

Abstract
The cAMP response protein (CRP) is a transcription factor known to regulate many genes in Escherichia coli. Computational studies of transcription factor binding to DNA are usually based on a simple matrix model of sequence-dependent binding energy. For CRP, this model predicts many binding sites that are not known to be functional. If they are indeed spurious, the underlying binding model is called into question. We use a species comparison method to assess the functionality of a population of such predicted CRP sites in E. coli. We compare them with orthologous sites in Salmonella typhimurium identified independently by clustalw alignment, and find a dependence of mutation probability on position in the site. This dependence increases with predicted site binding energy. The positions where mutation is most strongly suppressed are those where mutation would have the biggest effect on predicted binding energy. This finding suggests that many of the novel sites are functional, that the matrix model correctly estimates their binding strength, and that calculated CRP binding strength is the quantity that is conserved between species. The analysis also identifies many new E. coli binding sites and genes likely to be functional for CRP.
The binding of transcription factors (TFs) to specific sites is a central mechanism of transcriptional regulation (1). Powerful computational techniques for finding putative binding sites in genomes and for characterizing TF binding on whole-genome scales are becoming available (2–6). An energy matrix of size 4 × L (where L is the site length) is often used to capture the binding profile of a particular TF, under the simplifying assumption that the contribution of a particular position in a binding site is independent of neighboring positions (7–9). This assumption is of unknown accuracy, and even if it is a good physical approximation, the particular choice of energy matrix could be inadequate, leading to false identification of sites.
The cAMP response protein (CRP) is a dimeric DNA binding and bending protein that binds in multiple Escherichia coli promoters to 22-bp sites with a core consensus sequence GTGANNNNNNTCAC (10); the DPInteract database (11) contains 48 such sites, and RegulonDB (12) contains more than 88. Computational studies typically predict many more CRP binding sites than are found in either of these databases (13–15). Although the databases are certainly not complete, the apparent large overprediction of binding sites undermines the credibility of the matrix model of binding specificity on which these studies are based. To sharpen the issue, we contrast the situation for CRP with that for LacI, a TF that has a small number of known binding sites and is thought to be highly specific. In fact, an energy matrix that cleanly discriminates the known binding sites from the rest of the genome can be found for LacI. To decide whether CRP binding can successfully be described by a similar matrix model, we need to know whether the overpredicted CRP binding sites are functional. We look for evidence on this issue by identifying partner sites in orthologous regions in Salmonella typhimurium, a close relative of E. coli, and examining mutations between site pairs. The mutation probability depends on position in the site in a way that ensures rough conservation of CRP binding energy between species. We take this as evidence that the novel CRP sites have real biological function. We do not know why experimental methods have not picked them up, but the evidence that evolution cares about them is striking. We conclude that the simple matrix model works as well for CRP as for LacI, despite their very different binding profiles. We also make specific predictions of many new CRP binding sites (and regulated genes).
Methods
Genomes. We work with the genomes of E. coli K12 [National Center for Biotechnology Information (NCBI) accession no. NC004431, 4,639,221 bp] and S. typhimurium LT2 (NCBI accession no. NC003197, 4,857,432 bp). Genes and intergenic regions are identified by comparison with protein tables available from NCBI. Genomes and tables are included in the supporting information on the PNAS web site and at www.princeton.edu/~ccallan/binding (see Software and Software Availability).
Matrix Construction. A given TF contacts the genome at a site of length L (of order 20 bp in typical bacterial examples). To estimate the sequence-dependent affinity, we assign to each TF a matrix σb,i, which is used to score a site (b(i), i = 1... L) according to the additive rule
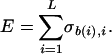 |
We will refer to σ as the energy matrix of the TF, and E is meant to approximate the binding energy (in units of kT) of the TF to the site. The usual method for constructing σ (7) starts from a list of known binding sites for a TF and applies the following algorithm: for each position i in a site, the number of occurrences Ni(b) of each DNA base b in the list of sites is counted, and the matrix elements are assigned by the rule
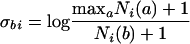 |
(see ref. 16). The +1 pseudocount regularizes the divergence that arises if any of the Ni(a) happen to vanish in the finite sample of known sites. The matrix is normalized to assign σb,i = 0 to the most common base pair at site i and σbi > 0 to all others; thus, the consensus sequence has E = 0, and all others have E > 0. For an implementation of this algorithm, see the openfill program distributed with the software (see Software and Software Availability). This method for assigning values to σbi has a sound physical and evolutionary rationale (7) when all of the input site sequences have roughly the same binding energy to the TF. When this is not the case, a more sophisticated algorithm may be needed (a concrete example will be given shortly). In short, the validity of the matrix model is distinct from the validity of the algorithm for evaluating the matrix itself.
The work reported here specifically concerns two TFs: CRP and LacI. We used the known sites in the E. coli K12 genome as listed in DPInteract (11). LacI contacts 21 bp and has three listed binding sites (all in close proximity to the lac operon); CRP contacts 22 bp and has 48 listed binding sites (widely dispersed in the genome). Binding-site files suitable for use with the openfill program are available. These files include twice as many sequences as cited above (i.e., 6 for LacI and 96 for CRP). This is because CRP and LacI, like many bacterial TFs, are symmetric dimers and can be regarded as reading either the top or the bottom strand of the DNA. The two strand reads are not usually identical and can legitimately be cited as independent site data. However, as is appropriate for CRP and LacI, openfill creates a symmetrized energy matrix that assigns the same energy to a sequence and its reverse complement (and therefore to both reads of a site).
Binding-Site Search. Binding site search was done with the program scangen (see Software and Software Availability), which takes as input the energy matrix for a TF, a genome, and a file containing the bounding coordinates and names of all coding regions in that genome (the latter two obtained from the NCBI database as described above). The scangen program assigns an energy score to each site in the genome and produces two types of output: cumulative histograms of number of binding sites versus E value (both total number of sites and the number of sites in noncoding regions) and a list of all sites below a chosen cutoff E value, giving for each site its coordinate and, if located in a noncoding region, the names of the flanking genes. The data concerning location with respect to coding regions is a useful diagnostic because functional sites are mostly located in noncoding regions.
Orthology and Alignment of Intergenic Regions and Site Pairs. We declare two intergenic regions to be orthologous if the flanking genes in both E. coli and S. typhimurium have the same gene names (according to the NCBI protein tables) and if the regions align well with clustalw (discussed below). The two genomes have extensive orthology: there are 3,475 intergenic regions in E. coli and 3,660 in S. typhimurium, of which 1,533 are orthologous by this definition. This is a rather restrictive notion of orthology; other workers (6, 17) find an additional ≈500 orthologous intergenic regions based on orthology of downstream proteins. With either definition, the mean difference rate (disregarding end gaps) between orthologous intergenic region pairs is comparable (≈25%).
For an E. coli site lying in an intergenic region having an S. typhimurium ortholog, we identify the sequence of the orthologous site by alignment of the relevant pair of intergenic regions. We used clustalw v.1.83 (18) to align intergenic region pairs, keeping 30 bp of coding sequence on either side (typically a few hundred base pairs of sequence in all); the default parameters for clustalw were used. The S. typhimurium sequence aligned to the E. coli site is then defined to be its orthologous partner site, provided that the alignment places no gaps in either sequence. We place no limitation on the number of mutations between the two sites.
Calculation of Expected Energy Change Under Mutation. We calculate the expected energy change for a site by first calculating the average rate of transitions and transversions for each region in E. coli from the clustalw alignment between the region and its orthologous region in S. typhimurium. We adjust the rate of transitions by a factor of two to account for null and back mutations. Then, for each purine in the site we calculate the expected energy change to be
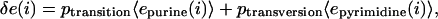 |
where 〈epurine〉 and 〈epyrimidine〉 are the average contributions of a purine and a pyrimidine at that position in the matrix. The contribution of each pyrimidine is calculated similarly, and the δ e values are summed over all positions to calculate the total expected change in energy. We did not introduce gapping into our model because gaps in sites in either genome disqualify a site for comparison and, moreover, occur infrequently (<5% of the aligned sequence consists of gaps).
Software and Software Availability. All software was developed in c++ and/or python 2.3 under linux. The software was developed ab initio by the authors; a package that can be used to reproduce all of our results is available at www.princeton.edu/~ccallan/binding. The software is copyright of Princeton University and the California Institute of Technology and is freely available and redistributable under an Open Source-compatible license.
Results
Binding Profiles of CRP and LacI Differ. LacI and CRP have very different profiles: binding-site catalogs indicate that LacI has three known sites in the immediate vicinity of the lac operon, whereas CRP affects the transcription of many genes and has 48 listed sites. Although it may be too strong to say that LacI regulates only the lac operon, it does seem clear that CRP affects many more genes than LacI. Because it is not obvious that the linear additive model for binding affinity can encompass both extreme behaviors, a comparative study of these two cases should be instructive.
We created energy matrices LacI-naive and CRP-naive by using the known sites as described in Methods and then used scangen to create histograms of the energy distribution of all sites in the E. coli genome. The results are presented in Tables 1 and 2. Each line gives the cumulative number of sites with E less than the indicated cutoff, the cumulative percentage of sites that are in genes, the cumulative percentage in intergenic regions, and the cumulative number of the known (or input) sites that have been captured. We count sites in coding and noncoding regions separately because functional sites are expected to lie mostly in the noncoding 15% of the E. coli genome and the statistics of site location could convey useful evidence for or against functionality.
Table 1. Sites isolated from the genome at several energy cutoffs with LacI-naive, a matrix representing LacI binding.
| Cutoff | No. of sites | Coding, % | Intergenic, % | Known |
|---|---|---|---|---|
| 3.00 | 1 | 0 | 100 | 1/3 |
| 5.00 | 2 | 50 | 50 | 2/3 |
| 7.00 | 26 | 85 | 15 | 3/3 |
| 9.00 | 483 | 86 | 14 | 3/3 |
The matrix was constructed from the three binding sites implicated in transcriptional regulation of the lac operon. The mean (SD) of energy over all sites in the genome is 20.9 (3.2).
Table 2. Sites isolated from the genome at several energy cutoffs with CRP-naive, a matrix constructed from the list of 48 known CRP binding sites.
| Cutoff | No. of sites | Coding, % | Intergenic, % | Known |
|---|---|---|---|---|
| 5.00 | 31 | 3 | 97 | 4/48 |
| 7.00 | 105 | 9 | 91 | 10/48 |
| 9.00 | 375 | 26 | 74 | 27/48 |
| 11.00 | 1,495 | 53 | 47 | 39/48 |
| 15.00 | 26,873 | 72 | 28 | 48/48 |
The mean (SD) of energy over all sites in the genome is 26.9 (4.8).
The tables display a potential problem with the linear additive model: using E value as the discriminant, the model seems to overpredict the number of TF binding sites. For LacI, note that although the top two bins in Table 1 capture only known sites, the next bin captures the remaining known site in addition to 23 others. The novel sites, although competitive in E value, are probably not true LacI sites because they are randomly located in the genome, with no preference for noncoding regions. Even so, the LacI-naive matrix discriminates the known sites from the rest of the genome surprisingly well, given how few input data are used. The situation for CRP, displayed in Table 2, is less good: although the known sites are assigned small E values compared to a random site in the genome, they have a large range in E (ΔE ∼ 12, or five orders of magnitude in calculated affinity) and many sites not in the known site list have comparable or better E values. Because CRP regulates many genes, it is less clear than for LacI that the novel sites are spurious. In contrast to LacI, the novel sites are not randomly distributed: the lower the E value, the more likely they are to lie in noncoding regions. This finding suggests that many of the novel CRP sites may be real. We will develop other lines of evidence for this in what follows.
A Modified LacI Matrix Accurately Discriminates Known Sites. It is important to understand whether the above problems are due to a nonoptimal choice of the matrix or instead to a failure of the linear additive model for sequence-dependent binding affinity. To explore this, we also ran scangen on two other matrices, LacI-relax and CRP-relax, both derived by using a relaxation method (C.T.B. and C.G.C., unpublished work). The relaxation method takes as input the known sites and their relative binding affinities (if known) as well as the background genome. The use of the relative binding affinities is crucial: it has the effect that the sequences of subsidiary weak binding sites, should any exist, can be used as input data to refine the matrix without destabilizing it. The relaxation implements the notion that for proper function, not only must binding to the known sites be strong, but net binding to the rest of the genome must be small. The matrices produced by this method differ significantly from the “naive” matrices and lead to the site histograms displayed in Tables 3 and 4 (the matrices used are available on-line as discussed in Software and Software Availability).
Table 3. Site list for LacI-relax, a matrix constructed to pick out known LacI sites preferentially.
| Cutoff | No. of sites | Coding, % | Intergenic, % | Known |
|---|---|---|---|---|
| 3.00 | 1 | 0 | 100 | 1/3 |
| 5.00 | 1 | 0 | 100 | 1/3 |
| 7.00 | 3 | 33 | 67 | 3/3 |
| 9.00 | 7 | 71 | 29 | 3/3 |
Note that two of the three known LacI binding sites are located in coding regions.
Table 4. Site list for CRP-relax, a matrix constructed to pick out known CRP sites preferentially.
| Cutoff | No. of sites | Coding, % | Intergenic, % | Known |
|---|---|---|---|---|
| 5.00 | 103 | 10 | 90 | 11/48 |
| 7.00 | 524 | 38 | 62 | 33/48 |
| 9.00 | 3,545 | 66 | 34 | 43/48 |
| 11.00 | 21,386 | 74 | 26 | 48/48 |
| 15.00 | 275,384 | 79 | 21 | 48/48 |
Comparing Table 3 with Table 1, we see that LacI-relax, in contrast with LacI-naive, succeeds in creating a gap in energy between the known sites and the rest of the genome, thus realizing the picture that LacI acts primarily on the lac operon. This is not a prediction, just the (nontrivial) statement that a linear model matrix that fits the qualitative facts about LacI can be found. Whether this matrix correctly predicts finer details like the affinity distribution of weaker (presumably nonfunctional) sites below the gap is an interesting question. If other genes than the lac operon were to be found to be regulated by LacI, this discussion would have to be revisited.
Table 4 shows the result of an attempt to improve the matrix for CRP by a similar relaxation method: apart from a possible rescaling of the E value, Table 4 looks similar to Table 2. In both tables, the known sites span a wide range of E values and are accompanied by a large number of novel sites of comparable E values. In what follows, we will look for evidence concerning the functionality of these sites via interspecies comparisons. This is important because, if the novel sites are spurious, it will be hard to avoid concluding that the linear additive energy model fails as a framework for predicting functionality, at least for CRP. Because relaxation did not have much effect, we will revert to assessing CRP sites by using the matrix CRP-naive.
Novel CRP Sites Have Similar Binding Energies in S. typhimurium. To assess the status of the novel E. coli sites found by the CRP energy matrix, we performed comparisons with orthologous sites in the closely related organism S. typhimurium. Partners of strong E. coli sites lying in intergenic regions are constructed by alignment of the orthologous S. typhimurium intergenic region (see Methods). Because the CRP energy matrix is not used in generating the alignment, we have no reason to expect the aligned sites in S. typhimurium to be scored as strong binding sites. (Note that S. typhimurium CRP differs from E. coli CRP in only one of 210 aa and can be expected to have the same sequence-specific binding affinity.)
What actually happens is shown in Fig. 1 where we compare the energy of strong CRP sites in E. coli with the energy of orthologous sites in S. typhimurium. The graph shows the line of equality (no mutations), a scatter plot of the actual orthologous pair energies (actual mutations), and a scatter plot of the same sites with the S. typhimurium site energy replaced by its expected value under random mutations. Actual mutated energies lie well below what is expected on the random mutation model for E. coli site energies up to E ∼ 7 (and a population difference exists up to higher values of site energy). The fixation of energy is not due to a general suppression of mutation: for the 34 site pairs with E. coli site energy less than 7.0, the average number of mutations per site is 4.1, not very different from the background rate of 5.5 for 22-bp sequences. The actual number of mutations per site runs from 0 to 11, and highly mutated sequences often have nearly the same energy (in one case, 7 of 22 positions are mutated, yet the change in E is only 0.22). The conservation of CRP binding energy (as distinct from sequence conservation) suggests that these sites are functional for CRP in some way useful to the organism.
Fig. 1.

Binding energy is strongly correlated between orthologous site pairs.
Novel CRP Site Pairs Have Strongly Biased Mutational Patterns. The most striking evidence of conservation is obtained by computing population averages of the position dependence of the probability of mutation (20). We did this for populations of orthologous site pairs defined by several energy cutoffs. The results, displayed in Fig. 2, show a strong positional bias within the site (the data have been reflected about the center position to smooth out statistical noise). The bias washes out abruptly when the cutoff exceeds Ecut ≃ 11. This is beyond the energy where, according to Fig. 1, the close correlation between individual site energies in the two species begins to wash out.
Fig. 2.

Paired site populations show a position-dependent pattern of mutation rates.
For all cutoffs, novel sites greatly outnumber input sites. Thus, the positional bias in mutation frequency is a property of the novel sites. The same analysis for the input sites alone (38 of the 48 input sites have S. typhimurium orthologs) gives a result indistinguishable from the Ecut = 9 curve in Fig. 2 (see supporting information). The positional bias in mutation frequency is essentially the same for the novel sites and the input sites and presumably has a common cause.
The positional bias has the shape expected if the sites are constrained to strongly bind a dimeric TF in both species. For example, only two of the site pairs found with Ecut = 5 have even a single mutation in the core positions 4–7 and their complements at positions 14–17. This pattern of fixation suggests that these eight bases dominate the site energy, consistent with the usual picture of a core region of the TF contacting the DNA and selecting a central motif. An examination of the CRP-naive matrix does show that these positions should be the most strongly fixed. We emphasize that the S. typhimurium sites were chosen with no consideration of their CRP binding energy: the nonrandom pattern of the mutations between them and their E. coli orthologs is evidence that some biologically meaningful aspect of the site sequence is being conserved. A plausible interpretation is that the sites are functional and that their CRP binding energy is approximately conserved.
Fig. 2 also confirms that the correlation of binding energies between E. coli and S. typhimurium shown in Fig. 1 is not due to strict sequence conservation: in the population of sites with Ecut = 7, only 8 of the 22 bases have a mutation rate of <5%, whereas the 14 remaining bases vary at a rate of 18% or higher. The total number of mutations between orthologous site sequences is often large, and it would be difficult to identify the orthologous pairs by looking for local patches of stronger-than-background sequence conservation.
Flanking Bases Are Required for Discrimination Ability. The pattern of base fixation shown in Fig. 2 suggests that there is a strong constraint on eight core positions within each site. This raises the question whether the flanking sequence lends useful discriminatory ability. To study this, we constructed a CRP-core-only matrix from CRP-naive by setting to zero all of the entries in the rows corresponding to the 14 flanking positions. The site energy histogram produced by scanning the E. coli genome with this modified matrix is given in Table 5. The high-scoring sites clearly constitute a very different population from the high-scoring sites under the CRP-naive matrix. There are many ways to see this, but an examination of the sites in the first bin (E < 1) makes the point: these sites all have E ≡ 0, i.e., they match the consensus sequence for the eight core positions. But half of these “perfect” sites lie in coding regions, whereas the sites that our evolutionary analysis suggests are under control of CRP are very heavily concentrated in the noncoding regions. The situation gets progressively worse for higher cutoffs. We conclude that the core-only matrix cannot sharply discriminate the sites of interest. Individual flanking positions contribute limited information but, taken together, they dramatically enhance the ability of the matrix to discriminate functional sites (or at least the sites shown by our evolutionary comparison to be under control of CRP). An evolutionary analysis of the population of sites that bind strongly according to CRP-core-only and also lie in coding regions is instructive: no significant position-dependent mutation pattern is seen, further confirming that these sites are spurious (see supporting information).
Table 5. Scangen list for CRP binding, using a matrix modified to ignore contributions from any but the eight core base pairs of the site.
| Cutoff | No. of sites | Coding, % | Intergenic, % | Known |
|---|---|---|---|---|
| 1.00 | 166 | 51 | 49 | 70/375 |
| 3.00 | 2508 | 82 | 18 | 217/375 |
| 5.00 | 17,411 | 86 | 14 | 325/375 |
| 7.00 | 78,995 | 86 | 14 | 372/375 |
| 9.00 | 271,661 | 86 | 14 | 375/375 |
The list of known sites in this table is chosen as all those sites that score with an energy of 9 or less using CRP-naive (see Fig. 2).
Discussion
Novel CRP Sites Are Probably Functional. Attempts to describe the sequence-specific DNA binding affinity of CRP by a linear additive energy model always lead to the prediction of many more strong binding sites (and regulated genes and operons) than are verified by direct experimental methods. We used the DPInteract site compendium (11) to demonstrate this, but the same conclusion would have been reached had we used other databases such as RegulonDB (12) (see supporting information). It is important to obtain independent information about the status of the many predicted novel binding sites: if they are spurious, the validity of the linear additive binding energy model is called into question; if they are real, the validity of that model is reaffirmed in a challenging context, and something new is learned about how CRP functions.
This issue could in principle be addressed experimentally, although that approach has drawbacks: in vivo tests may fail to expose the role of a given CRP motif because the test conditions are incorrect, and in vitro tests such as gel shifts or DNase footprinting can score pseudosites as functional CRP binding sites. We have instead sought evidence of their functionality via a computational study of binding-site evolution between closely related bacterial species. We have presented several lines of evidence suggesting that the novel sites are mostly functional, with a likelihood that increases with the strength of the predicted binding (as measured by the CRP energy matrix). They are, in increasing order of importance, as follows:
The novel CRP sites are overwhelmingly located in noncoding regions, exactly where regulatory sites should lie. Because the E. coli genome is only 15% noncoding and because spurious sites should be randomly located, this is an improbable chance occurrence. This observation is independent of evolutionary considerations.
If a novel strong E. coli site lies in an intergenic region with an S. typhimurium ortholog, an orthologous site pair can be defined by aligning the two regions. The difference in predicted binding energy between the two sites is systematically less than would be expected on the hypothesis of random mutations.
Within a population of orthologous pairs of novel strong binding sites, the probability of mutation depends strongly on position within the site; random mutations would have led to a position-independent profile. The actual position-dependent pattern is consistent with the way CRP is known to contact the DNA.
The cross-species comparison shows that populations of orthologous sites defined by strong predicted CRP binding energy have a nonrandom mutation pattern consistent with conservation of CRP binding energy. Conservation makes sense if the sites are functional for CRP and this evidence suggests that the primary determinant of functionality is the CRP binding energy of the site sequence. We have focused on sites with orthologs, but that was a device to select a subpopulation of sites on which functionality could be observed via its effect on mutations. Therefore, although the evolutionary evidence applies directly only to sites with orthologs, we suggest that strong sites in intergenic regions without orthologs are also likely to be functional for CRP.
The specific outcome of these considerations is a list of computational predictions of novel CRP binding sites in E. coli. We include all sites with E < 9 (the cutoff at which the mutation profile of the site pair population becomes indistinguishable from that of the starting databases; see supporting information) whether or not they have a companion species ortholog. A short list, generated with the stringent cutoff E < 4, is given in Table 6. The long list, containing >190 novel sites, is available on-line (see Software and Software Availability). The genes downstream from these sites would be interesting targets for investigation of the influence of CRP on their expression levels. The new genes do not, by and large, appear in the most comprehensive databases of regulatory information (see supporting information), suggesting that their regulation by CRP is subtle. On the other hand, the evolutionary evidence that such effects are important to the organism is strong. Understanding how and why will be an enlightening enterprise.
Table 6. List of genes with putative CRP sites upstream of the operon.
| Upstream site | Downstream gene(s) | Aligned site |
|---|---|---|
| 2.59 | tsr (b4355); yjiY (b4354) | — |
| 2.63 | b1904 (b1904) | — |
| 3.14 | yjcB (b4060); yjcC (b4061) | 3.33 |
| 3.19 | nupG (b2964) | 9.14 |
| 3.42 | mtlA (b3599); yibI (b3598) | — |
| 3.43 | b1458 (b1458) | — |
| 3.49 | tnaL (b3707) | — |
| 3.66 | qseA (b3243); yhcR (b3242) | — |
| 3.74 | yeaA (b1778); b1777 (b1777); gapA (b1779) | 5.87 |
| 3.77 | ydeA (b1528) | — |
| 3.77 | hpt (b0125); ged (b0124) | 4.47 |
| 3.83 | yefQ (b1111); ycfR (b1112) | 6.70 |
| 3.87 | proP (b4111) | — |
| 3.94 | ygiG (b3073); aer (b3072) | — |
An energy cutoff of 4.0 was used. Where an orthologous site exists in S. typhimurium, the energy of that site is also given. An extended list is available on our web site (see Software and Software Availability).
Binding Energy, Not Sequence, Is Conserved. Fig. 1 shows that the binding energies of sites in E. coli correlate well with the binding energy of independently aligned sites in S. typhimurium. Fig. 2 shows that this is not, however, due to strict sequence conservation: outside the eight core positions, the individual site pairs differ significantly at the sequence level. Others (19) have also noted the position-dependent mutation rate between orthologous binding sites. We have observed that this position dependence becomes more and more pronounced as the binding energy computed from the CRP-naive matrix becomes stronger. This finding suggests that the underlying cause of the mutation pattern is conservation of site binding energy between the two species. It also suggests that the matrix model binding energy is closely related to the actual in vivo binding energy. This agrees with the prediction made by Berg and von Hippel (16) on theoretical grounds.
Lessons for Practical Motif Hunting. Whereas these observations are useful for considering the actual nature of genomic binding of TFs, we can also try to draw some conclusions regarding the utility of various types of computational searches for novel TF binding sites on a whole-genome scale.
A promising technique for locating functional regulatory regions de novo identifies as putative binding sites elements of genomic sequence conserved between two or more closely related species (6, 20). However, we have found that sites may diverge at the sequence level without significantly changing the binding energy, as shown in Figs. 1 and 2. Indeed, our comparisons of E. coli and aligned S. typhimurium regions containing predicted CRP binding sites suggest that many of the sites would not be identifiable on the basis of local sequence conservation.
Another prevalent technique for binding site searches is to use a “consensus sequence,” consisting of the bases that appear most frequently in the list of known sites. In the case of CRP, searches for even the very general consensus sequence GTGANNNNNNTCAC would fail to discriminate between our novel (putatively functional) sites and nonfunctional sites, because of the lack of weighting from the flanking sequence. Searches with more restricted sequences would necessarily recover only a subset of our novel sites.
These observations suggest that a full matrix model will be needed to give optimum discrimination in separating actual binding sites from the rest of the genome. Unfortunately, there is usually only a limited number of known sequences from which to infer the matrix and a large error in its construction. Our results suggest that statistics could be improved by using aligned sites from orthologous intergenic regions as additional inputs. Beyond that, it may be fruitful to pursue optimization methods for tuning matrices to better represent TF binding. The results of using one such method (developed by us in unpublished work) to improve the LacI energy matrix were presented earlier in the paper. An independent study of CRP binding in E. coli that uses an optimization procedure (QPMEME) to refine the energy matrix has recently appeared (15). It also finds many novel predicted CRP sites and addresses the issue of whether they are real on the basis of their positions relative to transcription initiation sites. The quite interesting results of applying our species comparison assessment of functionality to the QPMEME site list are presented in the supporting information.
Concluding Remarks. We have demonstrated that many computationally identified CRP binding sites have nonrandom mutation patterns, strongly suggesting that they are functional. Sites that have strong predicted binding energy but no ortholog in S. typhimurium are also likely to be actual CRP sites, although our method gives no direct evidence of their functionality. Combined, we find that there are >190 novel CRP binding sites in the E. coli genome, none of them known from experimental evidence. Although this is important information about CRP itself, we would like to emphasize the equally important conclusion that the linear energy matrix model for TF binding works well in a case where it had been thought to generate far too many false positives. It will be interesting to explore this issue across a wider range of TFs and organisms.
Supplementary Material
Acknowledgments
We thank Erich M. Schwarz, Paola Oliveri, and Saeed Tavazoie for useful discussions, Nikolaus Rajewsky and Leonid Kruglyak for careful reading of the manuscript and helpful suggestions, and Eric H. Davidson, R. Andrew Cameron, and the Beckman Institute Center for Computational Regulatory Genomics at the California Institute of Technology for access to their computational resources (supported by National Institutes of Health Grant RR15044). C.T.B. was supported by National Institutes of Health Grant GM61005 to E. H. Davidson.
Abbreviations: CRP, cAMP response protein; TF, transcription factor.
References
- 1.Ptashne, M. (1992) A Genetic Switch (Blackwell Scientific, Oxford).
- 2.Sinha, S. & Tompa, M. (2003) Nucleic Acids Res. 31, 3586–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rajewsky, N., Vergassola, M., Gaul, U. & Siggia, E. D. (2002) BMC Bioinformatics 3, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lenhard, B., Sandelin, A., Mendoza, L., Engstrom, P., Jareborg, N. & Wasserman, W. W. (2003) J. Biol. 2, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Markstein, M., Markstein, P., Markstein, V. & Levine, M. S. (2002) Proc. Natl. Acad. Sci. USA 99, 763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCue, L. A., Thompson, W., Carmack, C. S. & Lawrence, C. E. (2002) Genome Res. 12, 1523–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berg, O. G. & von Hippel, P. H. (1988) Trends Biochem. Sci. 13, 207–211. [DOI] [PubMed] [Google Scholar]
- 8.Benos, P. V., Lapedes, A. S. & Stormo, G. D. (2002) BioEssays 24, 466–475. [DOI] [PubMed] [Google Scholar]
- 9.Benos, P. V., Bulyk, M. L. & Stormo, G. D. (2002) Nucleic Acids Res. 30, 4442–4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cashel, M., Gentry, V. J., Hernandez, V. J. & Vinella, D. (1996) in Escherichia coli and Salmonella: Cellular and Molecular Biology, ed. Neidhardt, F. C. (Am. Soc. Microbiol., Washington, DC), pp. 1458–1496.
- 11.Robison, K., McGuire, A. M. & Church, G. M. (1988) J. Mol. Biol. 284, 241–254. [DOI] [PubMed] [Google Scholar]
- 12.Salgado, H., Santos-Zavaleta, A., Gama-Castro, S., Millan-Zarate. D., Diaz-Peredo, E., Sanchez-Solano, F., Perez-Rueda, E., Bonavides-Martinez, C. & Collado-Vides, J. (2001) Nucleic Acids Res. 29, 72–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berg, O. G. & von Hippel, P. H. (1987) J. Mol. Biol. 193, 723–750. [DOI] [PubMed] [Google Scholar]
- 14.Tan, K., Moreno-Hagelsieb, G., Collado-Vides, J. & Stormo, G. D. (2001) Genome Res. 11, 566–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Djordjevic, M., Sengupta, A. M. & Shraiman, B. I. (2003) Genome Res. 13, 2381–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berg, O. G. & von Hippel, P. H. (1988) J. Mol. Biol. 200, 709–723. [DOI] [PubMed] [Google Scholar]
- 17.Rajewsky, N., Socci, N. D., Zapotocky, M. & Siggia, E. D. (2002) Genome Res. 12, 298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chenna, R., Sugawara, H., Koike, T., Lopez, R., Gibson, T. J., Higgins, D. G. & Thompson, J. D. (2003) Nucleic Acids Res. 31, 3497–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moses, A. M., Chiang, D. Y., Kellis, M., Lander, E. S. & Eisen, M. B. (2003) BMC Evol. Biol. 3, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Nimwegen, E., Zavolan, M., Rajewsky, N. & Siggia, E. D. (2002) Proc. Natl. Acad. Sci. USA 99, 7323–7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.