

Abstract
We recently used a positional cloning approach to identify a nonconservative lysine to alanine substitution (K232A) in the bovine DGAT1 gene that was proposed to be the causative quantitative trait nucleotide underlying a quantitative trait locus (QTL) affecting milk fat composition, previously mapped to the centromeric end of bovine chromosome 14. We herein generate genetic and functional data that confirm the causality of the DGAT1 K232A mutation. We have constructed a high-density single-nucleotide polymorphism map of the 3.8-centimorgan BULGE30–BULGE9 interval containing the QTL and show that the association with milk fat percentage maximizes at the DGAT1 gene. We provide evidence that the K allele has undergone a selective sweep. By using a baculovirus expression system, we have expressed both DGAT1 alleles in Sf9 cells and show that the K allele, causing an increase in milk fat percentage in the live animal, is characterized by a higher Vmax in producing triglycerides than the A allele.
Aquantitative trait locus (QTL) with major effect on milk fat composition has been mapped to the centromeric end of bovine chromosome 14 (1, 2). Linkage disequilibrium (LD) was used to refine the map position of the QTL to a 3.8-centimorgan interval bounded by microsatellite markers BULGE30 and BULGE9 (3, 4). A bacterial artificial chromosome (BAC) contig spanning this interval was constructed and shown to contain a very strong positional candidate: diacylglycerol acyl transferase 1 (DGAT1) (5). DGAT1 indeed catalyzes the last step in triglyceride synthesis (6) and abrogates milk yield when knocked out in the mouse (7). By sequencing the DGAT1 gene from individuals with known QTL genotype, a nonconservative lysine to alanine substitution was identified at position 232, and shown to be associated with a major effect on milk yield and composition in several dairy cattle populations and breeds (5, 8, 9). The DGAT1 K232A mutation was therefore considered to be the likely quantitative trait nucleotide underlying the BTA14 QTL effect.
However, based on the available data, we could not formally exclude that the effect observed with the K232A polymorphism was, in fact, caused by another mutation, located in the same or in another gene mapping to the BULGE30–BULGE9 interval that would be in strong LD with K232A. To resolve this issue, we herein (i) describe the development of a high-density map of single-nucleotide polymorphisms (SNP) of the BULGE30–BULGE9 interval and show that the association with milk yield and composition is strongest for the DGAT1 SNPs, thereby strongly incriminating that gene; (ii) present evidence that the K allele has been under positive selection supporting its effect on the functionality of DGAT1; and (iii) demonstrate that the K232A mutation increases the activity of the enzyme in a way that is in agreement with its effect on phenotype.
Materials and Methods
Pedigree Material and Phenotypic Data. The pedigree material was composed of a series of black-and-white Holstein–Friesian sires sampled respectively in The Netherlands (1,818 bulls) and in New Zealand (227 bulls). They correspond to three distinct “granddaughter designs” (i.e., series of paternal half-sib families) that have been described as data sets I–IV in Farnir et al. (4). The phenotypes analyzed in this work were daughter yield deviations for milk fat percentage (4). Phenotypes and pedigree information were obtained directly from Cr-Delta (data sets I, II, and IV) (Arnhem, The Netherlands) and LIC (data set III) (Hamilton, New Zealand).
SNP Genotyping Using an Oligonucleotide Ligation Assay (OLA). OLA were essentially performed as described (10). Sequence-tagged sites (STS) corresponding to the 19 SNPs analyzed were amplified in two multiplex PCRs including 10 and 9 STS. Aliquots from these PCRs were then used to perform multiplex OLA reactions allowing for the genotyping of 6, 5, 4, and 4 SNPs (Table 1, which is published as supporting information on the PNAS web site). Products of the OLA reactions were separated on an ABI3100 capillary sequencer. The resulting electropherograms were analyzed with a program designed for automatic genotyping.
Measuring LD Using r2. LD between two polyallelic loci A and B was measured as:
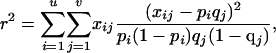 |
[1] |
where u and v are the respective number of alleles at the two marker loci, pi and qj are the population frequencies of alleles Ai and Bj, respectively, and xij is the observed frequency of gamete AiBj. The most likely marker linkage phase in the BULGE30–BULGE9 interval was determined for each bull, and LD was measured by using the pool of bull chromosomes inherited from the dam as described (11).
Association Studies. The effect of individual markers on phenotype was studied by using a linear model including a random marker effect and a random individual polygenic effect (“animal model”; ref. 12). Maximum likelihood solutions were obtained by using aireml (13) and results expressed as logarithm of odds (LOD) scores by comparison with the reml solutions obtained with the reduced model devoid of the marker effect. A detailed description of the method is given in Supporting Text, which is published as supporting information on the PNAS web site.
Extended Haplotype Homozygosities (EHH) (14). As for the computation of LD, EHH were computed by using the pool of maternally inherited bull chromosomes. The chromosomes were first sorted by DGAT1 core haplotype [polymorphisms K232A, Nt984 + 8(A-G), Nt984 + 26(C-T), Nt1501(C-T)]: haplotypes sHQ-D (K-A-C-C) and sHq (A-G-T-T) in the Dutch population and haplotypes sHQ-NZ (K-G-C-C), sHQ-D (K-A-C-C) and sHq (A-G-T-T) in the New Zealand population. For each of these groups, we then sequentially computed EHH toward the left (positions –1, –2,...) and right (positions +1, +2,...) of DGAT1 (position 0), essentially as described in ref. 14. A detailed description of the method is given in Supporting Text.
Expression of Recombinant DGAT1 in Sf9 Cells. DGAT1 cDNA was obtained by RT-PCR from mammary gland RNA extracted from a K232A heterozygous K/A cow. Reverse transcription was performed by using a DGAT1 3′ UTR-specific primer (5′-TTGCACAGCACTTTATTGACACA-3′). The complete coding sequence plus 11 bp of upstream sequence were amplified under long-range PCR conditions (Roche Diagnostics) using the following primers: 5′-CGCGGATCCGAACTAAGGCCATGGGCGACCGCGG-3′ and 5′-GCGGGTACCTCAGGTGCCGGCTGCCGGCG-3′. The cDNA was digested with BamHI (recognition sequences are underlined in the primers) and ligated into the BamHI linearized pFastBac-1 vector (Invitrogen). Resulting clones were screened by restriction analysis to determine the orientation of the insert, and selected clones were completely sequenced to determine the integrity of the DGAT1 coding sequence, the genotype at the K232A mutation, as well as the splicing status of exon VIII. Homologous recombination into DH10Bac as well as multiplication and titration of recombinant virus using Sf9 cells were performed following the recommendations of the manufacturer (Bac-To-Bac Baculovirus expression systems, Invitrogen). For expression experiments, monolayers of Sf9 cells (8.5 × 106 cells per 9-cm2 culture dish) were infected at a multiplicity of infection of 2. Cells were recovered after 48 h of culture at 28°C.
DGAT1 Activity Assay. Assaying DGAT1 activity was essentially performed as described (6). Sf9 cells obtained from a single dish were centrifuged at 800 × g for 10 min and resuspended in 10 ml of a solution containing 100 mM sucrose, 50 mM KCl, 40 mM KH2PO4, and 30 mM EDTA, pH 7.2. Cell membranes were disrupted by 10 strokes of a Teflon homogenizer. Nuclei and large cellular debris were pelleted by a first centrifugation at 10,000 × g. The supernatant was then ultracentrifuged for 1 h at 100,000 × g. The pellet containing the microsomes was resuspended in 250 μl of the solution described above. The protein content of the microsomal preparations was determined by using the bicinchoninic acid assay (Pierce). The DGAT activity was measured in a final volume of 200 μl of a solution containing 250 mM sucrose, 1 mM EDTA, 20 mM MgCl2, 100 mM Tris·HCl (pH 7.5), 25 μg of free fatty acid serum albumin, 40 nmol of diacylglycerol resuspended in acetone and 5 nmol [14C]oleoyl CoA (at specific activity of 37,000 dpm/nmol). Reactions were initiated by adding this assay mixture to microsomes preheated at 37°C for either 2, 4, or 8 min using 50, 25, or 12.5 μg of total microsomal protein. The assays were stopped by adding 1 ml of chroloform/methanol (1:1) containing 15 μg of triolein per ml. Chloroform (660 μl) was added to the assay that were then kept overnight at –20°C. After mixing with 330 μl of acidified H2O (17 mM NaCl/1 mM H2SO4), the organic phase was recovered and evaporated. The chloroform extractable material was separated by TLC on silica-gel 60 by using hexane/ethyl-acetate (9:1 vol/vol). TLC were exposed overnight on a PhosphorImager screen.
Posttranscriptional Effects. Total RNA was extracted from 100 mg of mammary gland tissue by using TRIzol (Invitrogen). Reverse transcription was performed as described above. The sequences of the primers labeled according to Fig. 3 are given in the supporting information.
Fig. 3.

Posttranscriptional effect associated with the DGAT1 K232A mutation. (A) Schematic representation of part of the DGAT1 gene showing exons III to XVII in red, the 3′ UTR in green, the position of the K232A and Nt1501(C-T) mutations in yellow, the alternative splicing pattern, and the different primers (P1–P5) used for PCR and RT-PCR amplification in blue. Primer combinations corresponding to the G-I, G-II, G-III, RT-I, RT-II, and RT-III products are given. (B) Ethidium bromide staining of the RT-PCR products (RT-IV) obtained from mammary gland mRNA of K/K and A/A individuals by using primers P1 and P4 and size-separated by agarose gel eletrophoresis. The most prominent band corresponds to the major splicing variant; the minor band marked by the arrow corresponds to the alternatively spliced variant. (C) HEX/6-FAM fluorescence ratios obtained by OLA genotyping of PCR products G-I, G-II, and G-III obtained from genomic DNA of heterozygous K/A individuals, and RT-PCR products RT-I, RT-II, and RT-III obtained from mammary gland mRNA of the same.
Results
Generation of a High-Density SNP Map of the BULGE30–BULGE9 Interval. To develop SNPs, we selected 12 BACs composing a minimum tiling path spanning the BULGE30–BULGE9 interval (85D12, 2I10, 259I10, 252I10, 231O03, 70E17, P118R6C2, 258E13, P88R4C4, 156I10, 46H2, and 8M13) (5). We subcloned Sau3AI fragments of individual BACs, and sequenced the inserts of 216 randomly selected clones. From these sequences, we developed 69 working bovine STS. To identify SNPs, we amplified and sequenced the corresponding STS from genomic DNA of the same panel of nine individuals with known BTA14 QTL genotype that were previously used to detect SNPs in the DGAT1 gene (5). We identified 19 polymorphic STS for a total of 29 SNPs, in addition to four previously described SNPs in the DGAT1 gene (5), and one SNP described in the CHRP gene (15) (see Table 1). The order of the corresponding STS was determined on the basis of the STS content of all BACs available in the region and is shown in Fig. 1A. Assuming that the BAC contig spans ≈1.4 Mb based on its alignment with the orthologous human region (5), this corresponds to one polymorphic STS every 72 Kb or one SNP every 48 Kb, on average.
Fig. 1.

(A) BAC contig (horizontal lines; see ref. 5), STS content (box), and linkage map of the BULGE30–BULGE9 interval. Polymorphic STS are given in red, and monomorphic STS are given in black. Dots on the linkage map correspond to blocks of nonrecombining markers; distance between adjacent marker blocks is given in centimorgan. (B and D) LD map in the Dutch and New Zealand populations, respectively. Linkage disequilibrium between all marker pairs was measured by using r2 values (see Materials and Mehtods), which are shown on a black-to-white scale. The heterozygosity (“Het”) of each marker in the respective populations, measured as (1 – ∑ p2 i i), where pi corresponds to the frequency of allele i, is given on the right of the LD square. Markers with more than two alleles are underlined. These are either microsatellite markers or DGAT1 for which the four SNPs [K232A, Nt984 + 8(A–G), Nt848 + 26(C–T) and Nt1501(C–T)] were considered separately when computing r2 but as haplotypes when computing Het. (C and E) Statistical significance (measured as a LOD score) of the effects of each individual marker on “milk fat percentage” computed by using a reml model, without (vertical red bars) or with (vertical blue bars) the K232A genotype as fixed effect in the mixed model (see Materials and Methods), computed, respectively, for the Dutch (C) and New Zealand (E) samples. EHH was computed at each marker position for the two and three major DGAT1 core haplotypes that are encountered, respectively, in the Dutch and New Zealand dairy cattle populations. ▪ corresponds to the sHq haplotype, ▴ corresponds to the sHQ-D haplotype, and Δ corresponds to the sHQ-NZ haplotype. For ▪ and ▴, EHH values are flanked by error bars corresponding to ± 1.96, the standard error of the estimate computed as 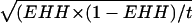 , where t is the number of usable chromosomes carrying the considered DGAT1 core haplotype. In the New Zealand population, EHH values for the sHQ-NZ haplotype (Δ) were significantly different from neither those of the sHQ-D haplotype nor those of the sHq haplotype; error bars have thus been omitted for clarity.
, where t is the number of usable chromosomes carrying the considered DGAT1 core haplotype. In the New Zealand population, EHH values for the sHQ-NZ haplotype (Δ) were significantly different from neither those of the sHQ-D haplotype nor those of the sHq haplotype; error bars have thus been omitted for clarity.
Construction of a Linkage and Linkage Disequilibrium Map of the BULGE30–BULGE9 Interval. We used multiplex OLA to genotype a total of 1,818 progeny tested Holstein–Friesian bulls sampled in The Netherlands, and 227 progeny tested Holstein–Friesian bulls sampled in New Zealand. The heterozygosities for the corresponding SNPs in the Dutch and New Zealand populations are reported in Fig. 1 B and D, respectively.
The obtained genotypes were used in conjunction with already available microsatellite genotypes (BULGE9, BULGE11, BULGE13, ILSTS39, and BULGE30) (5) to construct the linkage map shown in Fig. 1 A. Genotyped individuals were sorted by sire, yielding 91 paternal half-sib families, and the paternal gametes were used to estimate the distance between adjacent markers by using crimap (16). The map measures 3.85 centimorgan (cM) and comprises six “blocks” comprising 5, 2, 1, 2, 3, and 11 nonrecombining markers, respectively. Distance between adjacent markers blocks in cM are: (block 1)–0.2–(block 2)–1.2–(block 3)–0.8–(block 4)–0.05–(block 5)–1.6–(block 6).
We measured the extent of LD between SNP and microsatellite markers located in the BULGE30–BULGE9 interval separately for the Dutch and New Zealand populations. Pairwise LD was measured as (i) the P value of the observed allelic association under the null hypothesis of random assortment computed by Monte Carlo approximation of Fisher's exact test as described (11), and (ii) r2 computed as described in Materials and Methods. The first observation is that the whole region exhibits highly significant LD (data not shown). In the Dutch population, for instance, all but 10 of the 190 possible pairwise comparisons were yielding P values of 0 after >10,000 Monte Carlo simulations. Given the previous demonstration of long-range LD across the genome in the same population (11), this finding was not really surprising. The second observation is that, generally speaking, there is no simple decay of r2 with increasing distance between markers, whether genetic or physical (data not shown). The third is that the most distal marker bin (block 6; including the DGAT1 SNPs) is characterized by an exceedingly high level of intermarker LD when compared to the rest of the BULGE30–BULGE9 interval. Nineteen of the 21 (Dutch population) and 10 of the 11 (New Zealand population) marker pairs showing r2 values >0.9 correspond to markers located in this block (Fig. 1 B and D). Although we cannot exclude that this is only caused by lower levels of recombination within this block, it might also reflect the effect of selection for increased fat production, assuming that the QTL is located within this region.
Association Studies Support the Causality of DGAT1 in the Determinism of the BTA14 QTL. We then tested the effect of individual markers on milk fat percentage. The QTL was indeed shown previously to have the most pronounced effect on this milk composition trait (5). In the Dutch population, we obtained LOD scores superior to 200 for 10 of the 12 markers located in block 6 (Fig. 1C). This finding strongly suggests that the QTL is located within this segment. Although the highest LOD score was obtained for the K232A DGAT1 mutation, LOD scores obtained with other markers were nearly as high. Therefore, these results did not allow us to discriminate between a direct effect of DGAT1 or an effect of a linked gene within the region.
In the New Zealand population, however, the highest LOD scores (z = 16) were clearly restricted to the cluster of DGAT1 SNPs (Fig. 1E). Markers CHRP and BULGES.14.001–BULGES.14.004, which were yielding very high LOD scores in the Dutch population in which they are virtually perfectly associated with some of the DGAT1 polymorphisms, were giving much lower LOD scores in the New Zealand population in which their association with the DGAT1 polymorphisms is much weaker. These results strongly incriminate DGAT1 as being the causal gene underlying the QTL effect.
Although LOD scores are highest with the DGAT1 SNPs, other markers nevertheless show nonnegligible effects on milk fat percentage, which might indicate that additional genes contribute the BULGE30–BULGE9 QTL effect. To test this hypothesis, we added the K232A genotype as a fixed effect in the mixed model analysis. This completely erases the effect of all other markers in the BULGE30–BULGE9 interval, strongly suggesting that DGAT1 alone accounts for the entire QTL effect in the region (Fig. 1 C and E).
EHH Supports Positive Selection on the DGAT1 Gene. Because increased milk fat yield has been one of the major breeding objectives for many years, the fat-increasing K allele must have been under recent positive selection. Assuming a limited pool of K-bearing chromosome at the onset of selection, the ensuing selective sweep may have caused the K allele to be in LD with flanking markers over unusually long chromosome distances. To test this prediction, we performed the long-range haplotype test (LRH) as described (14). The LRH test is based on the EHH, which measures the probability that two chromosomes carrying a “core haplotype i” at the gene of interest are identical-by-descent at a given distance x, as assayed by homozygosity at all intervening SNPs. We have previously shown that the four DGAT1 SNPs [K232A, Nt984 + 8(A-G), Nt848 + 26(C-T) and Nt1501(C-T)] associate in two major haplotypes in the Dutch population (sHQ-D: K-A-C-C and sHq: A-G-T-T) and three major haplotypes in the New Zealand population (sHQ-NZ: K-G-C-C; sHQ-D: K-A-C-C and sHq: A-G-T-T). EHH. are computed for each core haplotype i and compared at increasing distances x from the core markers. A statistically significant superiority of EHH for a given core haplotype i at a given position x may be indicative of a selection operating on a mutation strongly associated with (or included in) this haplotype.
It can be seen from Fig. 1 C and E that, on the telomeric side of DGAT1, the EHH of the fat-increasing core haplotype sHQ-D is always significantly superior to the EHH of the sHq core haplotype, and this holds true in both populations. On the centromeric side, the same observation holds at the most distant BULGE9 marker position. In between DGAT1 and BULGE9, EHH values are very high for both core haplotypes, and are either not significantly different (New Zealand population) or are superior for the sHq core haplotype. EHH values computed for the sHQ-NZ core haplotype in the New Zealand population are neither significantly different from the EHH values of the sHQ-D nor sHq haplotype (because of the limited number of analyzed chromosome), but are generally superior to the EHH values of the corresponding sHq haplotype.
Altogether these observations are in agreement with the fat-increasing sHQ haplotypes (sHQ-D and sHQ-NZ) having undergone positive selection as predicted, and therefore support the causality of the DGAT1 gene in the determinism of the BTA14 QTL.
The K232A Mutation Affects the Enzymatic Activity of Recombinant DGAT1. If DGAT1 is responsible for the QTL effect, as evidenced by the previous analyses, it can be due to a structural mutation resulting in a difference in the amino acid sequence and hence functionality of the “Q” and “q” alleles, a regulatory mutation causing the amount of DGAT1 synthesized from the “Q” and “q” alleles to differ, or a combination of both. We previously demonstrated that the “Q” and “q” alleles differ by the nonconservative substitution of a lysine residue by an alanine residue at position 232. All other identified DNA sequence polymorphisms were either located in poorly conserved segments of introns or the 3′ UTR of the DGAT1 mRNA. The K232A mutation stood out as a prime candidate for the causative mutation.
To test the effect of the K232A mutation on the functionality of the enzyme, we expressed two variants of bovine DGAT1 differing only by the K232A mutation in Sf9 insect cells by using a baculovirus expression system. Five replicates of Sf9 cells were infected at a fixed multiplicity of infection (MOI = 2) with recombinant virus harboring either the K or A allele. Microsomes were prepared and DGAT activity assayed as described in Materials and Methods. Substrate concentrations were chosen to work under apparent Vmax conditions (6). For each membrane preparation, we performed three enzymatic reactions differing by the amount of incubated microsomes (50, 25, and 12.5 μg of total microsomal protein) and inversely proportionate time of incubation (2, 4, and 8 min), expected to yield identical amounts of product assuming enzymatic stability and absence of inhibition. The amounts of synthesized triglycerides (TG) were analyzed by two-way analysis of variance including (i) a “construct-effect” (no virus, nonrecombinant virus, DGAT1Kallele,DGAT1A allele) and (ii) an“amountofmicrosome/incubation time-effect.” The amount of microsome/incubation time“effect was not significant (P < 0.64), indicating that we were indeed working in steady-state conditions as required. The”construct effect“on the contrary was highly significant (P < 0.0001). Contrast analyses indicated that (i) the amount of TG synthesized without virus or with nonrecombinant virus did not differ significantly from each other (P = 0.69), (ii) the amount of TG synthesized with the constructs harboring the K and A alleles differed very significantly from those obtained without virus or with the nonrecombinant virus (P < 0.0001 for the four pairwise contrasts), and (iii) the amount of TG synthesized with the constructs harboring the K and A alleles differed very significantly from each other (P < 0.0001), the amount synthesized with the K allele being ≈1.5 times the amount synthesized with the A allele. This experiment was repeated twice, including once with different batches of virus, and always yielded very similar results. The results of one such experiment are shown in Fig. 2.
Fig. 2.

Effect of the K232A mutation on the Vmax of DGAT1. Amounts of synthesized triglycerides (TG) were estimated from the intensity of the TG spot on a TLC plate by phosphorimaging. Decreasing amounts of total microsomal protein (50, 25, and 12.5 μg) were incubated for increasing lengths of time (2, 4, and 8 min) with diacylglycerol and 14C-labeled oleoyl-CoA to assay DGAT1 activity under apparent Vmax conditions. Total microsomal protein was prepared from uninfected Sf9 cells (SF9: •), Sf9 cells infected with wild-type pFastBac-1 baculovirus (WTFASTBAC1: ♦), Sf9 cells infected with pFastBac-1 baculovirus expressing the alternative spliced DGAT1 form (DGAT1–AS: ▪), Sf9 cells infected with pFast-Bac1 baculovirus expressing the DGAT1 K allele (DGAT1–K: ▴), and Sf9 cells infected with pFastBac-1 baculovirus expressing the DGAT1 A allele (DGAT1–A: ▪). Small open symbols correspond to individual measurements; large filled symbols correspond to the mean of the corresponding group.
These experiments therefore strongly suggest that the Vmax of the DGAT1 K allele is superior to the Vmax of the A allele, which is in perfect agreement with the in vivo effect of the K232A mutation. Indeed, in the Dutch Holstein–Friesian dairy cattle population, the A to K substitution effect was shown to correspond to ≈0.35% of milk fat percentage, and to ≈10 kg of milk fat. These results therefore strongly suggest that the K232A substitution is indeed the causal mutation.
Transcriptional and Posttranscriptional Effects Associated with the K232A Mutation. It is worthwhile noting that the screening of bovine EST databases reveals the existence of alternatively spliced DGAT1 products (e.g., GenBank accession no. AW446985). Major and alternatively spliced mRNAs differ by the utilization in the latter of an intron 8 splice donor site located 6 bp upstream of the K232A mutation, which results in the “intronification” of most of exon VIII. This alternatively spliced mRNA has the potential to code for a protein suffering an internal deletion of 22 amino acid residues when compared to the full-length form (Fig. 3).
We performed RT-PCR experiments using bovine mammary gland mRNA and primers flanking exon VIII (P1 and P4 in Fig. 3) to determine the relative importance of these two isoforms. As can be seen from Fig. 3, the alternatively spliced product represents a minor fraction of the total DGAT1 mRNA in this tissue. Even if minor in all three K232A genotypes, the percentage of alternatively spliced product seemed higher in KK than in AA animals in these experiments.
To more accurately measure the effect of the K232A mutation on alternative splicing, we performed RT-PCR experiments using mammary gland mRNA from 24 heterozygous “K/A” cows by using primer sets that would (i) amplify both mRNA isoforms (RT-I; primers P1–P8), (ii) be specific for the major form (RT-II; primers P2–P8), and (iii) be specific for the alternatively spliced form (RT-III; primer P3–P8). An OLA (14) was used to determine the proportion of RT-PCR product originating from the K versus A allele. This could not be done by probing the K232A mutation directly because the corresponding sequence is eliminated from the mRNA by the alternative splicing event. The OLA assay was therefore designed to interrogate the Nt1501(C-T) SNP located in the 3′UTR region (5). Nt1501(C-T) is known to be in perfect association with the K232A polymorphism in the Dutch Holstein–Friesian population, Nt1501(C-T) C and T being associated with K232A K and A, respectively. Alternative allele-specific OLA products were fluorescently labeled by using hexachlorofluorescein (HEX) for the C allele, and 6-carboxyfluorescein (6-FAM) for the T allele, and separated on an ABI3100 automatic capillary sequencer. The proportion of mRNA originating from the K versus A allele was estimated as RK/A = FRRT/FRG. In this, FRRT is the HEX-to-6-FAM fluorescence ratio in the RT-PCR product, and FRG is the HEX-to-6-FAM fluorescence ratio in a control PCR product obtained from genomic DNA of a heterozygous “K/A” individual. FRG values were estimated for three distinct PCR products [G-I (P5–P8), G-II (P6–P8), and G-III (P7–P8)] designed to match the sizes of the corresponding RT-PCR products (RT-I, RT-II, and RT-III). Because the three corresponding FRG values did not differ significantly from each other (Fig. 3), all data were pooled to yield an overall FRG value of 0.41 ± 0.02.
FRRT values obtained for the RT-I product (0.39 ± 0.02) did not differ significantly from FRG, yielding a RK/A ratio of 0.96, i.e., essentially equal to 1. The simplest interpretation of this observation is that the promoters of both the K and A allele have equal strength. Therefore, it supports the suggestion that the observed QTL effect results from a qualitative, structural difference between the “Q” and “q” DGAT1 alleles (resulting from the K232A mutation), rather than from a qualitative difference in amount of enzyme resulting from an hypothetical, as of yet not identified, mutation in a distant control element.
FRRT values for the RT-III product (0.50 ± 0.02), on the contrary, were significantly superior to FRG (P < 0.01). This indicates that a higher proportion of alternatively spliced product originates from the K than from the A allele. However, dividing these fluorescence ratios indicates that the magnitude of the effect (although very significant) is rather modest, the K allele producing only 1.21 times more alternatively spliced product than the A allele.
If the K allele produces more alternatively spliced product than the A allele, the proportion of K allele in the major product must necessarily be reduced when compared to the A allele. FRRT values for RT-II (0.382 ± 0.03) were indeed lower than FRG (albeit nonsignificant), corresponding to an RK/A of 0.93 versus 1.21 for RT-III. The extent of this reduction informs us about the proportion of major versus alternatively spliced variant, which can be computed as described in Supporting Text. This yields estimates of RII/III of 6.5, 8.5 and 7.4 for K/K, A/A, and K/A individuals corresponding respectively to 13.2%, 10.5%, and 11.8% alternatively spliced product.
As a consequence, the K232A seems to significantly increase the amount of alternatively spliced DGAT1 mRNA produced by a factor of 1.2. However, the actual proportion of the alternatively spliced product barely varies between genotype species and remains relatively minor (on the order of 10%) in animals of all three K232A genotypes.
The alternatively spliced cDNA was cloned in pFastBac-1 and expressed in Sf9 cells. A protein of the expected size was detected by SDS/PAGE analysis of the corresponding cell extracts (data not shown). However, when using the corresponding microsome preparations in the DGAT1 activity assay, we only detected a very modest, nonsignificant increase in DGAT1 activity when compared to the uninfected Sf9 cells or Sf9 cells infected with nonrecombinant virus (Fig. 2). This finding suggests that the alternatively spliced mRNA, if translated, produces a protein that is essentially devoid of DGAT1 activity.
Discussion
As recently discussed by Glazier et al. (17), formally demonstrating that a given polymorphism is the causal quantitative trait nucleotide, i.e., directly contributing to the genetic variation underlying a complex phenotype, is often much more difficult than for Mendelian traits. This is due to a number of factors, including the fuzzy relationship between genotype and phenotype, the probabilistic location of the QTL, and the more subtle effects on gene function of the underlying mutations. At least some of the mutations underlying QTL are expected to be regulatory, affecting control elements for which the relationship between structure and function remains poorly understood. The ultimate test, analogous to Koch's postulate in microbiology, would be to produce transgenic animals that differ only for the tested polymorphism in an appropriate polygenic background. With the exception of the mouse, this remains essentially impractical in all other mammalian species. Therefore, this has to be compensated for by a series of “additional classes of evidence that together make a compelling case” (17).
We believe that our work in conjunction with that of others has now provided a substantial amount of concurring support (both genetic and functional) for the causality of the K232A DGAT1 mutation in the determinism of the proximal BTA14 QTL on milk yield and composition:
The map location of the QTL assessed by using both linkage and linkage disequilibrium analysis coincides with that of DGAT1.
The K allele segregates with two distinct marker haplotypes predicted to harbor the fat-increasing “Q” allele in two different dairy cattle populations.
The DGAT1 K232A mutation exhibits the strongest association with milk yield and composition of all 24 markers studied in the BULGE30–BULGE9 interval and the allele substitution effect corresponds very well with the QTL effect estimated by linkage analysis.
LRH tests suggest that the K allele has undergone positive selection, which would be expected given the breeding objectives prevalent in the two studied populations during the last decades.
The known enzymatic activity of the DGAT1 enzyme, i.e., the catalysis of the last step in triglyceride synthesis, is perfectly compatible with the most pronounced effect of the QTL, i.e., an increase in milk fat percentage.
Knocking-out DGAT1 in the mouse abrogates lactation, demonstrating the importance of DGAT1 in lactation physiology.
The K232A mutation is a nonconservative amino acid substitution affecting a highly conserved residue.
Expression of recombinant DGAT1 protein differing only at the K232A mutation demonstrates that this mutation affects the Vmax of the enzyme in a direction that is in perfect agreement with the observed phenotypic effect.
We also show that the alternate DGAT1 alleles associated with the “Q” and “q” QTL alleles do not exhibit quantitative differences in mRNA expression levels, which might have been indicative of the involvement of a regulatory mutation instead of or in addition to the K232A structural mutation. However, we do show that the K232A mutation modestly increases (1.2 times) alternative splicing and the generation of a minor mRNA (≈10%) that has the potential to code for a DGAT1 isoform that is essentially devoid of DGAT1 activity. The functional significance of this finding, if any, remains unknown. Assuming that major and alternatively spliced products are produced in the same cells, the corresponding protein product might integrate in some DGAT1 tetramers (18), thereby influencing the activity of the complex. This remains to be established.
We cannot exclude the existence of other as of yet uncharacterized DGAT1 alleles that might contribute to the genetic variance in milk yield and composition. In the Holstein–Friesian population, at least, the importance of such hypothetical alleles is predicted to be modest at best. Indeed, we cannot explain a higher proportion of the trait variance (e.g., 41% of the variance for milk fat percentage daughter yield deviations) when considering multiple markers spanning the BULGE30–BULGE9 interval by using an approach that simultaneously mines linkage and linkage disequilibrium than when considering the K232A mutation only (19).
It has been suggested that positive selection on QTL may result in detectable marker signatures around the QTL. This should theoretically allow for the mapping of QTL underlying the genetic variation for traits under selection (including artificial selection), without the need for the actual phenotypic data. The most powerful test designed for that purpose to date is the recently described LRH test (14). We have applied this test to the DGAT1 gene and have, indeed, found evidence of haplotype footprints, which are likely to result from the intense selection for increased milk fat yield that has been applied to dairy cattle populations. It remains to be established, however, whether the LRH test will have sufficient power and resolution to be useful for ab initio QTL detection. Preliminary analyses in the BULGE30–BULGE9 interval are not considered very encouraging (data not shown).
In conjunction with a previous publication (5), this work describes the second example in livestock (20) in which compelling evidence is presented supporting the identification of the causal mutation or quantitative trait nucleotide underlying a truly quantitative trait. These results illustrate the power of livestock population for the molecular dissection of complex quantitative traits.
Supplementary Material
Acknowledgments
We are grateful to Richard Spelman, Russell Snell, Chris Schrooten, and Erik Mullaert for fruitful discussions, and to Cr-Delta (Arnhem, The Netherlands) and Livestock Improvement Corporation (Hamilton, New Zealand) for providing us with pedigree and phenotypic data. This work was funded by research grants from Vialactia Biosciences (Auckland, New Zealand), Cr-Delta, Livestock Improvement Corporation, the Vlaamse Rundvee Vereniging (Belgium), the Belgian Ministry of Agriculture, and the European Union.
Abbreviations: QTL, quantitative trait locus; LD, linkage disequilibrium; BAC, bacterial artificial chromosome; SNP, single-nucleotide polymorphism; OLA, oligonucleotide ligation assay; STS, sequence-tagged site; LOD, logarithm of odds; EHH, extended haplotype homozygosity; LRH, long-range haplotype.
References
- 1.Coppieters, W., Riquet, J., Arranz, J.-J., Berzi, P., Cambisano, N., Grisart, B., Karim, L., Marcq, F., Simon, P., Vanmanshoven, P., et al. (1998) Mamm. Genome 9, 540–544. [DOI] [PubMed] [Google Scholar]
- 2.Heyen, D. W., Weller, J. I., Ron, M., Band, M., Beever, J. E., Feldmesser, E., Da, Y., Wiggans, G. R., VanRaden, P. M. & Lewin, H. A. (1999) Physiol. Genomics 1, 165–175. [DOI] [PubMed] [Google Scholar]
- 3.Riquet, J., Coppieters, W., Cambisano, N., Arranz, J.-J., Berzi, P., Davis, S., Grisart, B., Farnir, F., Karim, L., Mni, M., et al. (1999) Proc. Natl. Acad. Sci. USA 96, 9252–9257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farnir, F., Grisart, B., Coppieters, W., Riquet, J., Berzi, P., Cambisano, N., Karim, L., Mni, M., Simon, P., Wagenaar, D. & Georges, M. (2002) Genetics 161, 275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grisart, B., Coppieters, W., Farnir, F., Karim, L., Ford, C., Cambisano, N., Mni, M., Reid, S., Spelman, R., Georges, M. & Snell, R. (2002) Genome Res. 12, 222–231. [DOI] [PubMed] [Google Scholar]
- 6.Cases, S., Smith, S. J., Zheng, Y. W., Myers, H. M., Lear, S. R., Sande, E., Novak, S., Collins, C., Welch, C. B., Lusis, A. J., et al. (1998) Proc. Natl. Acad. Sci. USA 95, 13018–13023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith, S. J., Cases, S., Jensen, D. R., Chen, H. C., Sande, E., Tow, B., Sanan, D. A., Raber, J., Eckel, R. H. & Farese, R. V., Jr. (2000) Nat. Genet. 25, 87–90. [DOI] [PubMed] [Google Scholar]
- 8.Winter, A., Kramer, W., Werner, F. A., Kollers, S., Kata, S., Durstewitz, G., Buitkamp, J., Womack, J. E., Thaller, G. & Fries, R. (2002) Proc. Natl. Acad. Sci. USA 99, 9300–9305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spelman, R. J., Ford, C. A., McElhinney, P., Gregory, G. C. & Snell, R. G. (2002) J. Dairy Sci. 85, 3514–3517. [DOI] [PubMed] [Google Scholar]
- 10.Karim, L., Coppieters, W., Grobet, L., Valentini, A. & Georges, M. (2000) Anim. Genet. 31, 396–399. [DOI] [PubMed] [Google Scholar]
- 11.Farnir, F., Coppieters, W., Arranz, J. J., Berzi, P., Cambisano, N., Grisart, B., Karim, L., Marcq, F., Moreau, L., Mni, M., et al. (2000) Genome Res. 10, 220–227. [DOI] [PubMed] [Google Scholar]
- 12.Lynch, M. & Walsh, B. (1997) Genetics and Analysis of Quantitative Traits (Sinauer, Sunderland, MA).
- 13.Johnson, D. L. & Thompson, R. (1995) J. Dairy Sci. 78, 449–456. [Google Scholar]
- 14.Sabeti, P. C., Reich, D. E., Higgins, J. M., Levine, H. Z., Richter, D. J., Schaffner, S. F., Gabriel, S. B., Platko, J. V., Patterson, N. J., McDonald, G. J., et al. (2002) Nature 419, 832–837. [DOI] [PubMed] [Google Scholar]
- 15.Looft, C., Reinsch, N., Karall-Albrecht, C., Paul, S., Brink, M., Thomsen, H., Brockmann, G., Kuhn, C., Schwerin, M. & Kalm, E. (2001) Mamm. Genome 12, 646–650. [DOI] [PubMed] [Google Scholar]
- 16.Lander, E. & Green, P. (1987) Proc. Natl. Acad. Sci. USA 84, 2363–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glazier, A. M., Nadeau, J. H. & Aitman, T. J. (2002) Science 298, 2345–2349. [DOI] [PubMed] [Google Scholar]
- 18.Cheng, D., Meegalla, R. L., He, B., Cromley, D. A., Billheimer, J. T. & Young, P. R. (2001) Biochem. J. 359, 707–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim, J. J. & Georges, M. (2002) Asian-Aust. J. Anim. Sci. 15, 1250–1256. [Google Scholar]
- 20.Van Laere, A. S., Nguyen, M., Braunschweig, M., Nezer, C., Collette, C., Moreau, L., Archibald, A., Haley, C., Buys, N., Andersson, G., et al. (2003) Nature 425, 832–836. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.