

Abstract
Pilicides and curlicides are compounds that block the formation of the virulence factors pili and curli, respectively. To facilitate studies of the interaction between these compounds and the pili and curli assembly systems, fluorescent pilicides and curlicides have been synthesized. This was achieved by using a strategy based on structure–activity knowledge, in which key pilicide and curlicide substituents on the ring-fused dihydrothiazolo 2-pyridone central fragment were replaced by fluorophores. Several of the resulting fluorescent compounds had improved activities as measured in pili- and curli-dependent biofilm assays. We created fluorescent pilicides and curlicides by introducing coumarin and 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) fluorophores at two positions on the peptidomimetic pilicide and curlicide central fragment. Fluorescence images of the uropathogenic Escherichia coli (UPEC) strain UTI89 grown in the presence of these compounds shows that the compounds are strongly associated with the bacteria with a heterogeneous distribution.
Keywords: antivirulence, biological activity, coumarin, fluorescence, structure–activity relationships
Introduction

Multidrug resistant (MDR) bacterial strains present a growing global health problem. As a consequence, the search for new antibacterial agents and new methods to deal with bacterial resistance is urgent.1 Toward this end, understanding the details of the uropathogenic E. coli (UPEC) pathogenic cascade is revealing ways to target critical pathways to develop anti-virulence therapeutics. We have discovered that type-1 pili, an adhesive pili assembled by the chaperone/usher pathway (CUP), play an essential role in invasion of bladder cells and in the formation of biofilm-like intracellular bacterial communities (IBCs) that protect bacteria from host defenses and antibiotics.2–5 Further, CUP pili play a critical role in biofilm formation, mediating not only interactions with host tissue, but also colonization of catheters and other surfaces in nosocomial settings. Also, an amyloid fiber called curli is critical in UPEC biofilm formation and the molecular machine that mediates curli assembly has been dissected.6–8 Our understanding of the structural basis of CUP pili biogenesis has led to the design of pilicides that bind to the chaperone and block critical functions thus preventing pilus assembly.9–11 Numerous (>100) CUP systems are now known to be encoded in gram-negative genomes12 but little is known about their function. A typical E. coli genome encodes approximately 10 such systems.13 Further detailed studies of the function and regulation of CUP pili and other extracellular fibers are an important route to understanding the bacterial adaptation and survival strategies that may be particularly relevant to human infections and providing targets for the development of new therapeutics. Here we describe the development of new compounds that will serve as a strong foundation to support investigation of novel anti-fiber therapeutics targeting critical assembly and adhesion functions of fibers required for the determination of tropism and the organization of bacterial communities during infection.
Ring-fused 2-pyridones are peptidomimetics that can target protein–protein interactions in macromolecular assembly. We have previously shown that ring-fused dihydrothiazolo 2-pyridones (1) provide an excellent central fragment for design and synthesis of compounds that block the formation of pili and curli.10, 11, 14 Pilicides (2 a,b) are compounds that block pilus biogenesis (exemplified in UPEC), whereas curlicides (such as 3) prevent curli fiber biogenesis. Development of traceable pilicides and curlicides could potentially be obtained using a biomolecular labeling strategy, for example, by radiolabeling or by the introduction of a fluorescent label. In the latter case, a fluorescent probe is usually attached by using a linker to the biomolecule to avoid any interference with the biomolecular interactions. The low molecular weight of ligands such as the pilicides 2 a,b and curlicide 3, implies that this technique could change the overall molecular composition to a great extent and thereby potentially reduce the bioactivity of these compounds. An alternative approach would be to replace key substituents for bioactivity by a fluorophore. To increase the likelihood of succeeding by using this approach, and thus both retain the biological effect and gain fluorescent properties, the structure–activity knowledge on the central fragment could be used for both the choice of fluorophore and its positioning on the central fragment. One potential problem with this method is a higher probability for fluorescent quenching of the fluorophore due to its close proximity to the bioactive central fragment. Even so, exchange of one of the substituents on the peptidomimetic central fragment 1 to a fluorophore could render attractive compounds for uptake/distribution studies, development of competition-based assays, Förster resonance energy transfer (FRET) studies on binding interactions, and to specifically image conserved pili and curli assembly machineries in bacterial populations. Initial studies of the structure–activity relationships on the ring-fused dihydrothiazolo 2-pyridone central fragment have shown that the C7 and C8 positions are highly important for bioactivity and should preferably carry larger lipophilic substituents.10, 11, 15–17 Consequently, we have in the present study exchanged the substituents in the C7 and C8 positions on the central fragment 1 with coumarin (4) or BODIPY (5) fluorophores (Figure 1). The use of these particular fluorophores could be justified by their good (compound 4) to high (compound 5) quantum yields, absorption/emission wavelengths, lipophilicity, lack of net ionic charge, photostability, different emission colors and relatively small size.18–20
Figure 1.
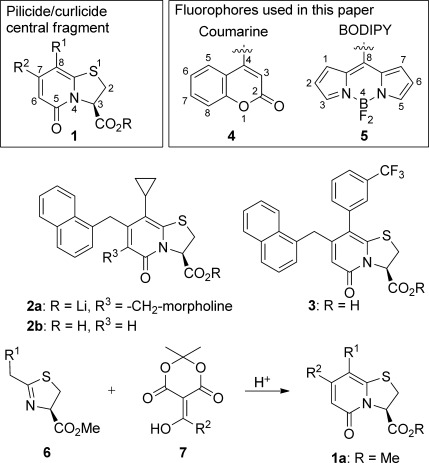
The dihydrothiazolo ring-fused 2-pyridone central fragment 1 (R=H or Li for biological activity), pilicide (2 a,b), curlicide (3), coumarin fluorophore 4, BODIPY fluorophore 5. Synthesis of the thiazolo ring-fused 2-pyridone central fragment (1 a) is performed by using 2-thiazolines (6) and Meldrum’s acid derivatives (7).
The synthesis of the ring-fused dihydrothiazolo 2-pyridone central fragment is based upon an acyl-ketene imine cyclocondensation between Δ2-thiazolines 6 and acyl Meldrum’s acid derivatives 7 (Figure 1).21 Carboxylic acids can be used as starting materials in the synthesis of both 6 and 7 and carboxylic acid functionalized fluorophores have therefore been used as building blocks for further incorporation in the C7 and C8 positions on the central fragment. In addition to this approach, coumarins could also be introduced directly on a bromomethyl-substituted central fragment using deprotonated 4-methyl coumarins. In total, 14 new fluorophore-substituted derivatives of the central fragment 1 have been synthesized and the photophysical measurements of these compounds revealed compounds with high quantum yields. In addition, biological evaluation of these compounds as pilicides and curlicides showed a great biological effect of several compounds, with some being both potent inhibitors of pili- and curli-dependent biofilm formation and having fluorescent properties. Finally, treatment of the UPEC strain UTI89 with the compounds under pili producing conditions shows that the compounds are associated to the bacteria and seem to discriminate between different bacteria in a population.
Results and Discussion
Coumarins substituted with electron-donating groups in position 7 such as 7-methoxy and 7-diethylamino coumarins are frequently used fluorophores (Figure 1). The linker to the pilicide/curlicide central fragment was preferred through the 4 position on the coumarins to resemble the geometry of the C7 naphthyl substituent in 2 and 3. Consequently, the 7-methoxy coumarin-4-yl acetic acid was first coupled to its corresponding acyl Meldrum’s acid derivative 8 using standard conditions.10 On the basis of previous structure–activity relationships of the pilicide/curlicide central fragment, a phenyl,9, 15 a 3-trifluoromethylphenyl,10, 22 a 2-thiophenyl,17 and a cyclopropyl9, 15 were selected as substituents on the Δ2-thiazolines (9 a–d). Reacting 8 with the 9 a–d in the acyl ketene imine cyclocondensation gave coumarin-substituted thiazolo ring-fused 2-pyridones 10 a–d in 68–86 % yield (Scheme 1). Compounds 10 a–d were next subjected to hydrolysis to give the corresponding carboxylic acids 11 a–d in 53–66 % yield.
Scheme 1.

a) TFA, dichloroethane, microwave irradiation (MWI): 120 °C, 140 s (10 a–d: 77 %, 86 %, 77 and 68 %, respectively); b) i) 0.1 M aq LiOH, THF/MeOH (4:1); ii) AcOH (11 a–d: 66, 58, 53 and 66 %, respectively).
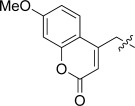
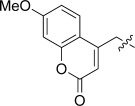
The photophysical evaluation of 11 a–d showed relatively low quantum yields of fluorescence (ΦF, Table 1). The C8 phenyl-substituted 11 a displayed highest quantum yield (ΦF=5 %), followed by the C8 3-trifluomethylphenyl-substituted 11 b (ΦF=1 %), the C8 2-thiophenyl-substituted 11 c (ΦF=0.7 %), and finally the C8 cyclopropyl-substituted 11 d (ΦF=0.4 %). These results may be due to a quenching effect of the fluorophore by the neighboring dihydrothiazolo ring-fused 2-pyridone pilicide/curlicide central fragment. In an attempt to circumvent this, the linker between the fluorophore and the 2-pyridone central fragment was increased by a one carbon extension. This was realized by using deprotonated 4-methyl coumarins as nucleophiles on bromomethyl-substituted 2-pyridone central fragments. By using this strategy the introduction of both a 7-methoxy-substituted coumarin and a 7-diethylamine-substituted coumarin could be accomplished (Scheme 2). The intermediate bromomethyl-substituted 2-pyridone central fragments (13 a and 13 b) were synthesized following a previously published procedure.23 Addition of lithiated coumarins (14 a and 14 b) to 13 a and 13 b generated the one carbon extended coumarin-substituted ring-fused 2-pyridones 15 a–d in 74–83 % yield. Subsequent hydrolysis rendered 16 a–d in high yields (81–91 %; Scheme 2).
Table 1.
The fluorophore-substituted compounds photophysical properties and abilities to inhibit pili- and curli-dependent biofilm formation. 
| ID | R1 (C8) | R2 (C7) | X | EC50[a] Pili [μM] | EC50[a] Curli [μM] | λabs[b,c] [nm] | λfl[b,d] [nm] | Quantum Yield[b] ΦF [%] (λex nm) |
|---|---|---|---|---|---|---|---|---|
| 11 a |
|
 |
-S- | >200 | NA[e] | 328 | 394 | 5 (330)[f] |
| 11 b | 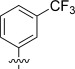 |
 |
-S- | >200 | NA[e] | 330 | 420 | 1 (330)[f] |
| 11 c |  |
-S- | >200 | NA[e] | 328 | 396 | 0.7 (330)[f] | |
| 11 d | 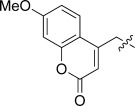 |
-S- | >200 | NA[e] | 328 | 413 | 0.4 (330)[f] | |
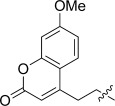
| 16 a | 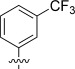 |
 |
-S- | 65 | 175 | 329 | 430 | 0.6 (355)[f] |
| 16 b | 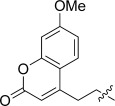 |
-S- | 156 | NA[e] | 327 | 393 | 0.5 (330)[f] | |
| 16 c | 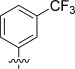 |
 |
-S- | 18 | 25 | 393 | 474 | 15 (390)[g] |
| 16 d | 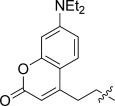 |
-S- | 12 | NA[e] | 392 | 484 | 6 (330)[f] | |
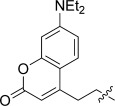
| 23 | 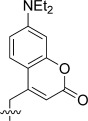 |
 |
-S- | 5 | 17 | 396 | 478 | 11 (346)[f] |
| 28 | 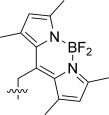 |
 |
-S- | 4 | 14 | 506 | 524 | 10 (470)[h] |
| 31 | 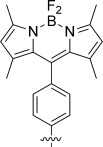 |
 |
-S- | 14 | 12 | 502 | 514 | 67 (470)[h] |
| 34 | 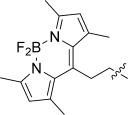 |
-S- | 10 | 24 | 498 | 509 | 11 (480)[h] | |
| 37 | 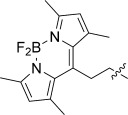 |
-O- | 13 | 14 | 497 | 531 | 27 (480)[h] | |
| 38 | 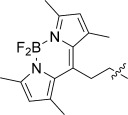 |
-S(O)- | 29 | 40 | 498 | 516 | 71 (480)[h] | |
| 3 |  |
 |
-S- | 17 | 38 | – | – | – |
[a] Estimated from 16–32 data points on every concentration. [b] All substances were dissolved in DMSO and subsequently diluted in phosphate buffer at pH 7.0. The samples DMSO concentrations never exceed 5 wt %. The sample concentrations in the DMSO stock solutions are adjusted so that the final samples never have a peak absorbance higher than 0.1. [c] Wavelengths of the peak absorption. [d] The peak fluorescence. [e] Not active. [f] Reference: POPOP in MeOH. [g] Reference: Perylene in cyclohexane. [h] Reference: Rhodamine 6G in water.
Scheme 2.
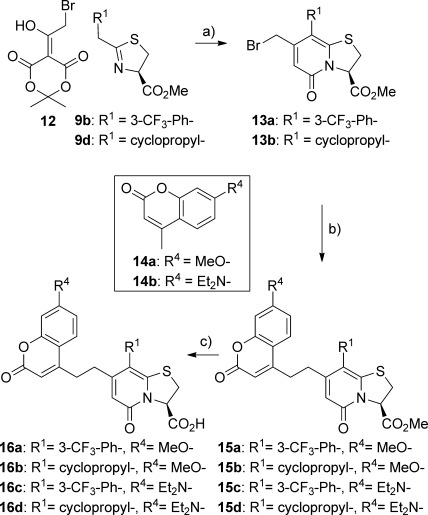
a) TFA, dichloroethane, MWI: 120 °C, 140 s (13 a,b: 92 and 87 %, respectively); b) 14 a or 14 b, LiHMDS, THF, −35 °C, 1 h (15 a–d: 74, 82, 83 and 75 %, respectively); c) For 16 a,c: 0.1 M aq LiOH, THF/MeOH (4:1) 1.5 h; ii) AcOH (16 a,c: 86 and 82 %, respectively), For 16 b,d: i) LiBr, TEA, MeCN (+2 v/v % H2O), RT, 3 h; ii) AcOH (16 b,d: 91 and 81 %, respectively).
The one carbon extended linker did not influence the quantum yield (16 a, ΦF=0.6 % and 16 b ΦF=0.5 %; Table 1). However, replacing the 7-(methoxy)coumarin by a 7-(diethylamino)coumarin as in 16 c and 16 d increased the quantum yields (ΦF=15 and 6 %, respectively, Table 1) and gave C7 coumarin-substituted compounds with useful fluorescent properties. As a consequence, the 7-(diethylamino)coumarin was used in the introduction of a coumarin in the C8 position of the pilicide/curlicide central fragment. The coumarin-substituted Δ2-thiazoline 20 was synthesized in three steps starting from 14 b and ethyl 2-bromoacetate (Scheme 3).24 Subsequent reaction with acyl Meldrum’s acid derivative 21 rendered the C8 coumarin-substituted central fragment 22 in 73 % yield (Scheme 3). After hydrolysis of 22 into the target compound 23 the photophysical properties were evaluated. From this a quantum yield comparable with the other 7-(diethylamino)coumarin-substituted compounds was observed for 23 (ΦF=11 %, Table 1). Thus, compounds with a coumarin fluorophore in both the C7 and C8 positions of the pilicide/curlicide central fragment have been synthesized.
Scheme 3.
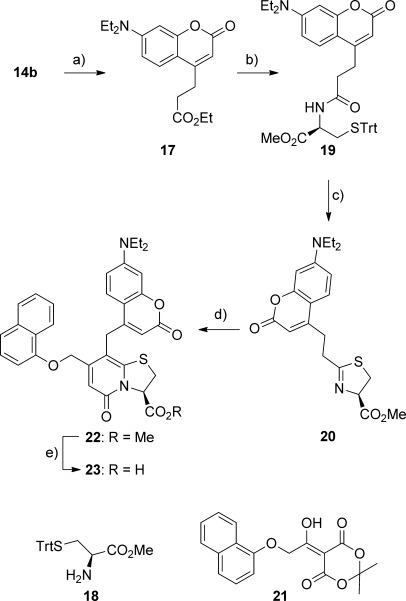
a) LiHMDS, DMPU, ethyl 2-bromoacetate, THF, −20 °C, 35 min, 77 %; b) i) 1 M (aq) LiOH, THF, RT, 16 h, ii) HBTU, 18, DMF, RT, 17 h, 60 %; c) TiCl4, CH2Cl2, 0 °C to RT, 16 h, 40 %; d) 21, TFA, dichloroethane, MWI: 120 °C, 3 min, 73 %; e) 0.1 M aqueous LiOH, THF, RT, 1 h; ii) AcOH, 91 %.
To increase the probability of identifying a bioactive compound with useful fluorescence properties, the possibility of introducing a different fluorophore on the central fragment was investigated. 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) is a known fluorophore that normally gives high quantum yields, carries no net charges, is relatively insensitive to the choice of solvent, and should give a different emission color as compared to the coumarins.20 The synthesis of the BODIPY core structure is often accompanied by low yields. The desired 8-propanoic acid-functionalized 1,3,5,7-tetramethyl-substituted BODIPY (24) has previously been synthesized in 21 % yield.25 From 24, the introduction of a BODIPY substituent in the C8 position of the pilicide/curicide central fragment could be pursued. Coupling 24 using a standard coupling procedure with methylester-protected cysteine gave the intermediate 25 in 64 % yield (Scheme 4). Ring closure of 25 to give 26 followed by acyl-ketene imine cyclocondensation with Meldrum’s acid derivative 21 gave the BODIPY-substituted dihydrothiazolo ring-fused 2-pyridone 27 in 64 % yield. The following hydrolysis proved to be problematic and the harsh conditions needed for this transformation ultimately gave 28 in 29 % yield as a racemate (Scheme 4). However, on the basis of previous reports showing that both enantiomers of the pilicide central fragment are biologically active, it was still entirely possible that racemic 28 could display interesting bioactivity.9, 26
Scheme 4.
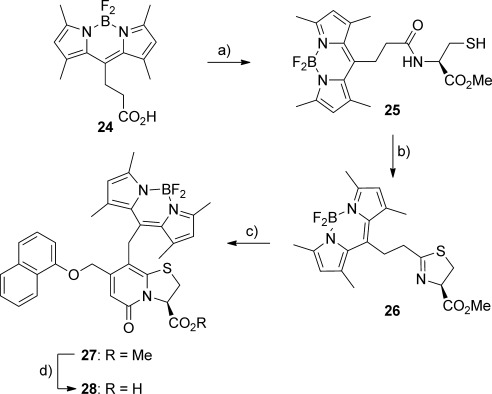
a) i) (COCl)2, CH2Cl2, RT, 17 h; ii) TEA, cysteine methyl ester, CH2Cl2, 0 °C to RT, 4.5 h, 64 %; b) i) TiCl4, CH2Cl2, 0 °C to RT, 4.5 h; ii) BF3⋅OEt2, CH2Cl2, RT, 40 min, 75 %; c) 21, TFA, dichloroethane, MWI: 120 °C, 140 s, 64 %; d) i) LiI, pyridine, MWI: 140 °C, 15 min; ii) TEA, BF3⋅OEt2, dichloroethane, 80 °C, 15 min, 29 %.
The photophysical evaluation of 28 gave a quantum yield of 10 %, which is surprisingly low for a BODIPY-substituted compound (Table 1). We hypothesized that photo-quenching was a possible reason for this and thus the use of an aryl linker between the fluorophore and the central fragment could circumvent the problem. This would not only increase the distance between the fluorophore and the pilicide/curlicide central fragment but it would also restrict the rotation of the 1,3,5,7-tetramethyl-substituted BODIPY, which is known to generate higher quantum yields.20 Exchange of the methylene linker for a phenyl results in the need for a revised synthetic approach. A recent publication shows that it is possible to introduce a benzoic acid in the C8 position of the pilicide/curlicide central fragment by Suzuki–Miyaura couplings.27 From this benzoic acid derivative, the transformation into the desired C8 BODIPY-substituted central fragment seemed feasible. Consequently, 2-pyridone 29 was treated with oxalyl chloride followed by reaction with 2,4-dimethylpyrrole in the presence of BF3⋅OEt2 and triethylamine to give the desired C8 BODIPY-substituted central fragment 30 in 15 % yield. Subsequent hydrolysis was straightforward for this BODIPY derivative, giving the corresponding carboxylic acid 31 in 84 % yield (Scheme 5). As hypothesized, introduction of the phenyl spacer in the C8 position increased the quantum yield of 31 to a satisfactory 67 % (Table 1).
Scheme 5.
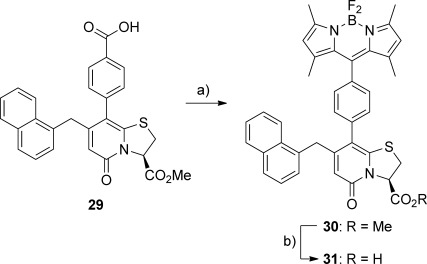
a) i) (COCl)2, DMF, CH2Cl2, RT, 1 h; ii) TEA, BF3⋅OEt2, 2,4-dimethylpyrrole, dichloroethane, MWI: 140 °C, 50 min, 15 %; b) 0.1 M aqueous LiOH, THF, RT, 1 h; ii) H+, 84 %.
With these two C8 BODIPY-substituted compounds (28 and 31) in hand, introduction of BODIPY in position C7 on the central fragment was next investigated. Coupling the BODIPY carboxylic acid 24 with Meldrum’s acid using standard coupling conditions and a simple MeOH trituration as purification gave the BODIPY-substituted acyl Meldrum’s acid derivative 32 in 82 % yield (Scheme 6). From this versatile intermediate the following acylketene imine cyclocondensation to the C7 BODIPY substituted pilicide/curlicide central fragment 33 was performed in 88 % yield. Sequential hydrolysis gave the corresponding carboxylic acid 34 (Scheme 6). The photophysical properties of 34 suggest, in analogy with the coumarins, that the BODIPY fluorophore also seems to be partially quenched in this position (ΦF=11 %, Table 1).
Scheme 6.
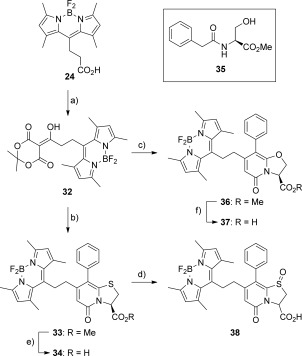
a) DCC, Meldrum’s acid, DMAP, CH2Cl2, 0 °C RT, 8 h, 82 %; b) 9 a, TFA, dichloroethane, MWI: 120 °C, 140 s, 85 %; c) i) 35, (NH4)2MoO4, toluene, reflux soxhlet (3 MS), 6 h; ii) 32, TFA, toluene, reflux soxhlet (3 MS), 2 h, 73 %; d) starting from 34, mCPBA, CH2Cl2, RT, 15 min, 80 %; e) i) 0.1 M aq LiOH, THF/MeOH (4:1); ii) AcOH, 88 %; f) i) LiBr, TEA, MeCN (+2 v/v % H2O), RT, 3 h; ii) AcOH, 88 %.
Even though the quantum yields for many of the synthesized compounds are acceptable and, in the case of 31, high, the underlying reason for the observed photo-quenching was still unknown. Thus, we further investigated the role of the sulfur in position 1 of the pilicide/curlicide central fragment on the photophysical properties. The sulfur in the central fragment was first replaced by an oxygen by reacting 32 and serine derivative 35 in a previously reported two-step one-pot procedure to give 36 in 73 % yield (Scheme 6).28, 29 Hydrolysis of 36 was straightforward and gave 37 in 88 % yield. The quantum yield of 37 (ΦF=29 %, Table 1) was almost three times greater than that of its sulfur-containing counterpart 34 (ΦF=11 %, Table 1). This suggests that the sulfur at position 1 of the pilicide/curlicide central fragment is at least partially responsible for the photo-quenching. To further verify this, the electronic properties of the sulfur were altered through oxidation. Treating 34 with mCPBA gave the sulfoxide 38 in 80 % yield as a 4:1 mixture of diastereomers (Scheme 6). Photophysical evaluation of sulfoxide 38 showed a great increase of quantum yield (ΦF=71 %, Table 1) compared to the parent sulfide 34 (ΦF=11 %, Table 1) thus supporting the theory that the sulfide in position 1 of the central fragment is involved in the observed partial photo-quenching.
The 14 fluorophore-substituted compounds were biologically evaluated for their ability to block pili and curli formation as measured by pili- or curli-dependent biofilm assays on polyvinylchloride surfaces.10, 11, 30 The amount of biofilm that is formed in the E. coli clinical isolate UTI89 grown in the presence of pilicide or curlicide, is related to the potency of the compound in blocking piliation or curliation. The results are summarized in Table 1.
The strategy of generating fluorescent pilicides by replacing key pilicide substituents with fluorophores based on structure–activity knowledge was successful and many of the tested compounds were both highly potent and fluorescent. Several of the compounds were able to inhibit formation of both pili- and curli-dependent biofilm, which is highly valuable from a therapeutic perspective. From the pili-dependent biofilm evaluation, C8 substitution with a methylene spacer using both coumarin (23) and BODIPY (28) fluorophores gave the most potent pilicides with estimated half-maximal effective concentrations (EC50) of 5 and 4 μM, respectively. Interestingly, compound 28 is also among the most potent inhibitors of curli-dependent biofilm formation with an estimated EC50 of 14 μM and is thus a potent dual pilicide–curlicide. This should be compared with the best reported di-substituted compound 3 (known as FN075), which exhibited EC50 values of 17 μM in pili-dependent biofilm inhibition and 38 μM in curli-dependent biofilm inhibition (Table 1). The phenyl spacer in 31 resulted in a slight decreased pilicide activity compared to the methylene linker in 28, but is still in the same range as 3. The C7 BODIPY-substituted derivatives all displayed good inhibitory properties of pili- and curli-dependent biofilm formation. The sulfide (34) and oxygen analogue (37) are more potent than the sulfoxide (39) both as pilicides and curlicides (Table 1). However, the fact that the sulfoxide retains much of the biofilm-inhibition activity might be of importance from a therapeutic and drug metabolism perspective. For the coumarin-substituted compounds with ethylene linker (16 a–d), the 7-(diethylamino)coumarins 16 c,d are more active pilicides than the 7-(methoxy)coumarins 16 a,b (Table 1). Furthermore, the compounds having an ethylene linker are more potent pilicides than the compounds with a methylene linker (16 a and 16 b compared with 11 b and 11 d). None of the four 7-(methoxy)coumarins with a methylene linker 11 a–d showed any activity (Table 1).
Finally, to study the compounds distribution in a bacterial population, UPEC strain UTI89 was grown under pili-producing conditions (24 h at 37 °C static) with compounds 28 and 34 and examined by fluorescence microscopy (Figure 2). Compounds 28 and 34 appeared to bind bacteria differently, with 34 appearing more punctate and 28 appearing more diffuse. Further experiments are working to examine the reason for these observed differences. We know that even under pilus-inducing conditions, not all bacteria are expressing pili. Thus it is likely that the pilicide is only binding bacteria expressing pili. Experiments are underway to confirm this hypothesis.
Figure 2.
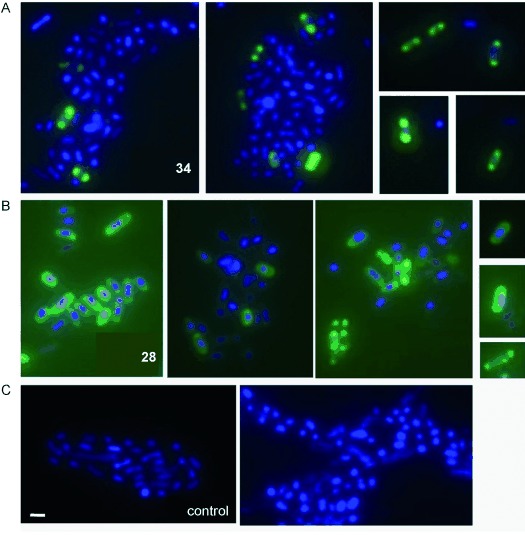
The UPEC strain UTI89 grown for 24 h with and without 100 μM of dual pilicide-curlicide 34 and 28. Blue is DAPI-nuclear stain, green is the compound. A) Bacteria grown in the presence of 100 μM of compound 34. B) Bacteria grown in the presence of 100 μM of compound 28. C) Bacteria grown without any added compound (scale bar is 2 μm).
Conclusion
We have herein developed compounds that are both fluorescent and highly active inhibitors of pili- and curli-dependent biofilm formation. The synthesis of these compounds was initiated to facilitate profound studies of the pilicides and curlicides and to gain information about the complex systems involved in the formation of pili and curli. To create these fluorescent and bioactive compounds we implemented a strategy based on structure–activity information, in which important substituents for bioactivity on the pilicide/curlicide central fragment were replaced by fluorophores. Synthetic methods were developed to enable a total of 14 compounds with either a coumarin or a BODIPY motif in the C7 or the C8 position. Consequently, new interesting intermediates and new reactions to introduce BODIPY fluorophores (e.g., via Meldrum’s acid derivatives and subsequent acyl ketene cyclocondensations) have been developed. Photo-quenching was frequently observed but its origin was elucidated and circumvented to give compounds with useful fluorescence properties. Biological evaluation using whole bacterial pili- and curli-biofilm assays revealed several compounds that are both fluorescent and highly active. Finally, treatment of UPEC strain UTI89 with the compounds under pili-producing conditions shows that the compounds are associated to the bacteria with a heterogeneous distribution over a population. The use of these compounds to study the biological systems they interact with, for example, the relation between the observed staining pattern of the compounds and the heterogeneous distribution of pili production in bacterial populations, is a matter of future studies within our laboratories.
Experimental Section
General synthesis: All reactions were carried out under an inert atmosphere with dry solvents under anhydrous conditions, unless otherwise stated. CH2Cl2 and 1,2-dichloroethane (DCE) and was distilled from calcium hydride and THF was distilled from potassium. DMF was distilled and dried over 3 Å molecular sieves. All microwave reactions were carried out in a monomode reactor (Smith Synthesizer, Biotage AB) using Smith process vials sealed with a Teflon septa and an aluminum crimp top. Reaction times refer to the irradiation time at the target temperature not the total irradiation time. The temperature was measured with an IR sensor. Flash column chromatography (eluent given in brackets) employed normal phase silica gel (Matrex, 60 Å, 35–70 μm, Grace Amicon). The 1H and 13C NMR spectra were recorded at 298 K with a Bruker DRX-400 spectrometer in CDCl3 (residual CHCl3 (δH=7.26 ppm) or CDCl3 (δC=77.0 ppm) as internal standard), [D6]DMSO (residual [D5]DMSO (δH=2.50 ppm) or [D6]DMSO (δC=40.0 ppm) as internal standard), [D6]Acetone (residual [D5]Acetone (δH=2.05 ppm) or [D6]Acetone (δC=29.9 ppm) as internal standard), and [D4]MeOH (residual [D3]MeOH (δH=3.30 ppm) or [D4]MeOH (δC=49.0 ppm) as internal standard). IR spectra were recorded on an ATI Mattson Genesis Series FTIR spectrometer. Optical rotations were measured with a PerkinElmer 343 polarimeter at 20 °C. MS data were recorded using electron spray (ES+) ionization on a Waters Micromass ZQ 2000 spectrometer. HRMS was obtained Micromass Q-Tof Ultima mass spectrometer and [M+H]+ molecular ions were generated by electrospray ionization.
(3R)-7-((7-Methoxy-2-oxo-2H-chromen-4-yl)methyl)-5-oxo-8-phenyl-3,5-dihydro-2H-thiazolo[3,2-a]pyridine-3-carboxylic acid (11 a): LiOH (0.1 M, 1.5 mL, 1 equiv) was added dropwise to a stirred solution of 10 a (71.3 mg, 0.15 mmol, 1 equiv) dissolved in THF/MeOH (2 mL, 4:1). The reaction mixture was stirred for two hours at room temperature before being concentrated. Purification by silica gel chromatography (CH2Cl2/MeOH/AcOH, 90:8:2) and then lyophilized from MeCN/H2O (1:3) to give 11 a (47 mg, 66 %). [α]D=−17 (c=0.25 in CHCl3); 1H NMR (400 MHz, [D6]DMSO): δ=3.49 (dd, J1=1.79 Hz, J2=11.82 Hz, 1 H), 3.72–3.88 (m, 6 H), 5.48 (dd, J1=1.63 Hz, J2=9.01 Hz, 1 H), 5.93 (s, 1 H), 6.01 (s, 1 H), 6.88 (dd, J1=2.50 Hz, J2=8.83 Hz, 1 H), 6.96 (d, J=2.49, 1 H), 7.20–7.28 (m, 1 H), 7.29–7.36 (m, 2 H), 7.36–7.44 ppm (m, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=32.4, 35.7, 56.8, 64.6, 101.9, 112.8, 113.1, 115.0, 115.3 (2C), 127.0, 129.2, 129.8 (2C), 130.8 (broad, 2C), 136.8, 149.6, 150.8, 154.0, 155.8, 160.79, 160.83, 163.3, 170.4 ppm; IR:  =1712, 1608, 1554, 1484, 1423, 1388, 1257, 1207, 1025, 836, 802, 701 cm−1; HRMS (EI): m/z: calcd for C25H20NO6S: 462.1011 [M+H]; found: 462.1018.
=1712, 1608, 1554, 1484, 1423, 1388, 1257, 1207, 1025, 836, 802, 701 cm−1; HRMS (EI): m/z: calcd for C25H20NO6S: 462.1011 [M+H]; found: 462.1018.
(3R)-7-((7-Methoxy-2-oxo-2H-chromen-4-yl)methyl)-5-oxo-8-(3-(trifluoromethyl)phenyl)-3,5-dihydro-2H-thiazolo[3,2-a]pyridine-3-carboxylic acid (11b): Prepared according to the procedure described for compound 11 a starting from 10 b (49 mg, 0.09 mmol, 1 equiv), giving 11 b (10 mg, 58 %). [α]D=−4 (c=0.51 in CHCl3); 1H NMR (400 MHz, [D4]MeOH/CDCl3 1:1): δ=3.60–3.68 (m, 1 H), 3.68–3.78 (m, 3 H), 3.83 (s, 3 H), 5.57 (d, J=8.11 Hz, 1 H), 5.97 (s, 1 H), 6.22 (s, 1 H), 6.73–6.81 (m, 2 H), 7.21 (d, J=8.73, 1 H), 7.39–7.61 ppm (m, 5 H); 13C NMR (100 MHz, [D4]MeOH/CDCl3 1:1): δ=33.1, 35.8, 56.2, 66.4, 101.5, 112.6, 112.8, 113.3, 115.6, 115.9, 124.3 (q, J=272.34 Hz), 125.9, 126.0, 127.4 (d, J=29.1 Hz), 130.4, 131.9 (q, J=32.42 Hz), 134.2 (d, J=34.73 Hz), 137.1, 150.9, 151.0, 154.1, 155.8, 162.3, 162.7, 163.8, 172.5 ppm; IR:
 =1716, 1609, 1484, 1434, 1388, 1330, 1288, 1211, 1126, 848, 705 cm−1; HRMS (EI): m/z: calcd for C26H19F3NO6S: 530.0885 [M+H]; found: 530.0880.
=1716, 1609, 1484, 1434, 1388, 1330, 1288, 1211, 1126, 848, 705 cm−1; HRMS (EI): m/z: calcd for C26H19F3NO6S: 530.0885 [M+H]; found: 530.0880.
(3R)-7-((7-Methoxy-2-oxo-2H-chromen-4-yl)methyl)-5-oxo-8-(thiophen-2-yl)-3,5-dihydro-2H-thiazolo[3,2-a]pyridine-3-carboxylic acid (11c): Prepared according to the procedure described for compound 11 a starting from 10 c (72.2 mg, 0.15 mmol, 1 equiv) to give 11 c (38 mg, 53 %). [α]D=−17 (c=0.69 in CHCl3); 1H NMR (400 MHz, [D6]DMSO): δ=3.51 (dd, J1=1.77 Hz, J2=11.61 Hz, 1 H), 3.78–3.83 (m, 1 H), 3.85 (s, 2 H), 5.43 (dd, J1=1.52 Hz, J2=9.16 Hz, 1 H), 5.91 (s, 1 H), 6.04 (s, 1 H), 6.91 (dd, J1=2.56 Hz, J2=8.88 Hz, 1 H), 6.98 (d, J=2.54 Hz, 1 H), 7.03–7.07 (m, 2 H), 7.48 (d, J=8.88 Hz, 1 H), 7.56–7.60 ppm (m, 1 H). 13C NMR (100 MHz, [D6]DMSO): δ=32.6, 35.8, 56.9, 65.1, 101.9, 107.1, 112.78, 112.81, 113.1, 115.5, 126.9, 128.4, 128.9, 130.4, 136.8, 151.5, 152.4, 154.2, 155.8, 160.7, 160.9, 163.3, 170.4 ppm; IR:
 =1712, 1646, 1608, 1481, 1438, 1388, 1280, 1207, 1145, 1018, 987, 840, 705 cm−1; HRMS (EI): m/z: calcd for C23H18NO6S2: 468.0576 [M+H]; found: 468.0587.
=1712, 1646, 1608, 1481, 1438, 1388, 1280, 1207, 1145, 1018, 987, 840, 705 cm−1; HRMS (EI): m/z: calcd for C23H18NO6S2: 468.0576 [M+H]; found: 468.0587.
(3R)-8-Cyclopropyl-7-((7-methoxy-2-oxo-2H-chromen-4-yl)methyl)-5-oxo-3,5-dihydro-2H-thiazolo[3,2-a]pyridine-3-carboxylic acid (11 d): Prepared according to the procedure described for compound 11 a starting from 10 d (49.0 mg, 0.11 mmol, 1 equiv) to give 11 d (31.2 mg, 66 %). [α]D=−58 (c=0.20 in CHCl3); 1H NMR (400 MHz, [D6]DMSO): δ=0.51–0.71 (m, 2 H) 0.76–0.88 (m, 2 H), 1.56–1.66 (m, 1 H), 3.52 (dd, J1=1.76 Hz, J2=11.86 Hz, 1 H), 3.82 (dd, J1=9.08 Hz, J2=11.87 Hz, 1 H), 3.86 (s, 3 H), 4.10–4.27 (m, 2 H), 5.40 (dd, J1=1.75 Hz, J2=9.08 Hz, 1 H), 5.79 (s, 1 H), 6.03 (s, 1 H), 6.97 (dd, J1=2.55 Hz, J2=8.87 Hz, 1 H), 7.03 (d, J=2.51 Hz, 1 H), 7.66 ppm (d, J=8.88 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=7.6, 7.8, 11.2, 32.2, 34.9, 56.5, 63.8, 101.6, 111.8, 112.4, 112.7, 112.9, 114.5, 127.0, 149.6, 152.9, 154.3, 155.6, 160.4, 160.6, 163.0, 170.1 ppm; IR: ν<Ü=>1712, 1643, 1608, 1481, 1438, 1388, 1280, 1207, 1141, 1041, 1022, 987, 840, 705 cm−1; HRMS (EI): m/z: calcd for C22H20NO6S: 426.1011 [M+H]; found: 426.1016.
(3R)-7-(2-(7-Methoxy-2-oxo-2H-chromen-4-yl)ethyl)-5-oxo-8-(3-(trifluoromethyl)phenyl)-3,5-dihydro-2H-thiazolo[3,2-a]pyridine-3-carboxylic acid (16 a): LiOH (0.1 M, 0.36 mL, 1 equiv) was added drop wise to a stirred solution of 15 a (20 mg, 36 μmol, 1 equiv) dissolved in THF/MeOH (1 mL, 4:1). The reaction mixture was left for approximately one and a half hours at room temperature. The solution was then concentrated, and co-concentrated from MeOH three times. Purification by silica gel chromatography (CH2Cl2/MeOH, 92:8, 90:8 + 2 % AcOH), concentrated and co-concentrated from CH2Cl2 and chloroform and then lyophilized from MeCN/H2O (1:3) giving 16 a as a white powder (17 mg, 86 %). [α]D=2 (c=0.51 in CHCl3); 1H NMR (400 MHz, [D4]MeOH/CDCl3 11:9): δ=2.62–2.73 (m, 2 H), 2.75–2.84 (m, 2 H), 3.55–3.62 (m, 1 H), 3.75–3.84 (m, 1 H), 3.84 (s, 3 H), 5.64–5.70 (m, 1 H), 5.89 (s, 1 H), 6.34 (s, 1 H), 6.9 (dd, J1=2.19 Hz, J2=8.64 Hz, 1 H), 6.78 (d, J=2.42 Hz, 1 H), 6.91 (t, J=8.54 Hz, 1 H), 7.44–7.51 (m, 1 H), 7.52–7.64 (m, 2 H), 7.67–7.73 ppm (m, 1 H); 13C NMR (100 MHz, [D4]MeOH/CDCl3 11:9): δ=32.46, 32.49, 32.9 (split), 56.1, 64.7, 101.7, 111.4, 112.7, 113.3, 114.6, 115.7, 124.4 (q, J=272.22 Hz), 125.6 (split), 126.0 (split), 127.5 (split), 130.7 (d, J=5.08 Hz), 132.2 (q, J=32.56 Hz), 134.5 (d, J=30.19 Hz), 137.5, 150.0, 154.8, 156.05, 156.07, 162.8, 163.7, 169.9, 170.0 ppm; IR:
 =1712, 1612, 1484, 1438, 1388, 1284, 1207, 1145, 1045, 1022, 987, 840, 705 cm−1; HRMS (EI): m/z: calcd for C27H21F3NO6S: 544.1042 [M+H]; found: 544.1049.
=1712, 1612, 1484, 1438, 1388, 1284, 1207, 1145, 1045, 1022, 987, 840, 705 cm−1; HRMS (EI): m/z: calcd for C27H21F3NO6S: 544.1042 [M+H]; found: 544.1049.
(3R)-8-Cyclopropyl-7-(2-(7-methoxy-2-oxo-2H-chromen-4-yl)ethyl)-5-oxo-3,5-dihydro-2H-thiazolo[3,2-a]pyridine-3-carboxylic acid (16 b): By following a previously published procedure,31
15 b (25 mg, 0.056 mmol) was hydrolyzed to its corresponding carboxylic acid 16 b (91 % yield). [α]D=−4 (c=0.3 in CHCl3/MeOH 2:1); 1H NMR (400 MHz, CDCl3): δ=0.56–0.66 (m, 2 H), 0.85–1.02 (m, 2 H), 1.50–1.60 (m, 1 H), 2.96–3.16 (m, 4 H), 3.64–3.74 (m, 1 H), 3.75–3.83 (m, 1 H), 3.87 (s, 3 H), 5.65–5.73 (m, 1 H), 6.14 (s, 1 H), 6.41 (s, 1 H), 6.81–6.91 (m, 2 H), 7.51–7.59 (m, 1 H), 8.9–9.4 ppm (bs, 1 H); 13C NMR (100 MHz, CDCl3): δ=7.5, 7.9, 11.0, 30.7, 30.9, 31.0, 55.8, 64.2, 101.2, 111.0, 112.4, 112.6, 113.0, 116.2, 125.1, 150.0, 154.7, 155.5, 157.9, 161.3, 162.4, 162.8, 168.4 ppm; IR:
 =1714, 1609, 1486, 1207, 1145, 1024, 835 cm−1; HRMS (EI): m/z: calcd for C23H22NO6S: 440.1168 [M+H]; found: 440.1169.
=1714, 1609, 1486, 1207, 1145, 1024, 835 cm−1; HRMS (EI): m/z: calcd for C23H22NO6S: 440.1168 [M+H]; found: 440.1169.
(3R)-7-(2-(7-(Diethylamino)-2-oxo-2H-chromen-4-yl)ethyl)-5-oxo-8-(3-(trifluoromethyl)phenyl)-3,5-dihydro-2H-thiazolo[3,2-a]pyridine-3-carboxylic acid (16c): Prepared according to the procedure described for compound 16 a, starting from 15 c (20 mg, 0.036 mmol, 1 equiv) gave 16 c as a pale yellow powder (41 mg, 82 %). [α]D=3 (c=0.60 in CHCl3); 1H NMR (400 MHz, [D4]MeOH): δ=1.18 (t, J=6.99 Hz, 6 H), 2.59–2.78 (m, 4 H), 3.38 (q, J=7.03 Hz, 4 H), 3.52–3.80 (m, 2 H), 5.48–5.74 (m, 2 H), 6.27–6.50 (m, 3 H), 6.65–6.81 (m, 1 H), 7.42–7.75 ppm (m, 4 H); 13C NMR (100 MHz, [D6]DMSO): δ=12.2 (2C), 31.1, 32.0 (broad, split), 32.7 (broad, split), 43.9, 65.3 (broad), 79.2, 96.9, 106.7, 106.9, 108.3, 112.2, 113.7, 124.0 (q, J=272.59 Hz), 124.8, 125.1, 126.8 (d, J=9.48 Hz), 129.7 (q, J=31.61 Hz), 130.2, 134.7, 137.7, 148.7, 150.1, 152.0, 155.4, 155.8, 160.3, 160.7, 167.7 ppm; IR:
 =1708, 1596, 1527, 1484, 1415, 1330, 1272, 1160, 1122, 1072, 802, 705 cm−1; HRMS (EI): m/z: calcd for C30H28F3N2O5S: 585.1671 [M+H]; found: 585.1667.
=1708, 1596, 1527, 1484, 1415, 1330, 1272, 1160, 1122, 1072, 802, 705 cm−1; HRMS (EI): m/z: calcd for C30H28F3N2O5S: 585.1671 [M+H]; found: 585.1667.
(3R)-8-Cyclopropyl-7-(2-(7-(diethylamino)-2-oxo-2H-chromen-4-yl)ethyl)-5-oxo-3,5-dihydro-2H-thiazolo[3,2-a]pyridine-3-carboxylic acid (16 d): By following a previously published procedure,31
15 d (27.5 mg, 0.056 mmol) was hydrolyzed to its corresponding carboxylic acid 16 d (81 % yield). [α]D=5 (c=0.13 in MeOH); 1H NMR (400 MHz, CDCl3): δ=0.55–0.67 (m, 2 H), 0.86–1.03 (m, 2 H), 1.21 (t, J=7.10 Hz, 6 H), 1.51–1.61 (m, 1 H), 2.90–3.13 (m, 4 H), 3.41 (q, J=7.07 Hz, 4 H), 3.61–3.70 (m, 1 H), 3.82–3.89 (m, 1 H), 5.64–5.71 (m, 1 H), 5.94 (s, 1 H), 6.33 (s, 1 H), 6.49–6.53 (s, 1 H), 6.57–6.62 (m, 1 H), 7.38–7.43 ppm (m, 1 H); 13C NMR (100 MHz, CDCl3): δ=7.4, 7.9, 11.0, 12.4 (2C), 30.4, 30.6, 31.3, 44.7 (2C), 64.2, 97.9, 107.6, 107.8, 108.7, 113.3, 115.9, 124.9, 149.5, 150.6, 154.9, 156.3, 158.0, 162.2, 162.7, 168.4 ppm; IR:
 =1710, 1613, 1596, 1487, 1415, 1139, 825 cm−1; HRMS (EI): m/z: calcd for C26H29N2O5S: 481.1797 [M+H]; found: 481.1794.
=1710, 1613, 1596, 1487, 1415, 1139, 825 cm−1; HRMS (EI): m/z: calcd for C26H29N2O5S: 481.1797 [M+H]; found: 481.1794.
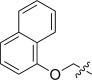
(3R)-8-((7-(Diethylamino)-2-oxo-2H-chromen-4-yl)methyl)-7-((naphthalen-1-yloxy)methyl)-5-oxo-3,5-dihydro-2H-thiazolo[3,2-a]pyridine-3-carboxylic acid (23): Compound 22 (0.34 mmol, 0.2 g) was dissolved in THF (20 mL) and LiOH (0.44 mmol, 0.1 M, 4.4 mL) was added, the reaction was stirred at RT for 1 h. The solvent was evaporated and the crude product was purified with column chromatography on silica gel (first pure EtOAc then CH2Cl2/MeOH/AcOH 90:5:5) to give 23 as a yellow solid (180 mg, 91 % yield). [α]D=−10 (c=0.5 in CHCl3); 1H NMR (400 MHz, [D6]DMSO): δ=7.99 (d, J=8.5 Hz, 1 H), 7.82 (d, J=8.1 Hz, 1 H), 7.60 (d, J=9.1 Hz, 1 H), 7.52–7.30 (m, 4 H), 6.96 (d, J=7.7 Hz, 1 H), 6.62 (dd, J=9.2, 2.4 Hz, 1 H), 6.48 (s, 1 H), 6.44 (d, J=2.4 Hz, 1 H), 5.55–5.49 (m, 1 H), 5.47 (s, 1 H), 5.17–5.06 (m, 2 H), 4.05–3.87 (m, 3 H), 3.65–3.58 (m, 1 H), 3.45–3.34 (m, 4 H), 1.10 ppm (t, 6 H); 13C NMR (100 MHz, [D6]DMSO): δ=169.4, 160.8, 160.1, 155.6, 153.1, 153.0, 150.3, 150.1, 149.5, 133.9, 127.4, 126.4, 125.8, 125.5, 125.3, 124.7, 121.1, 120.5, 113.2, 108.5, 107.3, 105.6, 105.4, 105.2, 96.8, 66.5, 63.3, 44.0 (2C), 31.6, 30.6, 12.3 ppm (2C); HRMS (EI): m/z: calcd for C33H30N2O6S: 583.1903[M+H+]; found: 583.1900.
7-((Naphthalen-1-yloxy)methyl)-5-oxo-8-((1,3,5,7-tetramethyl-4,4-difluoro-4-bora-3a,4a-diaza-s-indacene-8-yl)methyl)-3,5-dihydro-2H-thiazolo[3,2-a]pyridine-3-carboxylic acid (28): LiI (111 mg, 0.83 mmol) was added to a solution 27 (52 mg, 0.08 mmol) in dry pyridine (2 mL) while stirring. The reaction mixture was heated to 140 °C for 15 min by microwave irradiation. The reaction mixture was allowed to attain RT, diluted with CH2Cl2 and washed with 2 % KHSO4. The aqueous layer was extracted three times with CH2Cl2 and the combined organic layers were dried over Na2SO4, filtered and concentrated in vacuo. The resulting dark oil was diluted in 1,2-dichloroethane (2 mL) and TEA (58 μL, 0.42 mmol) was added dropwise while stirring. After 5 min BF3⋅OEt2 was added and the reaction mixture was heated to 80 °C for 15 min. After being allowed to attain RT the reaction mixture was diluted with CH2Cl2 and washed with water. The aqueous layer was extracted five times with CH2Cl2 and the combined organic layers were concentrated in vacuo. Purification by column chromatography (CH2Cl2, 2 % MeOH→CH2Cl2, 2 % MeOH, 1 % AcOH) gave 28 as an orange solid (15 mg, 29 %). [α]D=0; 1H NMR (400 MHz, [D6]DMSO): δ=2.21 (s, 3 H), 2.29 (s, 3 H), 2.43 (s, 6 H), 3.39 (dd, J1=1.26 Hz, J2=11.88 Hz, 2 H), 3.56 (dd, J1=8.76 Hz, J2=11.88 Hz, 2 H), 4.27 (d, J=16.73 Hz, 1 H), 4.35 (d, J=16.73 Hz, 1 H), 5.36–5.40 (m, 3 H), 6.18–6.24 (m, 2 H), 6.58 (s, 1 H), 7.17–7.23 (m, 1 H), 7.43–7.49 (m, 1 H), 7.53–7.61 (m, 3 H), 7.89–7.95 (m, 1 H), 8.24–8.29 ppm (m, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=14.7, 14.72, 15.3, 15.8, 28.53, 32.1, 62.0, 66.8, 105.1, 106.7, 113.4, 121.2, 121.7, 122.1, 122.1, 125.2, 126.2, 127.1, 128.1, 134.1 (broad), 134.6, 138.9, 142.4 (broad), 144.8, 150.6, 153.4, 154.5, 154.9 (broad), 160.5, 169.8 ppm; IR:
 =1647, 1553, 1506, 1198, 1159, 982 cm−1; HRMS (EI): m/z: calcd for C33H31BF2N3O4S: 614.2096 [M+H]; found: 614.2097.
=1647, 1553, 1506, 1198, 1159, 982 cm−1; HRMS (EI): m/z: calcd for C33H31BF2N3O4S: 614.2096 [M+H]; found: 614.2097.
(3R)-7-(Naphthalen-1-ylmethyl)-5-oxo-8-(4-(1,3,5,7-tetramethyl-4,4-difluoro-4-bora-3a,4a-diaza-s-indacene-8-yl)phenyl)-3,5-dihydro-2H-thiazolo[3,2-a]pyridine-3-carboxylic acid (31): Compound 30 (0.045 mmol, 30 mg) was dissolved in THF (5 mL) and LiOH (0.09 mmol, 0.1 M, 0.9 mL) was added, the reaction was stirred at RT for 1 h. The reaction mixture was diluted with water and pH was set to 1 with 1 M HCl (aq). The waterphase was extracted with EtOAc (2×50 mL), the combined organic phases was dried (Na2SO4), filtered and concentrated. The crude product was purified with HPLC, (C18, 250×21.2 mm, 5 μm, 0–100 % MeCN over 1 h) and lyophilized to give 31 as a red solid (25 mg, 84 % yield). [α]D=20 (c 0.05, CHCl3); 1H NMR (400 MHz, [D6]DMSO): δ=7.96–7.88 (m, 1 H), 7.85–7.78 (m, 1 H), 7.78–7.71 (m, 1 H), 7.64–7.34 (m, 7 H), 7.31–7.25 (m, 1 H), 6.18 (s, 1 H), 6.03 (s, 1 H), 5.70 (s, 1 H), 5.55–5.46 (m, 1 H), 4.09 (s, 2 H), 3.92–3.83 (m, 1 H), 3.57–3.49 (m, 1 H), 2.44 (s, 3 H), 2.41 (s, 3 H), 1.42 (s, 3 H), 0.93 ppm (s, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=169.5, 160.0, 154.9 (2C), 152.8, 148.2, 142.6 (2C), 141.4, 137.2, 134.0, 133.8, 133.3, 131.2 (4C), 130.6, 128.6, 128.2 (2C), 127.3, 126.9, 126.3, 125.8, 125.5, 123.4, 121.5 (2C), 114.2, 113.9, 63.4, 35.5, 31.4, 14.2 (2C), 13.8, 13.3 ppm; HRMS (EI): m/z: calcd for C38H32BF2N3O3S: 660.2304 [M+H+]; found: 660.2303.
(3R)-5-Oxo-8-phenyl-7-(2-(1,3,5,7-tetramethyl-4,4-difluoro-4-bora-3a,4a-diaza-s-indacene-8-yl)ethyl)-3,5-dihydro-2H-thiazolo[3,2-a]pyridine-3-carboxylic acid (34): Compound 34 was synthesized by following the same procedure described for 11 a starting from 33 (30 mg, 0.053 mmol). Purification by column chromatography (CH2Cl2/MeOH 97:3 to CH2Cl2/MeOH/AcOH 96:3:1) gave 34 as a red non-crystalline solid (25.5 mg, 88 %). [α]D=34 (c=0.05 in MeOH); 1H NMR (400 MHz, [D6]DMSO): δ=2.25 (s, 6 H), 2.36 (s, 6 H), 2.39–2.49 (m, 2 H), 3.14–3.25 (m, 2 H), 3.43–3.50 (m, 1 H), 3.73–3.82 (m, 1 H), 5.40–5.47 (m, 1 H), 6.19 (s, 2 H), 6.30 (s, 1 H), 7.18–7.24 (m, 1 H), 7.25–7.36 (m, 2 H), 7.36–7.43 ppm (m, 2 H); 13C NMR (100 MHz, [D6]DMSO): δ=14.1 (m, 2C), 15.7 (2C), 25.5, 31.7, 33.4, 64.0, 111.5, 113.8, 121.8 (broad), 128.2, 129.0 (2C), 129.9 130.1, 130.5, 136.2, 140.9 (broad), 144.9, 147.9, 152.0, 153.6 (broad), 160.2, 169.5 ppm; IR:
 =1636, 1548, 1509, 1489, 1407, 1198, 981, 704 cm−1; HRMS (EI): m/z: calcd for C29H29BF2N3O3S: 548.1991 [M+H]; found: 548.1972.
=1636, 1548, 1509, 1489, 1407, 1198, 981, 704 cm−1; HRMS (EI): m/z: calcd for C29H29BF2N3O3S: 548.1991 [M+H]; found: 548.1972.
(3R)-5-Oxo-8-phenyl-7-(2-(1,3,5,7-tetramethyl-4,4-difluoro-4-bora-3a,4a-diaza-s-indacene-8-yl)ethyl)-3,5-dihydro-2H-oxazolo[3,2-a]pyridine-3-carboxylic acid (37): Compound 37 was synthesized by following a previously published procedure,29 starting from 36 (51 mg, 0.094 mmol). Purification by column chromatography (CH2Cl2/MeOH 97:3 to CH2Cl2/MeOH/AcOH 96:3:1) gave 37 as a red non-crystalline solid (27 mg, 54 %). [α]D=−26 (c 0.1, CHCl3); 1H NMR (400 MHz, [D6]DMSO): δ=2.23 (bs, 6 H), 2.35 (s, 6 H), 2.44–2.68 (m, 2 H), 3.05–3.29 (m, 2 H), 4.58 (dd, J1=3.78 Hz, J2=8.92 Hz, 1 H), 4.78 (t, J=9.02 Hz, 1 H), 4.96 (dd, J1=3.72 Hz, J2=9.21 Hz, 1 H), 6.08 (s, 1 H), 6.16 (s, 2 H), 7.19–7.28 (m, 3 H), 7.29–7.37 ppm (m, 2 H); 13C NMR (100 MHz, [D6]DMSO): δ=14.1 (m, 2C), 15.6 (2C), 26.0, 33.3, 58.3, 72.8, 97.8, 107.1, 121.8 (broad), 127.3, 128.4 (2C), 130.5 (2C), 130.8 (broad), 132.8, 140.8 (broad), 145.0, 153.6 (broad), 153.9, 154.4, 158.3, 169.6 ppm; IR:
 =1659, 1549, 1510, 1197, 981, 703 cm−1; HRMS: m/z: calcd for C29H29BF2N3O4: 532.2219 [M+H]; found: 532.2213.
=1659, 1549, 1510, 1197, 981, 703 cm−1; HRMS: m/z: calcd for C29H29BF2N3O4: 532.2219 [M+H]; found: 532.2213.
1,5-Dioxo-8-phenyl-7-(2-(1,3,5,7-tetramethyl-4,4-difluoro-4-bora-3a,4a-diaza-s-indacene-8-yl)ethyl)-3,5-dihydro-2H-thiazolo[3,2-a]pyridine-3-carboxylic acid (38): mCPBA (70 %, 9 mg, 0.37 mmol) was added to 34 (18.6 mg, 0.034 mmol) in CH2Cl2 (1.5 mL) at RT. The solution was stirred for 15 min before quenching with Na2S2O5 (aq. saturated). The resulting solution was extracted three times with CH2Cl2, dried with Na2SO4, filtered, and concentrated. Purification by column chromatography (CH2Cl2/acetone 9:1 to CH2Cl2/acetone/AcOH 88:10:2) gave 38 as a red foam (15.4 mg, 80 % yield). [α]D=−10 (c=0.1 in DMSO); 1H NMR (400 MHz, [D6]DMSO): δ=2.26 (s, 6 H), 2.39 (s, 6 H), 2.54–2.64 (m, 2 H), 3.14–3.32 (m, 2 H), 3.53–3.62 (m, 2 H), 5.40–5.49 (m, 1 H), 6.02 (s, 2 H), 6.79 (s, 1 H), 7.30–7.49 ppm (m, 5 H); 13C NMR (100 MHz, [D6]DMSO): δ=14.1 (2C), 15.6 (2C), 25.4, 33.2, 51.0, 65.2, 119.9, 120.5, 121.9 (broad), 128.47, 128.54, 128.6 130.4, 130.5 (broad), 131.1, 133.4, 140.9 (broad), 144.6, 149.0, 151.8, 153.7 (broad), 159.0, 170.1 ppm; IR:
 =1650, 1549, 1509, 1407, 1197, 981, 704 cm−1; HRMS: m/z: calcd for C29H29BF2N3O4S: 564.1940 [M+H]; found: 564.1928.
=1650, 1549, 1509, 1407, 1197, 981, 704 cm−1; HRMS: m/z: calcd for C29H29BF2N3O4S: 564.1940 [M+H]; found: 564.1928.
Acknowledgments
This work was financially supported by the Swedish Research Council (621–2010–4730), the Knut and Alice Wallenberg Foundation, JC Kempe Foundation (SJCKMS), S.H. was supported by AI048689, AI049950, AI029549, and DK086378 grants.
Supporting Information
References
- 1.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. Clin. Infect. Dis. 2009;48:1. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 2.Anderson GG, Palermo JJ, Schilling JD, Roth R, Heuser J, Hultgren SJ. Science. 2003;301:105. doi: 10.1126/science.1084550. [DOI] [PubMed] [Google Scholar]
- 3.Mulvey MA, Lopez-Boado YS, Wilson CL, Roth R, Parks WC, Heuser J, Hultgren SJ. Science. 1998;282:1494. doi: 10.1126/science.282.5393.1494. [DOI] [PubMed] [Google Scholar]
- 4.Mulvey MA, Schilling JD, Hultgren SJ. Infect. Immun. 2001;69:4572. doi: 10.1128/IAI.69.7.4572-4579.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schilling JD, Lorenz RG, Hultgren SJ. Infect. Immun. 2002;70:7042. doi: 10.1128/IAI.70.12.7042-7049.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J, Hammar M, Normark S, Hultgren SJ. Science. 2002;295:851. doi: 10.1126/science.1067484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nenninger AA, Robinson LS, Hultgren SJ. Proc. Natl. Acad. Sci. USA. 2009;106:900. doi: 10.1073/pnas.0812143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robinson LS, Ashman EM, Hultgren SJ, Chapman MR. Mol. Microbiol. 2006;59:870. doi: 10.1111/j.1365-2958.2005.04997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aberg V, Almqvist F. Org. Biomol. Chem. 2007;5:1827. doi: 10.1039/b702397a. [DOI] [PubMed] [Google Scholar]
- 10.Cegelski L, Pinkner JS, Hammer ND, Cusumano CK, Hung CS, Chorell E, Aberg V, Walker JN, Seed PC, Almqvist F, Chapman MR, Hultgren SJ. Nat. Chem. Biol. 2009;5:913. doi: 10.1038/nchembio.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pinkner JS, Remaut H, Buelens F, Miller E, berg V, Pemberton N, Hedenström M, Larsson A, Seed P, Waksman G, Hultgren SJ, Almqvist F. Proc. Natl. Acad. Sci. USA. 2006;103:17897. doi: 10.1073/pnas.0606795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nuccio SP, Baumler AJ. Microbiol. Mol. Biol. Rev. 2007;71:551. doi: 10.1128/MMBR.00014-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen SL, Hung CS, Xu JA, Reigstad CS, Magrini V, Sabo A, Blasiar D, Bieri T, Meyer RR, Ozersky P, Armstrong JR, Fulton RS, Latreille JP, Spieth J, Hooton TM, Mardis ER, Hultgren SJ, Gordon JI. Proc. Natl. Acad. Sci. USA. 2006;103:5977. doi: 10.1073/pnas.0600938103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horvath I, Weise CF, Andersson EK, Chorell E, Sellstedt M, Bengtsson C, Olofsson A, Hultgren SJ, Chapman MR, Wolf-Watz M, Almqvist F, Wittung-Stafshede P. J. Am Chem Soc. 2012;134:3439. doi: 10.1021/ja209829m. DOI: 10.1021/ja209829m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Emtenäs H, Hlin K, Pinkner JS, Hultgren SJ, Almqvist F. J. Comb. Chem. 2002;4:630. doi: 10.1021/cc020032d. [DOI] [PubMed] [Google Scholar]
- 16.Svensson A, Larsson A, Emtenas H, Hedenstrom M, Fex T, Hultgren SJ, Pinkner JS, Almqvist F, Kihlberg J. ChemBioChem. 2001;2:915. doi: 10.1002/1439-7633(20011203)2:12<915::AID-CBIC915>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 17.E. Chorell, J. S. Pinkner, C. Bengtsson, Sainte-Luce Banchelin, T.; S. Edvinsson, A. Linusson, S. J. Hultgren, F. Almqvist. Bioorg. Med. Chem. 2012. DOI:
- 18.Gonçalves MST. Chem. Rev. 2009;109:190. doi: 10.1021/cr0783840. [DOI] [PubMed] [Google Scholar]
- 19.Lavis LD, Raines RT. ACS Chem. Biol. 2008;3:142. doi: 10.1021/cb700248m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loudet A, Burgess K. Chem. Rev. 2007;107:4891. doi: 10.1021/cr078381n. [DOI] [PubMed] [Google Scholar]
- 21.Emtenäs H, Alderin L, Almqvist F. J. Org. Chem. 2001;66:6756. doi: 10.1021/jo015794u. [DOI] [PubMed] [Google Scholar]
- 22.Åberg V, Norman F, Chorell E, Westermark A, Olofsson A, Sauer-Eriksson AE, Almqvist F. Org. Biomol. Chem. 2005;3:2817. doi: 10.1039/b503294f. [DOI] [PubMed] [Google Scholar]
- 23.Chorell E, Bengtsson C, Banchelin TS, Das P, Uvell H, Sinha AK, Pinkner JS, Hultgren SJ, Almqvist F. Eur. J. Med. Chem. 2011;46:1103. doi: 10.1016/j.ejmech.2011.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raman P, Razavi H, Kelly JW. Org. Lett. 2000;2:3289. doi: 10.1021/ol000178q. [DOI] [PubMed] [Google Scholar]
- 25.Wang DC, Fan JL, Gao XQ, Wang BS, Sun SG, Peng XJ. J. Org. Chem. 2009;74:7675. doi: 10.1021/jo901149y. [DOI] [PubMed] [Google Scholar]
- 26.Aberg V, Hedenstrom M, Pinkner JS, Hultgren SJ, Almqvist F. Org. Biomol. Chem. 2005;3:3886. doi: 10.1039/b509376g. [DOI] [PubMed] [Google Scholar]
- 27.Bengtsson C, Almqvist F. J. Org. Chem. 2010;75:972. doi: 10.1021/jo902458g. [DOI] [PubMed] [Google Scholar]
- 28.Chorell E, Edvinsson S, Almqvist F. Tetrahedron Lett. 2010;51:2461. [Google Scholar]
- 29.Pemberton N, Pinkner JS, Edvinsson S, Hultgren SJ, Almqvist F. Tetrahedron. 2008;64:9368. [Google Scholar]
- 30.O’Toole GA, Kolter R. Mol. Microbiol. 1998;28:449. doi: 10.1046/j.1365-2958.1998.00797.x. [DOI] [PubMed] [Google Scholar]
- 31.Mattsson S, Dahlstrom M, Karlsson S. Tetrahedron Lett. 2007;48:2497. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.