provides the first application of nanoparticle-derived NO as an antitumor therapy and supports the merit for future studies examining nanoparticle formulation for in vivo applications.
Keywords: Nanoparticle, silica, nitric oxide, ovarian cancer, Ras
Introduction
Nanotechnology has benefited a number of biomedical areas including drug delivery and magnetic resonance imaging contrast agents.1-5 The use of nanostructured materials (e.g., nanoparticles) as drug delivery systems has begun to impact medicine due to beneficial size-dependent physical and chemical properties.6 Due to their non-toxic nature and ability to form structures of tunable porosity, size, and surface functionalities, organically-modified silica nanoparticles have been explored as a carrier system for the controlled delivery of drugs, biocides, genes, and proteins.7-10 A limitation of cancer chemotherapy is the narrow therapeutic index of the majority of anti-neoplastic agents, with effective delivery to tumor cells, tumor cell resistance and normal cell toxicity the major concerns. The targeting capabilities available with nanoparticle-based drug delivery provide an attractive approach to overcome these limitations with improved site-specific delivery to the tumor.11,12
Nitric oxide (NO), a free radical bioregulator endogenously synthesized in the body, impacts multiple stages of tumor development, spanning cytostatic processes, cellular transformation, and formation of neoplastic lesions.13-15 Recent reports have highlighted the consequences of NO production on tumor biology including the inhibition of several classes of enzymes and iron metabolism proteins, cell apoptosis, and alternation of metastasis.16,17 Studies have shown that reactive nitrogen oxide species (e.g., NO2, NO2-, ONOO-, and N2O3) derived from NO impact the expression and activity of proteins critical to the cell cycle and apoptosis.16,18
To further understand the anti-cancer efficacy of NO, several classes of small molecule NO donors (e.g., NO-metal complexes, nitrosothiols, organic nitrites/nitrates, and diazeniumdiolates) have been developed and evaluated with respect to NO storage/release and efficacy against tumor cells. 19-235 For example, small molecule diazeniumdiolate NO donors have been used to demonstrate the efficacy of NO as a potent antitumor agent against ovarian cancer cells, the leading cause of death from gynecologic malignancies. 24 Indeed, O2-{2,4-dinitro-5-(4 -(N-methylamino)- benzoyloxy]phenyl})]-1-(N,N-dimethylamino)diazen-1-ium-1,2-diolate (PABA/NO) delayed human ovarian cancer tumor xenograft growth with potency similar to that of cisplatin, the mainstay of treatment for ovarian cancer.24 Another study demonstrated the efficacy of NCX-4016, a derivative of nitroaspirin, against cisplatin-resistant human ovarian cancer cells and xenograft tumors.25 Although small molecule NO donors have shown efficacy against tumors, their usefulness as anticancer agents is hindered clinically due to possible normal cell toxicity of the NO donor drug byproduct and the inability to target delivery of the drug selectively to cancer cells.
In this study, we have engineered NO-releasing silica nanoparticles as a therapeutic tool to determine if this would improve NO delivery to human ovarian cancer cells and have decreased cytotoxicity toward normal cells. We recently reported the synthesis of NO donor-modified silica nanoparticles that possess remarkably tunable nanoparticle size and NO storage/release properties.26,27 This delivery system allows us to control the therapeutic payload and visualize the nanoparticles through fluorescent tags. We compared the cytotoxicity of nanoparticle-derived NO to NO released from a small molecule NO donor (PYRRO/NO) and found increased anti-tumor activity of the nanoparticle-derived NO against ovarian tumor or transformed cell lines. Size-dependency studies suggest that selective cytotoxicity against tumor cells was enhanced by modulation of nanoparticle size. Our observations support the effectiveness of NO-releasing nanoparticles against ovarian cancer cells and merit future studies for further developing these materials for evaluation of anti-tumor activity in mouse models of ovarian cancer.
Materials and Methods
Cell lines, Drugs, and Antibodies
OVCAR-3, OVCAR-4, OVCAR-5, OVCAR-8, and SKOV-3 ovarian cancer cells were provided by Dr. R. Camalier of Division of Cancer Treatment and Diagnosis Tumor Repository at NCI (Frederick, MD). A2780 ovarian cancer cell line was provided by Dr. T. Fojo of Cancer Therapeutics Branch at NCI (Bethesda, MD). The HeyA8 ovarian cancer cell line was provided by Dr. G. Mills at MD Anderson Cancer Center (Houston, TX). The immortalized, nontransformed human ovarian surface epithelial cell lines (T29 and T80) and matched H-Ras(12V)- transformed counterparts (T29 H-Ras and T80 H-Ras) were described previously.28 All cell lines were maintained in either RPMI or DMEM medium supplemented with 10% fetal calf serum. Z-VAD-FMK and staurosporine were obtained from Calbiochem (Gibbstown, NJ). The caspase-3 and PARP antibodies were from Cell Signaling (Boston, MA), ERK1/2 antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA), Mito- and Lyso-Tracker from Molecular Probes (Eugene, OR), Alexa-594 secondary from Invitrogen (Eugene, OR), EEA1 from BD Transduction (Mississauga, ON, Canada), calnexin from Affinity BioReagents (Golden, CO), and LAMP1 from Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA).
Chemicals
Tetraethoxysilane (TEOS), 3-methylaminopropyltrimethoxysilane (MAP3), pyrrolidine (PYRRO), and sodium methoxide were purchased from Fluka (Buchs, Switzerland). N-(6-Aminohexyl)aminopropyltrimethoxysilane (AHAP3) and 3-aminopropyltrimethoxysilane (APTMS) were from Gelest (Tullytown, PA). Fluorescein isothiocyanate (FITC) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were from Sigma (St. Louis, MO). 4,5-Diaminofluorescein diacetate (DAF-2 DA) was purchased from Calbiochem (Gibbstown, NJ). Methanol (MeOH), ethanol (EtOH), 2-propanol (PrOH), toluene, and ammonia solution (28 wt% in water) were from Fisher Scientific (Fair Lawn, NJ). Nitric oxide (NO), carbon dioxide (CO2) (5%), argon (Ar), and nitrogen (N2) gases were from AGA Gas (Maumee, OH) or National Welders Supply (Raleigh, NC). Other solvents and chemicals were analytical-reagent grade and were used as received. A Millipore Milli-Q UV Gradient A10 System (Bedford, MA) was used to purify distilled water to a final resistivity of 18.2 MΩ·cm and a total organic content of ≤ 6 part per billion (ppb).
Synthesis of Nitric Oxide Donor Pyrrolidine and Organically-Modified Silica Nanoparticles
Sodium 1-(pyrrolidin-1-yl)diazen-1-ium-1,2-diolate (PYRRO/NO) was synthesized as previously reported.29 Silica nanoparticles were prepared by the co-condensation of tetraethyoxysilane (TEOS) with various ratios of an N-diazeniumdiolate-modified aminosilane (i.e. AHAP3 or MAP3) as described in depth previously.34 To form the N-diazeniumdiolate NO donor moiety, aminoalkoxysilane solution was prepared by dissolution of an appropriate amount of aminoalkoxysilane (i.e., AHAP3 or MAP3) in a mixture of ethanol, methanol, and sodium methoxide (NaOMe) base and then reacted with NO (5 atm) for 3 d. Tetraalkoxysilane (i.e., TEOS) was then mixed with different ratios of the N-diazeniumdiolate-modified aminoalkoxysilane (10–75 mol %, balance TEOS). Finally, the silane mixture was added to various solvent systems (e.g., 100% EtOH or 50/50% (v/v) EtOH/PrOH) in the presence of an ammonia catalyst (NH4OH) to form NO donor-modified silica nanoparticles via the sol-gel process. Nitric oxide-loaded nanoparticles were stored in sealed containers at -20 °C to protect the diazeniumdiolate moiety from thermal and proton-initiated decomposition.30 More recently, we have conducted experiments that indicate that storage in a closed container within an evacuated and sealed bag results in negligible NO loss for storage periods of several weeks at room temperature (data not shown). Control nanoparticles were prepared by exposing N-diazeniumdiolate-modified nanoparticles to aqueous media until all NO was depleted.
Characterization of N-diazeniumdiolate-Modified Silica Nanoparticles
NO release profiles of the N-diazeniumdiolate-modified silica nanoparticles were measured in deoxygenated phosphate buffered saline (PBS; 0.01 M, pH 7.4) at 37 °C using a Sievers NOA 280i chemiluminescence nitric oxide analyzer (Boulder, CO).32 Nanoparticle size was determined using atomic force microscopy (AFM) as previously described.26
Growth Assays
The MTT assay was performed as previously described.31 Cells were seeded in six replicates at 1000 to 5000 cells per well in 96-well plates, incubated overnight, and exposed to serial dilutions of pyrrolidine, PYRRO/NO, AHAP3/NO, AHAP3, MAP3/NO, or MAP3 in PBS continuously for 72 h. The drug-containing medium was then removed and replaced with MTT solution (Sigma, St. Louis, MO), after which the cells were incubated for 4 h at 37°C. After removal of the MTT, DMSO was added, and the absorbance was measured at 560 nm using a microplate reader. The results were expressed relative to the absorbance of cells grown in the absence of drug. The IC50 values were calculated from triplicate independent experiments. Anchorage-independent growth soft agar colony formation assays were performed by seeding single cell suspensions in growth medium at 10,000 cells/well supplemented with 0.4% bacto agar. Colonies formed within three weeks and were stained with MTT, then scanned on a Canon 9900F flat bed scanner (Canon, Lake Success, NY), and counted using Image J software (NIH, Bethesda, MD).
Synthesis of Fluorescently-labeled NO-Releasing Silica Nanoparticles
NO-releasing silica nanoparticles were fluorescently labeled via a “one-pot” co-condensation of three silane precursors including FITC-modified APTMS, N-diazeniumdiolated MAP3, and TEOS. The synthesis of FITC-APTMS conjugates was adapted from a previous report.26 Briefly, fluorescently-modified APTMS was prepared by reacting 321 μmol APTMS with 7.704 μmol FITC for 24 h in darkness. A 50 μL aliquot of the resulting product was then co-condensed with 3.24 mmol N-diazeniumdiolate-modified MAP3 and 1.39 mmol TEOS.
Confocal Fluorescence Microscopy
Cells were plated on glass coverslips coated with 0.01% gelatin. Cells were fixed in 4% paraformaldehyde, permeabilized in 0.2% Triton X-100, blocked in 0.5% bovine serum albumin, and incubated in primary [GM130 (1:50 dilution), calnexin (1:500), EEA1 (1:250), or LAMP1 (1:500)] followed by secondary Alexa-594 antibodies. To stain for mitochondria and lysosomes, cells were incubated in growth medium containing either 100 nM Mito-Tracker or 75 nM Lyso-Tracker for 30 min. To fluorescently monitor intracellular NO, cells were incubated in growth medium containing 100 μg/mL L-MAP3 NO-releasing nanoparticles alone (NO-MAP3 control) or 10 μM DAF-2 DA for 30 min. Next, cells treated with 10 μM DAF-2 DA were washed with PBS and incubated with medium alone (DAF-2 control), 100 μg/mL L-MAP3 NO-releasing nanoparticles, 100 μg/mL PYRRO/NO, or 1000 μg/mL PYRRO/NO for 1 h. Apoptotic and necrotic cells were labeled using the annexin V Fluos staining kit (Roche, Penzberg, Germany). Cells were mounted on coverslips with FluorSave (Calbiochem; Gibbstown, NJ) and imaged with a Zeiss Laser Scanning Microscope (LSM 510, Germany).
Statistical Analysis
Data were evaluated by SigmaPlot (Systat Software; San Jose, CA), and represented as means ± SD. P ≤ 0.001 were considered significant for all analyses.
Results
Synthesis and Characterization of NO-Releasing Silica Nanoparticles
The structure and concentration of the N-diazeniumdiolate-modified silane precursors as well as the solvent employed during the sol–gel polymerization was found to influence nanoparticle size and NO release properties (Table 1). Adjusting the type and concentration of NO donor-modified silane derivatives (i.e., N-diazeniumdiolated AHAP3 or MAP3) lead to nanoparticles ranging in diameter from 90 to 161 nm. As we observed previously, the diameter of the nanoparticle decreased with increasing N-diazeniumdiolated silane concentration.32 Other related studies have shown that the chain-length of the alcohol solvent affects the kinetics and degree of hydrolysis and condensation, ultimately resulting in the formation of nanoparticles of different diameters.33 In our analyses, we determined that the diameter of 75 mol% N-diazeniumdiolated MAP3 (balance TEOS) silica nanoparticles increased from 90 ± 10 nm for the silica prepared in 100% EtOH to 350 ± 50 when prepared in 50:50% EtOH:PrOH.
Table 1.
NO release properties of silica nanoparticles.
| Aminoalkoxysilane | Diameter | t[NO]a | |
|---|---|---|---|
| (type) | (mol%) | (nm) | (μmol/mg) |
| AHAP3 | 10 | 161 ± 23 | 0.56 |
| 25 | 142 ± 15 | 1.59 | |
| 35 | 128 ± 11 | 2.58 | |
| 45 | 136 ± 15 | 3.77 | |
| MAP3 | 45 | 155 ± 25 | 1.59 |
| 55 | 140 ± 20 | 2.90 | |
| 65 | 120 ± 15 | 4.30 | |
| 70 | 97 ± 15 | 6.21 | |
| 75 | 90 ± 10 | 7.35 | |
| 75b | 350 ± 50 | 12.52 | |
t[NO], total amount of NO released.
EtOH/PrOH (50:50 v/v%) solvent system used during sol–gel polymerization.
The NO storage and release properties of N-diazeniumdiolate-modified silica nanoparticles analyzed under physiological conditions (in PBS at pH 7.4 and 37 °C) are summarized in Table 1. The NO release profiles and total NO released from N-diazeniumdiolate-modified silica nanocomposites with different concentrations of MAP3 precursors (i.e., 45 to 75 mol%, balance TEOS) are shown in Figs. 1A and 1B. As expected, the initial NO release was characterized by a bolus of NO ranging from 3.3 × 104 to 10.3 × 104 ppb/mg depending on the MAP3 concentration. In addition, increasing the mol% of MAP3 from 45 to 75 mol% led to a corresponding increase in total NO release (t[NO]), ranging from 1.59 to 7.35 μmol/mg. Increasing nanoparticle diameter from 90 nm to 350 nm resulted in an increase in t[NO] from 7.35 to 12.52 μmol/mg The maximum concentration of NO release ([NO]m) for 45 mol% MAP3 was larger than that for AHAP3 of the same mol% (3.30 × 104 and 2.17 × 104 ppg/mg, respectively), while the total NO release was lower (1.59 μmol/mg and 3.77 μmol/mg).
Figure 1.
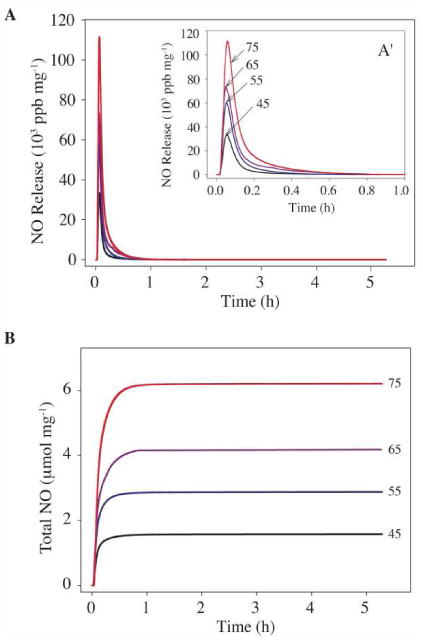
Real time NO release profiles (A) and total NO release amounts (B) for diazeniumdiolate-modified MAP3 silica nanoparticles at the indicated concentration (45, 55, 65, and 75 mol%; balance TEOS). Inset plot (A′) represents expansion of graph A from 0 to 1.0 h.
Cytotoxicity of NO Donor Silica Nanoparticles Against Ovarian Carcinoma Cells
To study the tumoricidal potential of NO donor silica, we first evaluated the cytotoxicity of control and N-diazeniumdiolate-modified silica nanoparticles on the A2780 human ovarian carcinoma cell line. The MTT viability assay was employed to determine the relative sensitivities of A2780 cells to growth inhibition by each nanoparticle treatment. The sensitivity (expressed as minus the logarithm base 10 of the 50% growth inhibitory concentration (IC50)) of the small molecule NO donor (PYRRO/NO) was 2 mM (Fig. 2A). We found that the NO-releasing silica (AHAP3/NO and MAP3/NO) exhibited greater cytotoxicity against A2780 cells than when treated with similar doses of NO from PYRRO/NO (Figs. 2B and 2C). A2780 cells were treated with varying doses of control and NO-releasing AHAP3 silica (13 – 1000 μg/mL) and viability was nearly completely inhibited at a dose of 500 μg/mL. Even the lowest concentration of NO-releasing silica nanoparticles tested proved cytotoxic against cancer cells, potentially resulting from a small population of the heterogeneous cancer cells with higher sensitivity to the nanoparticles. The concentrations of NO released indicated in Fig. 2 are theoretical concentrations calculated from those values listed in Table 1 based on the dose of PYRRO/NO, AHAP3/NO particle, or MAP3/NO particle. The actual concentration of NO available during the MTT assay, which was performed in open-air conditions, is expected to be slightly lower than the theoretical concentration determined from the analysis in a sealed vessel.
Figure 2.
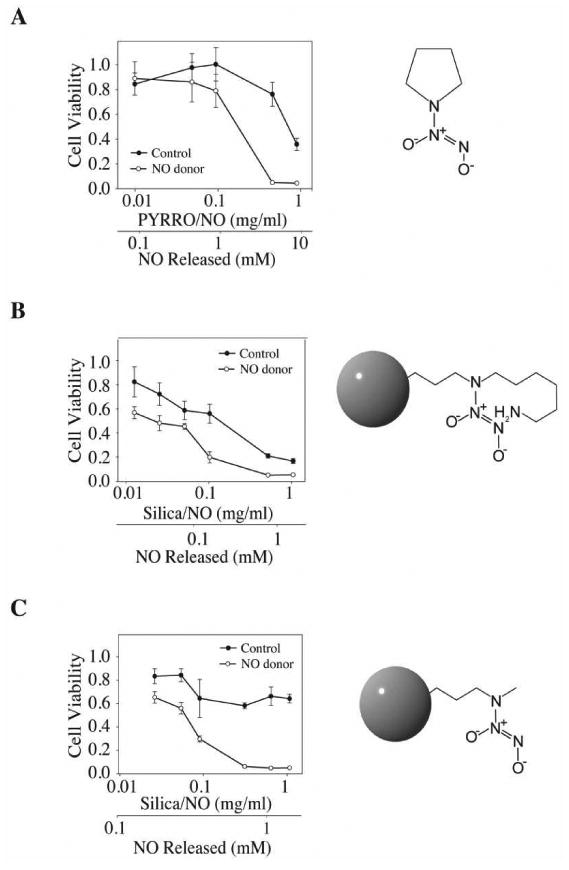
The MTT assay was performed to determine the cytotoxicity as a function of concentrations of: (A) PYRRO/NO; (B) 45 mol% AHAP3/NO, and (C) 75 mol% MAP3/NO silica nanoparticles on the A2780 ovarian carcinoma cell line: ●, controls (i.e. pyrrolidine, non-diazeniumdiolated AHAP3 and MAP3 silica nanoparticles) and ○, NO releasing. The results were plotted as cell viability, which was measured by absorbance at a wavelength of 560 nm (y-axis) versus drug concentration (x-axis). Amount of NO released was calculated from the values listed in Table 1.
We also observed that control AHAP3 silica nanoparticles (i.e., non-diazeniumdiolate modified) exhibited cytotoxic effects against the tumor cells (IC50 = 120 μg/mL), albeit less than that of their NO-releasing counterparts. We attributed this cytotoxicity of the AHAP3 controls to the primary amines available on the surface of the silica structures. To reduce the cytotoxicity of control and NO-releasing AHAP3 nanoparticles, MAP3 (containing only secondary amines) was employed as the aminosilane to create more biocompatible vehicles. As expected, the cytotoxicity of MAP3 controls against A2780 cells was significantly lower, whereas the NO release from NO donor-modified MAP3 silica remained cytotoxic against A2780 cells, with proliferation inhibited completely at 200 μg/mL(Fig. 2C).
To preliminarily investigate the effect of nanoparticle size on the cell cytotoxicity of our NO-releasing silica, we synthesized two sizes of 75 mol% MAP3 nanoparticles by varying the solvent system used during their preparation: 90 and 350 nm nanoparticles, designated as S- and L-MAP3, small and large MAP3 nanoparticles, respectively. We then treated untransformed immortalized human surface ovarian cells (HOSE), Ras-transformed HOSE, and a panel of ovarian carcinoma cell lines with control and NO-releasing S-MAP3 or L-MAP3 (400 μg/mL) nanoparticles and determined loss of anchorage-dependent cell viability. The IC50 values for these analyses are summarized in Table 2 and representative data are shown in Fig. 3. The NO-releasing S-MAP3 nanoparticles demonstrated comparable strong cytotoxicity against the entire panel of ovarian tumor cell lines as well as the paired immortalized and Ras-transformed HOSE cells (IC50 ∼60-100 μg/mL). In contrast, the NO-releasing L-MAP3 nanoparticles while significantly cytotoxic towards the ovarian carcinoma cell lines and the Ras-transformed T29H and T80H cells, showed markedly decreased cytotoxicity toward the non-transformed T29 and T80 cells (2.3- to 3.4-fold; p-value ≤ 0.001) than the smaller diameter particles. This size-dependent tumor cell cytotoxicity may allow for the development of NO-based tumor-selective chemotherapies with lessened toxicity to normal cells.
Table 2.
Effect of NO-releasing silica nanoparticle size on IC50 for inhibition of cellular viability.a
| Cell lineb | IC 50, μg/mL | |
|---|---|---|
| S-MAP3c | L-MAP3d | |
| HeyA8 | 100 ± 5.8 | 85 ± 5.5 |
| OVCAR-3 | 75 ± 4.3 | 60 ± 5.3 |
| OVCAR-4 | 87 ± 6.7 | 92 ± 8.8 |
| OVCAR-5 | 75 ± 3.2 | 65 ± 5.4 |
| OVCAR-8 | 90 ± 4.2 | 90 ± 5.8 |
| A2780 | 63 ± 5.2 | 30 ± 3.7 |
| SKOV-3 | 80 ± 8.9 | 63 ± 7.1 |
| T29e | 62 ± 8.2 | 150 ± 17 |
| T80e | 90 ± 8.6 | 220 ± 24 |
| T29Hf | 90 ± 7.7 | 62 ± 7.3 |
| T80Hf | 64 ± 5.1 | 61 ± 6.2 |
Values were determined by the MTT assay.
Human ovarian cancer cell lines, except where noted.
Diameter of S-MAP3 = 90 ± 10 nm; and
diameter of L-MAP3 = 350 ± 50 nm.
Immortalized normal human ovarian surface epithelial cell lines.
Transformed with constitutively activated mutant H-Ras(G12V).
Figure 3.
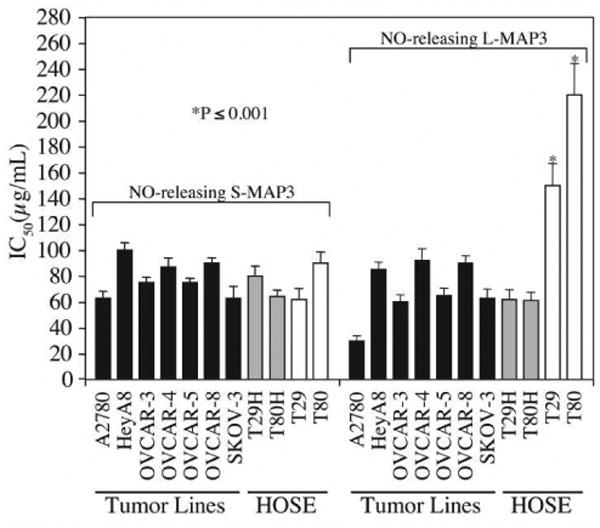
MAP3 NO-donor silica nanoparticles are cytotoxic to ovarian cells. (A) The MTT assay was used to screen the cytotoxicity of control MAP3 silica nanoparticles alone, S-MAP3 NO-donor silica nanoparticles (d = 90 ± 10 nm), or L-MAP3 NO donor-modified silica nanoparticles (d = 350 ± 50 nm) on a panel of ovarian cells. The cell lines tested include ovarian cancer patient cell lines: HeyA8, OVCAR-3, OVCAR-4, OVCAR-5, OVCAR-8, A2780, and SKOV-3; the genetically-engineered isogenic cell lines: immortalized normal ovarian cells T29 and T80 and their H-Ras(G12V)-transformed counterparts T29H and T80H. IC50 values and standard deviations were calculated from three independent experiments and the p-values were calculated for comparing T29 and T80 small versus large MAP3 NO-donor nanoparticle IC50 values.
NO-Releasing Silica Nanoparticles Induce Apoptosis
To determine whether the NO-releasing MAP3 nanoparticles cause inhibition of growth by inducing apoptosis in ovarian cells, we examined the levels of cleaved caspase-3 and cleaved PARP (biochemical markers of apoptosis). Western blot analysis demonstrated induction of apoptosis through an increase in cleaved caspase-3 and cleaved PARP levels in T80 and T80 H-Ras cells after treatment with 400 μg/mL of NO-releasing L-MAP3 nanoparticles for 6 h (Fig. 4A). Cells were treated with 10 μM staurosporine for 1 h as a positive control for induction of apoptosis. Cleaved caspase-3 and PARP protein levels increased to a greater extent in the T80 H-Ras cells compared to the T80 cells. Pre-treatment of T80 H-Ras cells with the broad spectrum caspase inhibitor, z-VAD-FM, followed by NO-releasing L-MAP3 nanoparticle treatment prevented caspase-3 and PARP cleavage. In addition, T80 H-Ras cells either treated with staurosporine or 400 μg/mL of L-MAP3 NO-donor nanoparticles for 6 h stained positive for the apoptotic marker, annexin V Fluor 647, and the necrotic marker, PI (Fig. 4B). These data suggest that NO-releasing L-MAP3 nanoparticles induced apoptosis more so in transformed ovarian cells compared to immortalized normal ovarian cells through a caspase-dependent mechanism.
Figure 4.
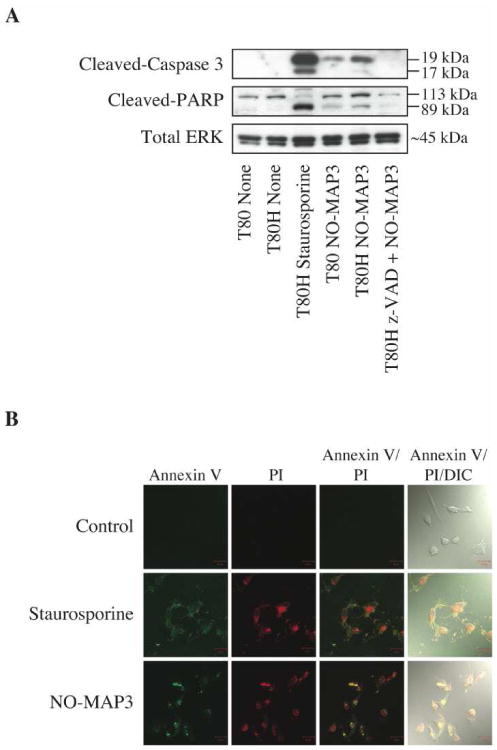
MAP3 NO-donor silica nanoparticles induce apoptosis in ovarian cells. T80 and T80H cells were untreated, treated with 10 μM staurosporine for 1 h, or treated with 400 μg/mL of NO-releasing L-MAP3 nanoparticles alone or in combination with 100 μM z-VAD-FMK for 6 h. Cell lysates were subjected to western blot analysis to probe for the apoptotic markers, cleaved-caspase 3 and cleaved-PARP, or loading control, total ERK1/2 (A). T80H cells were untreated or treated with either 10 μM staurosporine for 1 h or 400 μg/mL of NO-releasing L-MAP3 nanoparticles for 6 h. Cells were then stained with annexin V Alexa Fluor 647 to label apoptotic and PI to label necrotic cells and then imaged under the Zeiss laser scanning confocal microscope (B).
Anchorage-independent Growth is Inhibited in Transformed Ovarian Cells by Treatment with NO-releasing MAP3 Nanoparticles
We next investigated the ability of the NO-releasing nanoparticles to inhibit anchorage-independent growth by analyses of soft agar colony formation with cells treated with the small and large nanoparticles. Single cell suspensions in agar were maintained in growth medium supplemented with either vehicle (i.e., media), control MAP3 nanoparticles, NO-releasing S MAP3, or NO-releasing L-MAP3 nanoparticles (200 μg/mL) and the appearance of proliferating colonies of cells was monitored for two weeks (Fig. 3). Both S- and L-MAP3 significantly reduced colony formation compared to control MAP3 nanoparticle treatment. These results further indicate the therapeutic potential of NO-releasing nanoparticles.
Subcellular Localization of NO Donor Nanoparticles in Ovarian Cancer Cells
Previous studies have demonstrated uptake of nanoparticles such as silica and liposomes through the endocytic pathway.34 To determine cellular localization of the NO-releasing silica nanoparticles, we modified the nanoparticles to fluoresce by co-condensing FITC-modified APTMS, N-diazeniumdiolated MAP3, and TEOS. Incorporation of FITC-modified APTMS did not have a significant effect on nanoparticle size or NO release (data not shown). A2780 ovarian cancer cells were treated with FITC-labeled NO-releasing L-MAP3 (100 μg/mL) for either 1 or 24 h. Following treatment, the intracellular locations of mitochondria and lysosomes were determined by staining with either Mito-Traker or Lyso-Tracker, respectively. Cells were then fixed and stained for early and late endosomes, ER or Golgi, and visualized by confocal microscopy. FITC-labeled NO-releasing L-MAP3 demonstrated co-localization with the late endosomes and lysosomes after one and 24 h treatments (Figs. 6A and 6B). Furthermore, ovarian cancer cells treated for 1 h with NO-releasing L-MAP3 (100 μg/mL) were also treated with DAF-2 DA, a fluorescent marker for the presence of intracellular NO. As shown in Fig. 6C, no fluorescence was observed upon treatment with L-MAP3 (100 μg/mL), DAF-2 DA alone, or PYRRO/NO (100 μg/mL). Fluorescence did occur in cells treated with both L-MAP3 (100 μg/mL) and DAF-2 DA indicating the NO was present within the cell. A much larger concentration of PYRRO/NO (1000 μg/mL) was needed to observe similar fluorescence levels (NO concentration) in the cells.
Figure 6.
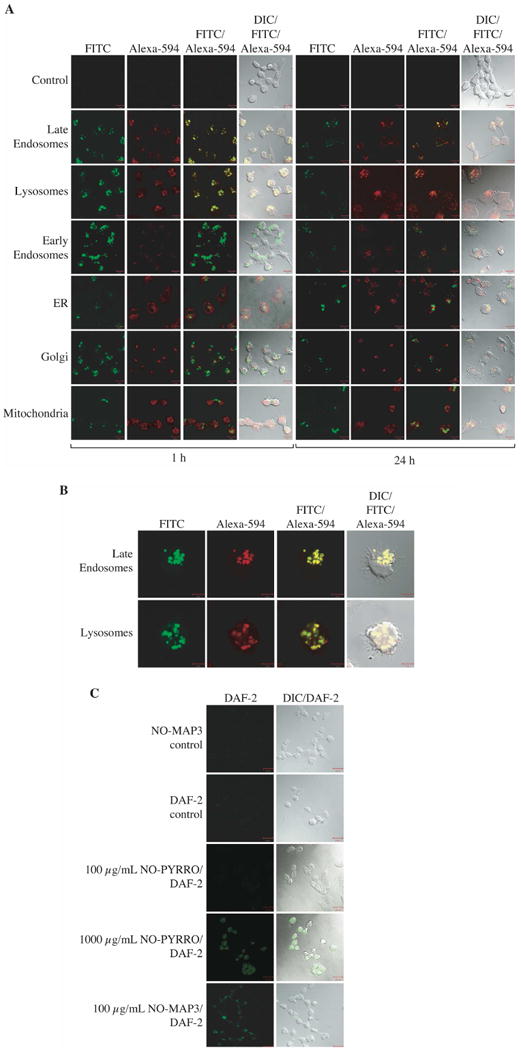
A2780 ovarian cancer cells were treated with 100 μg/mL L-MAP3 NO-releasing nanoparticles for either 1 or 24 h. The mitochondria or lysosomes were labeled through incubation of the cells with media containing either Mito-Tracker or Lyso-Tracker. Cells were fixed and stained for the remaining cellular compartments with GM130 (Golgi), calnexin (ER), EEA1 (early endosomes), or LAMP1 (late endosomes) primary antibodies. Cells were then stained with a secondary Alexa-594 antibody and imaged under the Zeiss laser scanning confocal microscope (A). The enlarged single cell view of A2780 cells treated with MAP3 NO-releasing nanoparticles for 1 h stained for late endosomes and lysosomes indicates co-localization (B). A2780 cells were treated with 100 μg/mL L-MAP3 NO-releasing nanoparticles alone (NO-MAP3 control) or 10 μM DAF-2 DA for 30 min. Next, cells treated with 10 μM DAF-2 DA were washed with PBS and incubated with medium alone (DAF-2 control), 100 μg/mL L-MAP3 NO-releasing nanoparticles, 100 μg/mL PYRRO/NO, or 1000 μg/mL PYRRO/NO for 1 h. Cells were imaged under the Zeiss laser scanning confocal microscope (C).
Discussion
Although a variety of small molecule NO donors have been reported effective against different tumor types including pancreatic, colon, and ovarian cancers,24,25,35,36 they have also suffered from highly toxic side effects of the drug byproduct and poor cellular permeability and retention. The development of novel delivery systems for NO donors that may overcome these limitations is of crucial importance to advance the feasibility of NO-based therapies. In this study, we employed NO-releasing silica scaffolds to assess the usefulness of nanoparticle-based NO delivery against ovarian cancer. When compared to control nanoparticles, the NO donor-modified nanoparticles demonstrated enhanced growth inhibition of ovarian tumor cells and preliminary size dependency support the anti-tumor efficacy of NO delivered from silica nanoparticles against ovarian cancer cells as well as the feasibility of materials based from these observations as a novel approach for cancer treatment.
When compared to the small molecule NO donor PYRRO/NO, we found greater anti-tumor activity with the N-diazeniumdiolated-modified NO-releasing silica nanoparticles. While other small molecule NO donors exist, PYRRO/NO was chosen for study due to its NO release mechanism allowing for a direct comparison between the NO-releasing silica nanoparticles and a previously reported small molecule NO donor. Without changing the structure of the NO donor functionality, we were able to prolong the NO using the silica nanoparticle. Although NO release may be further extended using more recent diazeniumdiolate chemistries (e.g., O2-arylated diazeniumdiolate), our goal was to keep this chemistry constant. (N-diazeniumdiolates are characterized by a one-step proton-initiated dissociation mechanism, while O2-arylated diazeniumdiolates follow a two-step dissociation mechanism (i.e., S-glutathionylation and protonation).24 The dose of NO required to inhibit tumor cell growth was significantly decreased for the nanoparticles relative to NO derived from the small molecule. Due to an extremely short half-life (∼3 s), the effective PYRRO/NO concentration is inherently larger because most of the NO is released prior to the small molecule being taken up by the target cell.29 Since the NO release from the nanocomposite scaffolds is prolonged (t1/2= 7 min), more NO is available for therapeutic use upon interaction between the nanoparticle and tumor cell. Presence of intracellular NO was confirmed using DAF-2 DA, a widely used NO-sensitive fluorescent probe.37,38 Upon treatment with NO-releasing silica particles and DAF-2 DA, we observed intracellular DAF-2 fluorescence, confirming that our silica particles are capable of delivering significant levels of NO within the cell. Due to the kinetics of NO release, it is likely that some of the payload is released prior to cellular uptake of the particles. However, the presence of DAF-2 fluorescence proves that the nanoparticles are successful in delivering NO to the cell.
Based on our results, NO-releasing silica nanoparticles containing MAP3 are preferable over those composed of AHAP3. Control MAP3 nanoparticles (i.e., non-diazeniumdiolated) demonstrated little to no cytotoxicity in our ovarian cell line panel, while control AHAP3 nanoparticles were slightly cytotoxic. The increased cytotoxicity of AHAP3 over MAP3 nanoparticles is attributed to the presence of the primary amine.39,40 Furthermore, greater aminosilane concentrations were achievable for MAP3 particle systems, thus allowing higher NO payloads. The primary amine of AHAP3 interrupts particle formation at higher concentrations due to hydrogen bonding interactions. These results validate the MAP3 NO-releasing silica nanoparticles as a non-toxic carrier system for large NO payloads.
Nanoparticle size is an important factor in determining the efficacy of a specific anti-cancer therapeutic approach. In the case of drug carriers, size may influence delivery volume, release characteristics, and accumulation site in the body such as tumor, liver, or bone marrow. Previous studies have shown size-dependent tumor-specific delivery of nanoparticles to the tumor microvasculator through the enhanced permeation and retention (EPR) effect.41,42 We found that increasing the size of the nanoparticle from d = 90 ± 10 nm to d = 350 ± 50 nm resulted in enhanced preferential cytotoxicity for tumor versus nontumor ovarian cells. Although our initial studies were done in vitro, with evaluation of only two normal immortalized cell lines and their transformed isogenic counterparts, we believe that these observations are quite significant. Future work will involve synthesizing a range of sizes and investigating not only efficiency of NO delivery but also how particle size affects uptake and distribution within cells.
Many groups have determined that the mechanism of internalization and localization of nanoparticles was influenced greatly by size, surface properties, and cell line-dependence differences.34,43-45 Silica nanoparticles are known to be taken up via endocytosis and localize to endosomes and lysosomes. Thus, as expected, our analyses found that the NO-releasing silica nanoparticles entered the cytosol of the treated cells and localized to late endosomes and lysosomes. Our results show clearly that larger NO-releasing nanoparticles demonstrated enhanced destabilization of mitochondrial function and caspase-induced apoptosis in tumor versus non-tumor cells. The role of NO in the mitochondrial-mediation of apoptosis has been established.46 The nitro-aspirin drug, NCX-4016, induced apoptosis by activation of caspase-3 and cleavage of the substrate PARP in ovarian cancer cells.47 Similarly, the large NO-releasing nanoparticles induced cell death in T80 and more so in T80 H-Ras cells as shown by caspase-3 and PARP cleavage and staining positive for annexin V/PI. The smaller diameter particles exhibited greater cytotoxicity toward T80 cells than the larger particles. Indeed, it has been shown that toxicity of amorphous silica is inversely related to particle size with diameters less than 100 nm showing an increase in cytotoxicity.48,49 This phenomenon is attributed to the fact that the surface area to volume ratio increases exponentially as particle size decreases. More atoms or molecules are expressed on the surface of smaller diameter particles per volume compared to larger particles therefore increasing the biological activity of the smaller nanoparticle. The enhanced cytotoxicity and caspase induced-apoptosis seen in T80 H-Ras cells after treatment with the large NO-releasing nanoparticles may be attributed to the greater concentration of NO released by these scaffolds and/or NO targeting Ras-driven apoptotic pathways.
While we have shown that NO-releasing silica nanoparticles are effective against ovarian tumor cells, modifications must be made to enhance the therapeutic potential due to the short half-life of NO release. Addition of an outer shell (i.e. TEOS) could temporarily protect the diazeniumdiolate functionalities and delay NO release. Alternatively, encapsulation of the NO-releasing silica particles within a liposome would suspend NO release until obliteration of the liposome. Future studies will need to determine whether size selectivity is maintained in the much more complex in vivo environment, where pharmacodynamic and phamacokinetic parameters will also influence nanoparticle-delivered NO anti-tumor activity and selectivity. Furthermore, prior to in vivo testing, the surface of the particles will be modified with polyethylene glycol (PEG) to increase biocompatibility and blood circulation times.50,51
Conclusion
In summary, our studies support the potential value of silica-based nanoparticles for delivery of NO for anti-cancer treatment. In addition to the future analyses discussed previously, another important direction for this work will be the use of NO-releasing nanoparticles for combination treatment with both conventional cytotoxic drugs and with molecularly targeted therapies. A recent study found that the in vitro and in vivo sensitivity of SW620 colon carcinoma cells to cisplatin-induced apoptosis was enhanced by co-treatment with the NO donor DETANONOate.23 For the treatment of ovarian cancer, it will be important to determine if nanoparticle-based NO delivery will synergistically enhance response to chemotherapeutic agents used in the first line treatment of ovarian cancer (e.g., cisplatin and paclitaxel).
Figure 5.
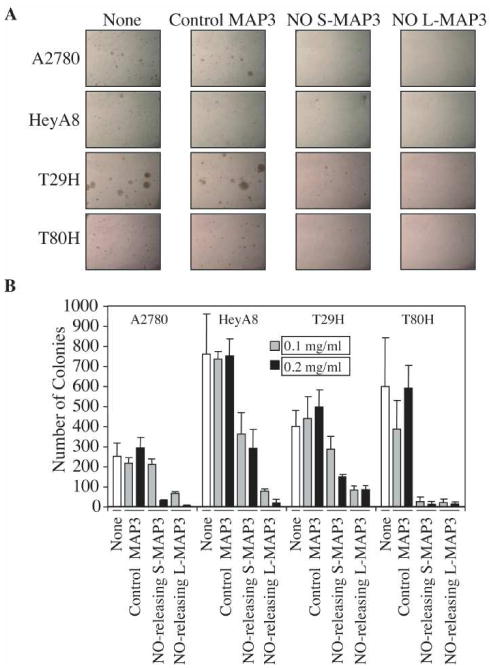
S- (d = 90 ± 10 nm) and L- (d = 350 ± 50 nm) NO donor-modified MAP3 nanoparticles inhibited anchorage-independent growth of ovarian cells compared to control MAP3 silica. The soft agar assay was performed to evaluate the ability of NO donor-modified nanoparticles to inhibit the anchorage-independent growth of ovarian cancer cells (A2780 and HeyA8) and transformed ovarian cells [T29 H-Ras(G12V) and T80 H-Ras(G12V)]. Cells were suspended in soft agar supplemented with growth medium that contained either vehicle, control MAP3 (non-NO releasing), S-MAP3, or L-MAP3 NO-releasing nanoparticles. Colonies formed within three weeks: pictures were taken under the light microscope (50×) (A) and colonies were stained with MTT and quantified using Image J software (B).
Acknowledgments
We thank Lanika DeGraffenreid for assistance in figure preparation, R. Camalier, T. Fojo and G. Mills for cell lines, and the Michael Hooker Microscopy Facility at UNC-CH for use of their confocal microscope. This research was supported by the National Institutes of Health (EB000708) and the Carolina Center of Cancer Nanotechnology Excellence (NCI CA119343).
Contributor Information
Channing J. Der, Email: cjder@med.unc.edu.
Mark H. Schoenfisch, Email: schoenfisch@unc.edu.
References
- 1.Reddy LH. Drug delivery to tumors: Recent strategies. J Pharm Pharmacol. 2005;57:1231–1242. doi: 10.1211/jpp.57.10.0001. [DOI] [PubMed] [Google Scholar]
- 2.Bruchez M, Moronne M, Gin P, Weiss S, Alivisatos AP. Semiconductor nanocrystals as fluorescent biological labels. Science. 1998;281:2013–2016. doi: 10.1126/science.281.5385.2013. [DOI] [PubMed] [Google Scholar]
- 3.Nam JM, Thaxton CC, Mirkin CA. Nanoparticle-based bio-bar codes for the ultrasensitive detection of proteins. Science. 2003;301:1884–1886. doi: 10.1126/science.1088755. [DOI] [PubMed] [Google Scholar]
- 4.Mahtab R, Rogers JP, Murphy CJ. Protein-sized quantum dot luminescence can distinguish between “straight”, “bent”, and “kinked” oligonucleotides. J Am Chem Soc. 1995;117:9099–9100. [Google Scholar]
- 5.Weissleder R, Elizondo G, Wittenburg J, Rabito CA, Bengele HH, Josephson L. Ultrasmall superparamagnetic iron oxide: Characterization of a new class of contrast agents for MR imaging. Radiology. 1990;175:489–493. doi: 10.1148/radiology.175.2.2326474. [DOI] [PubMed] [Google Scholar]
- 6.Zhang L, Gu FX, Chan JM, Wang AZ, Langer RS, Farokhzad OC. Nanoparticles in medicine: therapeutic applications and developments. Clin Pharmacol Ther. 2008;83:761–769. doi: 10.1038/sj.clpt.6100400. [DOI] [PubMed] [Google Scholar]
- 7.Barbe C, Bartlett J, Linggen K, Finnie K, Qiang LH, Larkin M, Calleja S, Bush A, Calleja G. Silica particles: A novel drug-delivery system. Adv Mater. 2004;16:1959–1966. [Google Scholar]
- 8.Stein A, Melde BJ, Schroden RC. Hybrid inorganic-organic mesoporous silicates-nanoscopic reactors coming of age. Adv Mater. 2000;12:1403–1419. [Google Scholar]
- 9.Brunner TJ, Wick P, Manser P, Spohn P, Grass RN, Limbach LK, Bruinink A, Stark WJ. In vitro cytotoxicity of oxide nanoparticles: Comparison to asbestos, silica, and the effect of particle solubility. Environ Sci Technol. 2006;40:4374–4381. doi: 10.1021/es052069i. [DOI] [PubMed] [Google Scholar]
- 10.Sayari A, Hamoudi S. Periodic mesoporous silica-based organic-inorganic nanocomposite materials. Chem Mater. 2001;13:3151–3168. [Google Scholar]
- 11.Cho K, Wang X, Nie S, Chen ZG, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14:1310–1316. doi: 10.1158/1078-0432.CCR-07-1441. [DOI] [PubMed] [Google Scholar]
- 12.Byrne JD, Betancourt T, Brannon-Peppas L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv Drug Deliv Rev. 2008;60:1615–1626. doi: 10.1016/j.addr.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 13.Bonavida B, Khineche S, Huerta-Yepez S, Garban H. Therapeutic potential of nitric oxide in cancer. Drug Resist Updat. 2006;9:157–173. doi: 10.1016/j.drup.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 14.Miller MR, Megson IL. Recent developments in nitric oxide donor drugs. Br J Pharmacol. 2007;151:305–321. doi: 10.1038/sj.bjp.0707224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huerta S, Chilka S, Bonavida B. Nitric oxide donors: novel cancer therapeutics (review) Int J Oncol. 2008;33:909–927. [PubMed] [Google Scholar]
- 16.Albina JE, Reichner JS. Role of nitric oxide in mediation of macrophage cytotoxicity and apoptosis. Cancer Metas Rev. 1998;17:19–53. doi: 10.1023/a:1005904704618. [DOI] [PubMed] [Google Scholar]
- 17.Fukumura D, Kashiwagi S, Jain RK. The role of nitric oxide in tumour progression. Nat Rev Cancer. 2006;6:521–534. doi: 10.1038/nrc1910. [DOI] [PubMed] [Google Scholar]
- 18.Lo HW, Hung MC. Nuclear EGFR signaling network in cancers: Linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Brit J Cancer. 2006;94:184–188. doi: 10.1038/sj.bjc.6602941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mimeault M, Jouy N, Depreux P, Henichart JP. Synergistic antiproliferative and apoptotic effects induced by mixed epidermal growth factor receptor inhibitor ZD1839 and nitric oxide donor in human prostatic cancer cell lines. Prostate. 2005;62:187–199. doi: 10.1002/pros.20138. [DOI] [PubMed] [Google Scholar]
- 20.Pervin S, Singh R, Chaudhuri G. Nitric oxide-induced cytostasis and cell cycle arrest of a human breast cancer cell line (MDA-MB-231): Potential role of cyclin D1. Proc Natl Acad Sci. 2001;98:3583–3588. doi: 10.1073/pnas.041603998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jia L, Schweizer J, Wang Y, Cerna C, Wong H, Revilla M. Effect of nitric oxide on cytotoxicity of Taxol: Enhanced Taxol transcellular permeability. Biochem Pharmacol. 2003;66:2193–2199. doi: 10.1016/j.bcp.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 22.Adami A, Crivellente F, De Prati AC, Cavalieri E, Cuzzolin L, Tommasi M, Suzuki H, Benoni G. Biotransformation and cytotoxic properties of NO-donors on MCF7 and U251 cell lines. Life Sci. 1998;63:2097–2105. doi: 10.1016/s0024-3205(99)80006-x. [DOI] [PubMed] [Google Scholar]
- 23.Huerta S, Baay-Guzman G, Gonzalez-Bonilla CR, Livingston EH, Huerta-Yepez S, Bonavida B. In vitro and in vivo sensitization of SW620 metastatic colon cancer cells to CDDP-induced apoptosis by the nitric oxide donor DETANONOate: Involvement of AIF. Nitric Oxide. 2009;20:182–194. doi: 10.1016/j.niox.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 24.Saavedra JE, Srinivasan A, Buzard GS, Davies KM, Waterhouse DJ, Inami K, Wilde TC, Citro ML, Cuellar M, Deschamps JR, Parrish D, Shami PJ, Findlay VJ, Townsend DM, Tew KD, Singh S, Jia L, Ji X, Keefer LK. PABA/NO as an anticancer lead: Analogue synthesis, structure revision, solution chemistry, reactivity toward glutathione, and in vitro activity. J Med Chem. 2006;49:1157–1164. doi: 10.1021/jm050700k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bratasz A, Weir NM, Parinandi NL, Zweier JL, Sridhar R, Ignarro LJ, Kuppusamy P. Reversal to cisplatin sensitivity in recurrent human ovarian cancer cells by NCX-4016, a nitro derivative of aspirin. Proc Natl Acad Sci USA. 2006;103:3914–3919. doi: 10.1073/pnas.0511250103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shin JH, Metzger SK, Schoenfisch MH. Synthesis of nitric oxide-releasing silica nanoparticles. J Am Chem Soc. 2007;129:4612–4619. doi: 10.1021/ja0674338. [DOI] [PubMed] [Google Scholar]
- 27.Shin JH, Schoenfisch MH. Inorganic/organic hybrid silica nanoparticles as a nitric oxide delivery scaffold. Chem Mater. 2008;20:239–249. doi: 10.1021/cm702526q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu J, Yang G, Thompson-Lanza JA, Glassman A, Hayes K, Patterson A, Marquez RT, Auersperg N, Yu Y, Hahn WC, Mills GB, Bast RC., Jr A genetically defined model for human ovarian cancer. Cancer Res. 2004;64:1655–1663. doi: 10.1158/0008-5472.can-03-3380. [DOI] [PubMed] [Google Scholar]
- 29.Saavedra JE, Billiar TR, Williams DL, Kim YM, Watkins SC, Keefer LK. Targeting nitric oxide (NO) delivery in vivo. Design of a liver-selective NO donor prodrug that blocks tumor necrosis factor-alpha-induced apoptosis and toxicity in the liver. J Med Chem. 1997;40:1947–1954. doi: 10.1021/jm9701031. [DOI] [PubMed] [Google Scholar]
- 30.Keefer LK, Nims RW, Davies KM, Wink DA. “NONOates” (1-substituted diazen-1-ium- 1,2-diolates) as nitric oxide donors: convenient nitric oxide dosage forms. Methods Enzymol. 1996;268:281–293. doi: 10.1016/s0076-6879(96)68030-6. [DOI] [PubMed] [Google Scholar]
- 31.Stevens EV, Nishizuka S, Antony S, Reimers M, Varma S, Young L, Munson PJ, Weinstein JN, Kohn EC, Pommier Y. Predicting cisplatin and trabectedin drug sensitivity in ovarian and colon cancers. Mol Cancer Ther. 2008;7:10–18. doi: 10.1158/1535-7163.MCT-07-0192. [DOI] [PubMed] [Google Scholar]
- 32.Polizzi MA, Stasko NA, Schoenfisch M. Water-soluble nitric oxide-releasing gold nanoparticles. Langmuir. 2007;23:4938–4943. doi: 10.1021/la0633841. [DOI] [PubMed] [Google Scholar]
- 33.Hong S, Bielinska AU, Mecke A, Keszler B, Beals JL, Shi X, Balogh L, Orr BG, Baker JR, Jr, Banaszak Holl MM. Interaction of poly(amidoamine) dendrimers with supported lipid bilayers and cells: hole formation and the relation to transport. Bioconjug Chem. 2004;15:774–782. doi: 10.1021/bc049962b. [DOI] [PubMed] [Google Scholar]
- 34.Sun W, Fang N, Trewyn BG, Tsunoda M, Slowing II, Lin VS, Yeung ES. Endocytosis of a single mesoporous silica nanoparticle into a human lung cancer cell observed by differential interference contrast microscopy. Anal Bioanal Chem. 2008;391:2119–2125. doi: 10.1007/s00216-008-2162-1. [DOI] [PubMed] [Google Scholar]
- 35.Rosetti M, Tesei A, Ulivi P, Fabbri F, Vannini I, Brigliadori G, Amadori D, Bolla M, Zoli W. Molecular characterization of cytotoxic and resistance mechanisms induced by NCX 4040, a novel NO-NSAID, in pancreatic cancer cell lines. Apoptosis. 2006;11:1321–1330. doi: 10.1007/s10495-006-6986-x. [DOI] [PubMed] [Google Scholar]
- 36.Ouyang N, Williams JL, Rigas B. NO-donating aspirin inhibits angiogenesis by suppressing VEGF expression in HT-29 human colon cancer mouse xenografts. Carcinogenesis. 2008;29:1794–1798. doi: 10.1093/carcin/bgn127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kojima H, Nakatsubo N, Kikuchi K, Kawahara S, Kirino Y, Nagoshi H, Hirata Y, Nagano T. Detection and imaging of nitric oxide with novel fluorescent indicators: diaminofluoresceins. Anal Chem. 1998;70:2446–2453. doi: 10.1021/ac9801723. [DOI] [PubMed] [Google Scholar]
- 38.Shao C, Stewart V, Folkard M, Michael BD, Prise KM. Nitric oxide-mediated signaling in the bystander response of individually targeted glioma cells. Cancer Res. 2003;63:8437–8442. [PubMed] [Google Scholar]
- 39.Luo Y, Prestwich GD. Cancer-targeted polymeric drugs. Curr Cancer Drug Targets. 2002;2:209–226. doi: 10.2174/1568009023333836. [DOI] [PubMed] [Google Scholar]
- 40.Stasko NA, Johnson CB, Schoenfisch MH, Johnson TA, Holmuhamedov EL. Cytotoxicity of polypropylenimine dendrimer conjugates on cultured endothelial cells. Biomacromolecules. 2007;8:3853–3859. doi: 10.1021/bm7008203. [DOI] [PubMed] [Google Scholar]
- 41.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 42.Leuschner C, Kumar C. Nanoparticles for cancer drug delivery. In: Kumar CSSR, Hormes J, Leuschner C, editors. Nanofabrication towards Biomedical Applications: Techniques, Tools, Applications, and Impact. 1st. Wiley-VCH Verlag GmbH & Co.; Weinheim, German: 2005. pp. 289–315. [Google Scholar]
- 43.Slowing I, Trewyn BG, Lin VS. Effect of surface functionalization of MCM-41-type mesoporous silica nanoparticles on the endocytosis by human cancer cells. J Am Chem Soc. 2006;128:14792–14793. doi: 10.1021/ja0645943. [DOI] [PubMed] [Google Scholar]
- 44.Lu J, Liong M, Zink JI, Tamanoi F. Mesoporous silica nanoparticles as a delivery system for hydrophobic anticancer drugs. Small. 2007;3:1341–1346. doi: 10.1002/smll.200700005. [DOI] [PubMed] [Google Scholar]
- 45.Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem J. 2004;377:159–169. doi: 10.1042/BJ20031253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boyd CS, Cadenas E. Nitric oxide and cell signaling pathways in mitochondrial-dependent apoptosis. Biol Chem. 2002;383:411–423. doi: 10.1515/BC.2002.045. [DOI] [PubMed] [Google Scholar]
- 47.Selvendiran K, Bratasz A, Tong L, Ignarro LJ, Kuppusamy P. NCX-4016, a nitro-derivative of aspirin, inhibits EGFR and STAT3 signaling and modulates Bcl-2 proteins in cisplatin-resistant human ovarian cancer cells and xenografts. Cell Cycle. 2008;7:81–88. doi: 10.4161/cc.7.1.5103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oberdorster G, Oberdorster E, Oberdorster J. Nanotoxicology: an emerging discipline evolving from studies of ultrafine particles. Environ Health Perspect. 2005;113:823–839. doi: 10.1289/ehp.7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu K, Grabinski C, Schrand A, Murdock R, Wang W, Gu B, Schlager J, Hussain S. Toxicity of amorphous silica nanoparticles in mouse keratinocytes. J Nanopart Res. 2009;11:15–24. [Google Scholar]
- 50.He X, Nie H, Wang K, Tan W, Wu X, Zhang P. In Vivo Study of Biodistribution and Urinary Excretion of Surface-Modified Silica Nanoparticles. Anal Chem. 2008;80:9597–9603. doi: 10.1021/ac801882g. [DOI] [PubMed] [Google Scholar]
- 51.Ryan SM, Mantovani G, Wang X, Haddleton DM, Brayden DJ. Advances in PEGylation of important biotech molecules: delivery aspects. Expert Opin Drug Deliv. 2008;5:371–383. doi: 10.1517/17425247.5.4.371. [DOI] [PubMed] [Google Scholar]