

Abstract
Background: Exposure to arsenic via drinking water is a global environmental health problem. In utero exposure to arsenic via drinking water increases the risk of lower respiratory tract infections during infancy and mortality from bronchiectasis in early adulthood.
Objectives: We aimed to investigate how arsenic exposure in early life alters lung development and pathways involved in innate immunity.
Methods: Pregnant BALB/c, C57BL/6, and C3H/HeARC mice were exposed to 0 (control) or 100 μg/L arsenic via drinking water from gestation day 8 until the birth of their offspring. We measured somatic growth, lung volume, and lung mechanics of mice at 2 weeks of age. We used fixed lungs for structural analysis and collected lung tissue for gene expression analysis by microarray.
Results: The response to arsenic was genetically determined, and C57BL/6 mice were the most susceptible. Arsenic-exposed C57BL/6 mice were smaller in size, had smaller lungs, and had impaired lung mechanics compared with controls. Exposure to arsenic in utero up-regulated the expression of genes in the lung involved in mucus production (Clca3, Muc5b, Scgb3a1), innate immunity (Reg3γ, Tff2, Dynlrb2, Lplunc1), and lung morphogenesis (Sox2). Arsenic exposure also induced mucous cell metaplasia and increased expression of CLCA3 protein in the large airways.
Conclusions: Alterations in somatic growth, lung development, and the expression of genes involved in mucociliary clearance and innate immunity in the lung are potential mechanisms through which early life arsenic exposure impacts respiratory health.
Keywords: gene expression, growth and development, innate immunity, mucociliary clearance, toxicity
Arsenic is a toxic metalloid contaminant in drinking water sources throughout the world, affecting hundreds of millions of people (Mandal and Suzuki 2002). Exposure to arsenic via drinking water in early life has been shown to increase the risk of mortality from bronchiectasis (Smith et al. 2006). Bronchiectasis unrelated to cystic fibrosis (CF) is a chronic, progressive lung disease characterized by cough and sputum production, periodic infectious exacerbations, and premature death in adulthood (Grimwood 2011). Bronchiectasis associated with CF is thought to arise from impaired innate immune pathways in the lung (Barker 2002), and although the pathology of non-CF bronchiectasis is poorly understood, similar mechanisms are likely to be involved (Grimwood 2011). The processes linking arsenic exposure in early life and the development of bronchiectasis are unclear.
There is evidence for an effect of in utero arsenic exposure on innate immunity. Arsenic exposure during pregnancy induces oxidative stress and increases inflammatory cytokine levels and reduces T-cell numbers in the placenta and impairs thymic development in the infant (Ahmed et al. 2011; Raqib et al. 2009). Arsenic exposure during pregnancy has also been shown to increase the risk of lower respiratory tract infections during infancy (Rahman et al. 2011). Recurrent lower respiratory tract infections in early childhood and defective pulmonary immune defenses are important risk factors for the development of non-CF bronchiectasis (King et al. 2006). In addition, in utero exposure to arsenic reduces birth weight, which is associated with worse respiratory health outcomes in later life (Huyck et al. 2007). Low birth weight infants are more susceptible to lower respiratory tract infections in infancy (McCall and Acheson 1968), have impaired lung function during childhood (Chan et al. 1989), and are at an increased risk of death from chronic lung diseases in adulthood (Barker et al. 1991). Taken together, these factors suggest that arsenic-induced impairments in growth and development of the lung, along with alterations in immune function, may contribute to the development of bronchiectasis in individuals exposed to arsenic in utero.
In the present study, we investigated the effects of in utero exposure to arsenic on postnatal lung mechanics, lung structure, and gene expression using mouse models. We hypothesized that in utero exposure to arsenic via drinking water impairs lung development and immune pathways in the lung.
Methods
Animals and exposure to arsenic. Animals were purchased from the Animal Resources Centre (Murdoch, Western Australia, Australia). Three strains of mice were used; BALB/c, C3H/HeARC [TLR4 (toll-like receptor 4) intact], and C57BL/6. Animals were treated humanely and with regard for alleviation of suffering. All studies were conducted according to the guidelines of the National Health and Medical Research Council Australia and approved by the institutional animal ethics committee. Pregnant mice were given drinking water containing 0 (control) or 100 µg/L of arsenic from gestation day (GD) 8 until birth of their offspring (at approximately GD20) in the form of sodium arsenite (NaAsO2). The arsenic concentration of drinking water was confirmed by inductively coupled plasma–mass spectrometry (ICP-MS) (Geotechnical Services, Perth, Western Australia, Australia). After their offspring were born, mothers were all given control drinking water. All mice received the same fixed formulation diet (Specialty Feeds, Glen Forrest, Western Australia, Australia). The arsenic content in the mouse chow was determined through ion chromatography ICP-MS to be 0.42 ± 0.02 µg/g total arsenic and 0.03 ± 0.001 µg/g inorganic arsenic (Centre for Environmental Risk Assessment and Remediation, University of South Australia, Mawson Lakes, South Australia, Australia). Outcomes were measured in the offspring at 2 weeks of age (BALB/c, n = 29 controls and 24 arsenic exposed; C3H/HeARC, n = 14 controls and 17 arsenic exposed; C57BL/6, n = 24 controls and 32 arsenic exposed).
Thoracic gas volume (TGV) and lung mechanics. To measure lung mechanics in vivo, mice were anesthetized, tracheotomized, and mechanically ventilated. Plethysmography was used to measure TGV as described previously (Janosi et al. 2006). Lung mechanics were measured using the forced-oscillation technique as described previously (Sly et al. 2003). [For further details, see Supplemental Material, p. 2 (http://dx.doi.org/10.1289/ehp.1205590).]
Stereological analysis of lung structure. Lung structure was assessed using stereology techniques according to ATS/ERS (American Thoracic Society/European Respiratory Society) guidelines (Hsia et al. 2010). [For further details, see Supplemental Material, p. 3 (http://dx.doi.org/10.1289/ehp.1205590).]
Gene expression profiling studies. Peripheral lung tissue was excised from mice and stabilized in RNALater (Qiagen, Hilden, Germany) (n = 6 litters per treatment group, per strain). The lung tissue was disrupted in TRIzol (Invitrogen, Life Technologies, Carlsbad, CA, USA) employing a rotor stator homogenizer and extracted with TRIzol followed by RNA purification using RNeasy (Qiagen). The quality of the RNA samples was assessed on the bioanalyzer (model 2100; Agilent Technologies, Santa Clara, CA, USA) [RNA integrity number (RIN) 8.30 ± 0.58]. Total RNA samples were labelled and hybridized to Mouse Gene ST1.0 microarrays (Affymetrix, Santa Clara, CA, USA) by the Molecular Genetic Research Services, at the Australian Neuromuscular Research Institute, QEII Medical Centre, Nedlands, Western Australia, Australia.
The raw microarray data were pre-processed employing the PLIER+16 algorithm (gcbg background subtraction, quantile normalization, iterPLIER summarization) in Expression Consol software (Affymetrix) (Bosco et al. 2009). Two low-quality microarrays were removed from the analysis. To identify differentially expressed genes, the data were analysed with moderated t-statistics (LIMMA R package; http://www.R-project.org) (Smyth 2004) and plotted as Volcano plots [see Supplemental Material, Figure S1 (http://dx.doi.org/10.1289/ehp.1205590)]. Genes with an absolute moderated t-statistic of > 3.5 and fold change > 2 were considered differentially expressed. The microarray data are available from the Gene Expression Omnibus repository (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE37831. A subset of differentially expressed genes was selected for quantitative real-time polymerase chain reaction quantitative real-time polymerase chain reaction (qRT-PCR) validation studies.
qRT-PCR. Total RNA was reverse-transcribed with QuantiTect Reverse Transcription Kit (Qiagen) according to the manufacturer’s instructions. Primer assays were purchased from Qiagen and qRT-PCR was performed with QuantiTect SYBR Green (Qiagen) on the ABI Prism 7900HT (Applied Biosystems, Life Technologies, Foster City, CA, USA). Relative standard curves were prepared from serially diluted RT-PCR products. Data were normalized to the Eef1a1 (eukaryotic translation elongation factor 1 alpha 1) gene and multiplied by a scaling factor to obtain whole numbers.
Quantification of mucous cells and protein in the airways. We performed airway histopathology and immunohistochemistry to determine the expression of mucus-producing cells and protein expression in the airways. After euthanasia, a tracheal cannula was instilled with 4% formaldehyde at 10 cmH20. Lungs were embedded in paraffin wax and the left lobe was sectioned for airway histology. Alcian blue–periodic acid–Schiff stain was used to detect mucus-producing cells, and immunohistochemistry, using an avidin–biotin–peroxidase complex method (Sabo-Attwood et al. 2005), was used to detect cells positive for CLCA3 (chloride channel calcium activated 3 protein), MUC5B (mucin 5, subtype B, tracheobronchial protein), and REG3γ (regenerating islet-derived 3 gamma protein). [For further details, see Supplemental Material, pp. 3–4 (http://dx.doi.org/10.1289/ehp.1205590).] The expression of mucus-producing and protein-positive epithelial cells was calculated as the percentage of positively stained epithelial cells divided by the total number of epithelial cells in the airway. Airways were classified by their basement membrane perimeter (large > 1,500 µm, medium > 1,000 µm, small < 1,000 µm).
Statistical analysis. All statistical analyses were conducted using STATA (version 9.2; StataCorp, College Station, TX, USA). Group means were compared using t-tests, analysis of variance, and analysis of covariance (where adjustment for a continuous variable was required). Data were transformed as necessary to satisfy the assumptions of normality and homoscedasticity. A p-value of ≤ 0.05 was considered statistically significant.
Results
Maternal and birth outcomes. There was no difference in water consumption between mothers given 100 μg/L arsenic compared with control mothers for any strain of mouse (BALB/c, p = 0.46; C3H/HeARC, p = 0.34; C57BL/6, p = 0.82) [see Supplemental Material, Table S1 (http://dx.doi.org/10.1289/ehp.1205590)]. There were no differences in litter size (BALB/c, p = 0.30; C3H/HeARC, p = 0.87; C57BL/6, p = 0.54) or gestational age at birth (BALB/c, p = 0.84; C3H/HeARC, p = 0.21; C57BL/6, p = 0.31) between arsenic-exposed and control mice. Arsenic-exposed C57BL/6 offspring were smaller in birth weight and birth length compared with control C57BL/6 offspring [p < 0.001 and p < 0.001, respectively (see Supplemental Material, Table S1)]. There were no differences in birth weight or length between arsenic-exposed mice and control BALB/c (p = 0.15 and p = 0.21) or C3H/HeARC offspring (p = 0.18 and p = 0.78). For all analyses reported there was no difference in responses between male and female offspring, so data were pooled.
TGV and lung mechanics. Arsenic-exposed C57BL/6 mice were significantly smaller in body weight (p < 0.001) than control offspring at 2 weeks of age and had significantly lower TGV than controls (p < 0.001), even after adjusting for snout-vent length (p = 0.04) [see Supplemental Material, Figure S2 (http://dx.doi.org/10.1289/ehp.1205590)]. Arsenic-exposed C57BL/6 mice had significantly higher tissue damping and tissue elastance (p < 0.001 in both cases) compared with C57BL/6 controls (Figure 1). These differences were maintained after adjusting for TGV (p = 0.006 and p = 0.004, respectively). There was no effect of arsenic on airway resistance (p = 0.29) in C57BL/6 mice, which was maintained after adjusting for TGV (p = 0.78).
Figure 1.
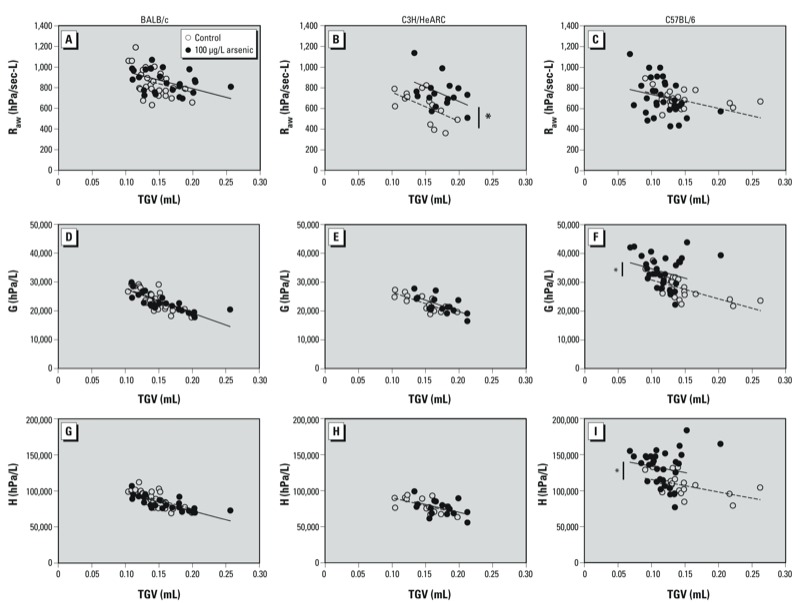
Airway resistance (Raw; A–C), tissue damping (G; D–F) and tissue elastance (H; G–I) plotted against TGV for 2-week-old BALB/c (A,D,G), C3H/HeARC (B,E,H), and C57BL/6 (C,F,I) mice exposed to 100 µg/L arsenic via drinking water or control water from GD8 to birth. *p < 0.05 compared with control.
In C3H/HeARC mice there was no difference in body weight (p = 0.38) at 2 weeks of age for arsenic-exposed mice compared with control mice. TGV was significantly higher in arsenic-exposed mice compared with controls (p = 0.02), which was maintained after adjusting for snout-vent length (p < 0.001) [see Supplemental Material, Figure S2 (http://dx.doi.org/10.1289/ehp.1205590)]. At 2 weeks of age, arsenic-exposed C3H/HeARC mice had significantly higher airway resistance compared with controls (p = 0.04), which was still higher after adjusting for TGV (p = 0.002). There was no effect of arsenic on tissue damping or tissue elastance (p = 0.46 and p = 0.34, respectively) even after adjusting for TGV (p = 0.15 and p = 0.65, respectively) (Figure 1).
In BALB/c mice there was no difference in body weight (p = 0.31) for arsenic-exposed mice compared with control mice at 2 weeks of age. TGV was significantly higher in mice exposed to arsenic in utero (p = 0.05); however, there was no difference after adjusting for snout-vent length (p = 0.20) [see Supplemental Material, Figure S2 (http://dx.doi.org/10.1289/ehp.1205590)]. In BALB/c mice there was no difference in airway resistance at 2 weeks of age between the groups (p = 0.82), which was also the case after adjusting for TGV (p = 0.28). There was evidence to suggest that both tissue damping and tissue elastance were lower in arsenic-exposed mice compared with controls (p = 0.07 and p = 0.02, respectively); however, these differences were negated after adjusting for TGV (p = 0.65 and p = 0.22, respectively) (Figure 1).
Stereological analysis of lung structure. In C57BL/6 mice, lung volume (calculated by stereology) was significantly smaller in arsenic-exposed offspring compared with controls (p = 0.03). The number of alveoli in the lung (p = 0.01) and the surface area of the lung (p = 0.04) were also smaller in arsenic-exposed C57BL/6 mice compared with control mice of the same strain; however, there was no difference in the volume of the alveoli (p = 0.41) (Figure 2). We found no differences in any of the structural parameters measured between arsenic-exposed mice and control BALB/c mice or C3H/HeARC.
Figure 2.
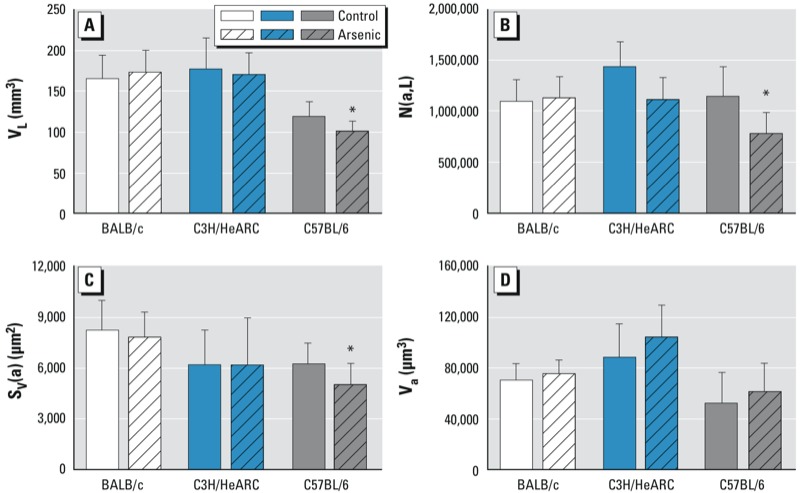
Lung volume (VL; A), number of alveoli in the lung [N(a,L); B], alveolar surface area [Sv(a); C], and alveolar volume (Va; D) for 2-week-old BALB/c, C3H/HeARC, and C57BL/6 mice exposed to 100 µg/L arsenic via drinking water or control water from GD8 to birth. *p < 0.05 compared with control.
Gene expression profiling studies. Using microarray analysis, we identified 10 annotated genes in the lung that were differentially expressed as a result of in utero exposure to arsenic (Table 1). C57BL/6 mice had 7 genes that were differentially expressed, whereas C3H/HeARC mice had 2 and BALB/c mice had only 1 differentially expressed gene(s). Only 1 gene was down-regulated in response to arsenic, Olfr1274 (olfactory receptor 1274), in the C57BL/6 strain. All of the other genes identified as differentially expressed were up-regulated in response to arsenic exposure.
Table 1.
Genes altered by arsenic exposure.
| Mouse strain/ gene symbol | Gene name | Function | Reference | Fold change |
|---|---|---|---|---|
| C57BL/6 | ||||
| Clca3 | Chloride channel calcium activated 3 | Regulation of mucus production and secretion | Zhou et al. 2002 | 15.05 |
| Lplunc1 | Long palate, lung, and nasal epithelium carcinoma-associated protein 1 | Innate immunity in mouth, nose, and lungs | Bingle and Craven 2002 | 13.72 |
| Reg3γ | Regenerating islet-derived 3 gamma | Innate immunity–antibacterial properties in the mucus secretions | McAleer et al. 2011 | 8.77 |
| Scgb3a1 | Secretoglobin, family 3A, member 1 | Secretary cell subtype in the lung | Roy et al. 2011 | 5.80 |
| Tff2 | Trefoil factor 2 (spasmolytic protein 1) | Innate immunity–allergen induced gene, expressed in mucous cells | Nikolaidis et al. 2003 | 3.26 |
| Muc5b | Mucin 5, subtype B, tracheobronchial | Major mucin in respiratory mucus | Kirkham et al. 2002 | 3.16 |
| Olfr1274 | Olfactory receptor 1274 | Olfaction | Zhang and Firestein 2002 | –2.07 |
| C3H/HeARC | ||||
| Dynlrb2 | Dynein light chain roadblock–type 2 | Regulation of TGF-B pathway | Jin et al. 2009 | 2.36 |
| Sox2 | SRY-box containing gene 2 | Lung branching morphogenesis and epithelial cell differentiation | Gontan et al. 2008 | 2.27 |
| BALB/c | ||||
| Ssxb10 | Synovial sarcoma, X member B, breakpoint 10 | Cancer | Chen et al. 2003 | 2.41 |
Three of the 10 genes that were up-regulated [Clca3, Muc5b, and Scgb3a1) (secretoglobin, family 3A, member 1] are involved in the regulation and/or secretion of mucus in the airways (Roy et al. 2011). These genes were all up-regulated in C57BL/6 mice. Four of the 10 genes {Lplunc1 [long palate, lung, and nasal epithelium carcinoma-associated protein 1], Tff2 [trefoil factor 2 (spasmolytic protein 1)], Reg3γ, and Dynlrb2 [dynein light chain roadblock–type 2]} that were up-regulated are involved in innate immune function (Bingle and Craven 2002; Jin et al. 2009; McAleer et al. 2011; Nikolaidis et al. 2003). Lplunc1, Tff2, and Reg3γ were up-regulated in C57BL/6 mice, and Dynlrb2 was up-regulated in C3H/HeARC mice. We also identified a gene involved in lung branching morphogenesis and epithelial cell differentiation, Sox2 (Gontan et al. 2008), which was up-regulated in C3H/HeARC mice. In BALB/c mice, an oncogene [Ssxb10 (synovial sarcoma, X member B, breakpoint 10)] was the only gene up-regulated (Chen et al. 2003).
Validation of array expression using qRT-PCR. Using qRT-PCR, we validated the expression of four genes in C57BL/6 mice (Clca3, Muc5b, Tff2, and Reg3g) and one gene in C3H/HeARC mice [Sox2 (SRY-box containing gene 2)]. In all cases, the expression patterns, normalized to the Eef1a1 gene, correlated to those obtained in the microarray analysis (Figure 3).
Figure 3.
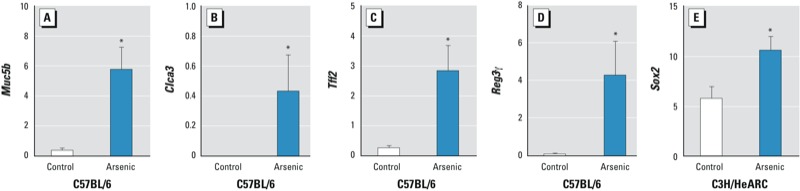
Validation of microarray gene expression in a subset of genes (Muc5b, Clca3, Tff2, Reg3γ, and Sox2) using qRT-PCR. All values were normalized to the Eef1a1 gene. *p < 0.05 compared with control.
Quantification of mucus and protein in the airways. We used Alcian blue–periodic acid–Schiff staining and immunohistochemistry to quantify mucus-producing cells and CLCA3, MUC5B, and REG3γ protein in the airways of BALB/c, C3H/HeARC, and C57BL/6 mice (Figure 4). We found that arsenic exposure in utero caused mucous cell metaplasia in the large (p < 0.001) and medium (p = 0.02) airways of C57BL/6 mice and increased the expression of CLCA3 (p < 0.01) and REG3γ (p = 0.02) proteins in the large airways of C57BL/6 mice (Figure 5). The levels of MUC5B protein were low and not differentially expressed between the airways of arsenic-exposed mice and control C57BL/6 mice. There were no differences in the number of mucus-producing cells or CLCA3, MUC5B, or REG3γ proteins between the airways of arsenic and control mice of either the BALB/c or C3H/HeARC strains (data not shown).
Figure 4.
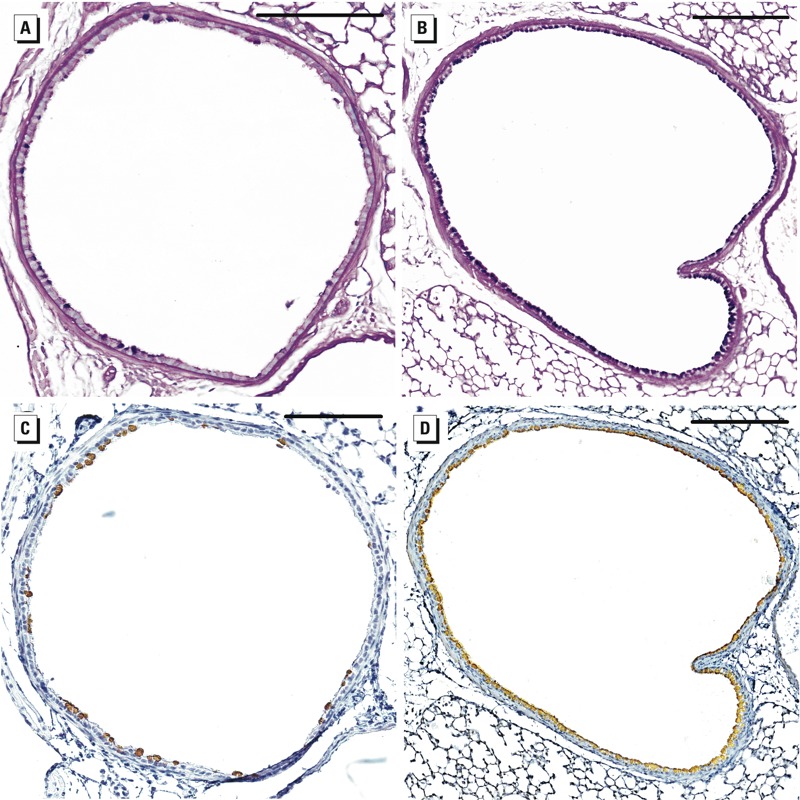
Airway histology showing that arsenic induces mucous cell metaplasia and increases CLCA3 protein expression in the airways of C57BL/6 mice. Lung sections stained with Alcian blue–periodic acid–Schiff (A,B) and an antibody to CLCA3 (C,D) show increased mucous cell (dark purple stain) and CLCA3 protein (brown stain) expression in the airways of C57BL/6 mice exposed 100 µg/L arsenic (B,D) via drinking water or control water (A,C) from GD8 to birth. Bars = 200 µm.
Figure 5.
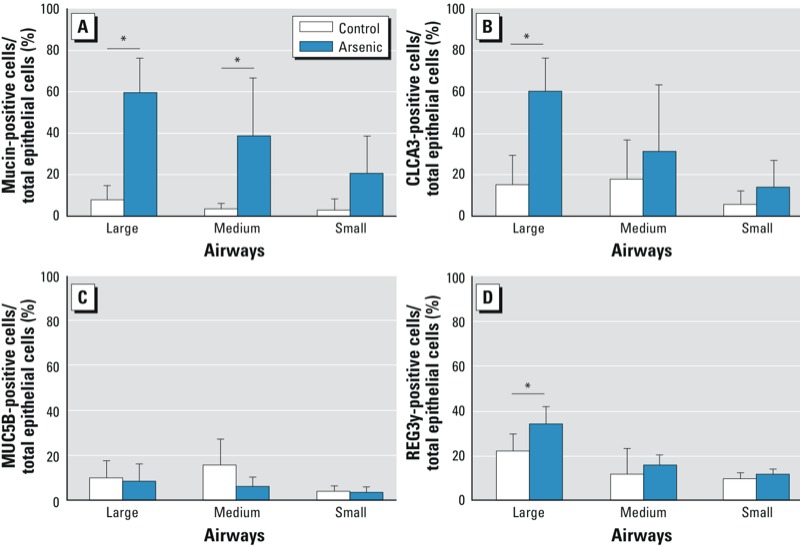
Quantitation of mucus-producing cells (A) and CLCA3-(B), MUC5B- (C), and REG3γ- (D)positive epithelial cells in the airways of C57BL/6 mice. Values are expressed as the percentage of mucus-producing epithelial cells and protein-positive epithelial cells ÷ total number of epithelial cells in large, medium, and small airways of C57BL/6 mice exposed to 100 µg/L arsenic via drinking water or control water from GD8 to birth. *p < 0.05 compared with control.
Discussion
Exposure to arsenic in utero has been shown to increase morbidity from lower respiratory tract infections in infancy and increase mortality from bronchiectasis in adulthood (Rahman et al. 2011; Smith et al. 2006). In the present study, our results show that the response to arsenic in mice is genetically determined and that the C57BL/6 strain is the most susceptible of the three strains studied. In utero exposure to arsenic impaired somatic growth, lung volume, and parenchymal lung mechanics in C57BL/6 mice. In addition, exposure to arsenic during gestation is capable of altering the expression of genes that function in mucociliary clearance, innate immunity, and lung growth in the offspring. Arsenic also induced mucous cell metaplasia and increased expression of CLCA3 protein in the airways of C57BL/6 mice. Increased mucus secretion and altered lung growth may impair the ability of the airways to clear pathogens and thus provide a potential mechanism by which exposure to arsenic increases the risk of developing chronic localized infections leading to bronchiectasis.
Exposure to arsenic in utero impaired intrauterine growth and lung development. Arsenic-exposed C57BL/6 mice were small for gestational age; had lower lung volume, lung surface area, and alveolar number; and had impaired lung mechanics compared with control mice of the same strain. This finding supports results from a recent study that also found that C57BL/6 mice exposed to arsenic in utero had deficits in growth in early life (Kozul-Horvath et al. 2012). Impairments to body size and lung size during infancy may result in airways and alveoli that are prone to closure and collapse, thus increasing the susceptibility to obstruction and increased pathogen load. Low birth weight is associated with impaired lung function and increased mortality from obstructive lung disease (Barker et al. 1991; Hulskamp et al. 2009). A potential mechanism for the observed intrauterine growth restriction may be oxidative stress caused by arsenic-induced production of reactive oxygen species inducing placental insufficiency (Vahter 2007). This proposed mechanism is supported by the knowledge that the mouse strain most sensitive to arsenic, C57BL/6, is highly susceptible to the oxidative stress effects of cigarette smoke and bleomycin compared with other strains (Chen et al. 2001a; Yao et al. 2008). There is evidence from cigarette smoke exposure models that intrauterine growth restriction alone can cause alterations in early life lung function (Larcombe et al. 2011). It is therefore possible that the effects of arsenic exposure on body weight, lung function, and lung size in the present study, in the C57BL/6 mice at least, were due to arsenic altering the nutritional status of the offspring. However, we also identified responses, such as alterations in lung gene expression and airway morphology that seem to be unique to arsenic exposure and have yet to be linked with altered nutritional status. It is clear that exposure to arsenic during pregnancy is having a significant effect on intrauterine growth, which may predispose infants to chronic lung diseases through impairments in lung growth and development.
We found that arsenic exposure during pregnancy was capable of altering the expression of genes in the lung at 2 weeks of age. The differentially expressed genes identified are involved in mucus production, innate immunity, and lung morphogenesis pathways. The gene that was most highly up-regulated was the calcium-activated chloride channel, Clca3 gene. This gene has been detected in goblet cells within the tracheal and bronchial epithelium of mice when metaplasia of mucous cells is present (Leverkoehne and Gruber 2002) and is thought to play a role in the mucus hypersecretion seen in chronic obstructive pulmonary disease (COPD) and CF in humans (Rogers 2003; Shale and Ionescu 2004). Blocking the production and/or activity of Clca3 can inhibit the production of mucus, whereas up-regulation can enhance mucus production (Zhou et al. 2002). Clca3 has been implicated in the regulation of mucus secretion by controlling the packaging and/or release of secreted mucins such as MUC5AC and MUC5B, which are the major gel-forming mucins in respiratory secretions (Nakanishi et al. 2001; Zhou et al. 2002). MUC5B levels are increased in sputum from the airways of patients with COPD and CF compared with sputum from healthy airways (Kirkham et al. 2002) and are associated with the overproduction of mucus in diseased airways (Chen et al. 2001b). In our study, Muc5b gene expression was also up-regulated and mucous cell metaplasia was present in the airways of C57BL/6 mice. Mucous cell metaplasia is a pathological feature of various respiratory diseases including both CF and non-CF bronchiectasis, COPD, and asthma (Kim 1997). Mucus hypersecretion can hinder the ability of the cilia to clear mucus from the airways, thus increasing the susceptibility to viral and bacterial colonization and infection (Shale and Ionescu 2004). Increased mucus in the airways may also lead to obstruction and contribute to abnormalities in lung mechanics (Rose et al. 2001). Increased mucus production resulting in increased susceptibility to respiratory infections combined with airway obstruction can lead to chronic localized inflammation: a hallmark of the bronchiectatic lung (Barker 2002).
As well as increasing the risk of infection, arsenic exposure in utero may compromise the immune response to respiratory infections. In the present study, we identified four genes that play a role in modulating the immune system that were up-regulated by exposure to arsenic. TFF2 protein has a role in mucosal epithelial restitution and wound healing in the gastrointestinal tract (Podolsky 2000). In the lung, TFF2 protein expression is increased in mucous cells of allergen-challenged mice and is up-regulated in airway epithelial cells in subjects with asthma (Kuperman et al. 2005; Nikolaidis et al. 2006). Tff2 has also been identified as a candidate gene associated with strain-dependent differences in lung function in mice (Ganguly et al. 2007). REG3γ is a protein with antimicrobial activity identified as playing a role in innate mucosal immunity in the gastrointestinal tract (Zheng et al. 2008) and more recently in the lung (McAleer et al. 2011). Dynlrb2 is differentially expressed in patients with primary ciliary dyskinesia, a condition that can lead to the development of bronchiectasis due to impairment of mucociliary clearance (Geremek et al. 2011). Dynlrb2 has also been shown to regulate the TGF-β (transforming growth factor, beta) signalling pathway (Jin et al. 2009). Increased production and activation of TGF-β has been linked to immune defects associated with the susceptibility to opportunistic infection (Letterio and Roberts 1998). LPLUNC1 is a protein suggested to play a role in innate immunity in the lung, mouth, and nose, through either direct antibacterial actions or by indirect neutralizing activity (Bingle and Craven 2002). Mucous cell metaplasia and altered immune pathways in the lung may partially explain the increased susceptibility to and exacerbated response to influenza infection seen in a mouse model of arsenic exposure (Kozul et al. 2009). Importantly, compromised innate immune pathways in the lung during infancy may increase the risk of respiratory infections in early life, when susceptibility to infection is high, and increase the risk of developing chronic lung disease in adulthood.
To put these observations into context, we recognize that there were some limitations to the present study. In the mouse chow given to all the mice in our study, there was a small amount (0.03 µg/g) of inorganic arsenic, which has the potential to alter gene expression in the liver and lung (Kozul et al. 2008). However, the concentrations of inorganic arsenic in our study were low, the same diet was given to all of the mice, and we still detected differences in a variety of outcomes, including gene expression, as a result of the exposure to 100 µg/L arsenic in drinking water. Another limitation of the study is that only a single concentration of arsenic (100 µg/L) was used in our study. This represents a relatively high concentration of arsenic [10 times higher than the current World Health Organization (WHO 2011a) maximum contaminant level], and the effects observed in the present study may not reflect changes that would occur at higher or lower doses. In the present study, we identified only a small number of genes that were differentially expressed as a result of arsenic exposure. Even by relaxing our criteria for differential expression, we were not able to identify any functionally coherent pathways using more complex methods of analysis. Because we assessed the lung tissue sometime after the arsenic exposure had ceased, it is possible that we missed some pathways and there is value in examining expression networks in lung tissue during exposure in the future (Petrick et al. 2009). Our study, however, has identified and validated a number of genes that we believe provide important clues to the underlying mechanisms and, given their function, are likely to be important in the lung disease phenotype induced by arsenic exposure in humans (e.g., infection/bronchiectasis). In addition, we examined the effects of arsenic exposure on the lung in offspring at only 2 weeks of age in the present study. The long-term effects of arsenic exposure on lung development may differ between strains whereby changes in parenchymal growth may manifest at different ages (Ngalame et al. 2012). An analysis of later time points may provide further information about the genetic basis of susceptibility to arsenic-induced lung disease.
Conclusions
In the present study, we have shown that in utero exposure to arsenic via drinking water impairs lung development, resulting in reduced lung size and impaired lung structure and function. The response to arsenic is genetically determined and the C57BL/6 strain is the most susceptible. Arsenic exposure in utero altered the expression of genes that regulate innate immunity, mucus production, and morphogenesis of the lung. Arsenic-induced alterations in the development of lung structure and innate immunity may reduce the ability to clear respiratory pathogens during a period of high susceptibility in infancy (Simoes 1999). Because respiratory infections are one of the leading causes of mortality in children under 5 years of age in developing countries (WHO 2011b), arsenic-induced increases in susceptibility to infection can have devastating effects on infant morbidity and mortality in affected areas. In addition, recurrent respiratory infections throughout life can contribute to the development of chronic lung diseases, such as bronchiectasis, in adulthood. The evidence from our mouse model is that in utero exposure to arsenic impairs lung development, resulting in altered structure, function, and gene expression in infancy. Further investigation into how arsenic affects lung development in humans will be essential to understand and prevent escalating morbidity and mortality from respiratory disease in arsenic-exposed populations.
Supplemental Material
Acknowledgments
We acknowledge A. James and R. Jones for their guidance and assistance in the stereological analysis of lung structure.
Footnotes
This work was supported by project grant 634420 from the National Health and Medical Research Council (Australia).
The authors declare they have no actual or potential competing financial interests.
References
- Ahmed S, Mahabbat-e Khoda S, Rekha RS, Gardner RM, Ameer SS, Moore S, et al. 2011Arsenic-associated oxidative stress, inflammation, and immune disruption in human placenta and cord blood. Environ Health Perspect 119258–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker A. Bronchiectasis. N Engl J Med. 2002;346:1383–1394. doi: 10.1056/NEJMra012519. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Godfrey KM, Fall C, Osmond C, Winter PD, Shaheen SO. Relation of birth weight and childhood respiratory infection to adult lung function and death from chronic obstructive airways disease. BMJ. 1991;303(6804):671–675. doi: 10.1136/bmj.303.6804.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingle CD, Craven CJ. PLUNC: a novel family of candidate host defence proteins expressed in the upper airways and nasopharynx. Hum Mol Genet. 2002;11(8):937–943. doi: 10.1093/hmg/11.8.937. [DOI] [PubMed] [Google Scholar]
- Bosco A, McKenna KL, Firth MJ, Sly PD, Holt PG. A network modeling approach to analysis of the Th2 memory responses underlying human atopic disease. J Immunol. 2009;182(10):6011–6021. doi: 10.4049/jimmunol.0804125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KN, Noble-Jamieson CM, Elliman A, Bryan EM, Silverman M. Lung function in children of low birth weight. Arch Dis Child. 1989;64(9):1284–1293. doi: 10.1136/adc.64.9.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ES, Greenlee BM, Wills-Karp M, Moller DR. Attenuation of lung inflammation and fibrosis in interferon-γ-deficient mice after intratracheal bleomycin. Am J Respir Cell Mol Biol. 2001a;24(5):545–555. doi: 10.1165/ajrcmb.24.5.4064. [DOI] [PubMed] [Google Scholar]
- Chen Y, Zhao YH, Di YP, Wu R. Characterization of human mucin 5B gene expression in airway epithelium and the genomic clone of the amino-terminal and 5´-flanking region. Am J Respir Cell Mol Biol. 2001b;25(5):542. doi: 10.1165/ajrcmb.25.5.4298. [DOI] [PubMed] [Google Scholar]
- Chen YT, Alpen B, Ono T, Gure AO, Scanlan MA, Biggs WH, III, et al. Identification and characterization of mouse SSX genes: a multigene family on the X chromosome with restricted cancer/testis expression. Genomics. 2003;82(6):628–636. doi: 10.1016/s0888-7543(03)00183-6. [DOI] [PubMed] [Google Scholar]
- Ganguly K, Stoeger T, Wesselkamper SC, Reinhard C, Sartor MA, Medvedovic M, et al. Candidate genes controlling pulmonary function in mice: transcript profiling and predicted protein structure. Physiol Genomics. 2007;31(3):410–421. doi: 10.1152/physiolgenomics.00260.2006. [DOI] [PubMed] [Google Scholar]
- Geremek M, Bruinenberg M, Zietkiewicz E, Pogorzelski A, Witt M, Wijmenga C. Gene expression studies in cells from primary ciliary dyskinesia patients identify 208 potential ciliary genes. Hum Genet. 2011;129(3):283–293. doi: 10.1007/s00439-010-0922-4. [DOI] [PubMed] [Google Scholar]
- Gontan C, de Munck A, Vermeij M, Grosveld F, Tibboel D, Rottier R. Sox2 is important for two crucial processes in lung development: Branching morphogenesis and epithelial cell differentiation. Dev Biol. 2008;317(1):296–309. doi: 10.1016/j.ydbio.2008.02.035. [DOI] [PubMed] [Google Scholar]
- Grimwood K. Airway microbiology and host defences in paediatric non-CF bronchiectasis. Paediatr Respir Rev. 2011;12(2):111–118. doi: 10.1016/j.prrv.2010.10.009. [DOI] [PubMed] [Google Scholar]
- Hsia CCW, Hyde DM, Ochs M, Weibel E. An official research policy statement of the American Thoracic Society/European Respiratory Society: Standards for Quantitative Assessment of Lung Structure. Am J Respir Crit Care Med. 2010;181:394–418. doi: 10.1164/rccm.200809-1522ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulskamp G, Lum S, Stocks J, Wade A, Hoo AF, Costeloe K, et al. Association of prematurity, lung disease and body size with lung volume and ventilation inhomogeneity in unsedated neonates: a multicentre study. Thorax. 2009;64(3):240–245. doi: 10.1136/thx.2008.101758. [DOI] [PubMed] [Google Scholar]
- Huyck KL, Kile ML, Mahiuddin G, Quamruzzaman Q, Rahman M, Breton CV, et al. Maternal arsenic exposure associated with low birth weight in Bangladesh. J Occup Environ Med. 2007;49(10):1097–1104. doi: 10.1097/JOM.0b013e3181566ba0. [DOI] [PubMed] [Google Scholar]
- Janosi TZ, Adamicza A, Zosky GR, Asztalos T, Sly PD, Hantos Z. Plethysmographic estimation of thoracic gas volume in apneic mice. J Appl Physiol. 2006;101(2):454–459. doi: 10.1152/japplphysiol.00011.2006. [DOI] [PubMed] [Google Scholar]
- Jin Q, Gao G, Mulder KM. Requirement of a dynein light chain in TGFβ/Smad3 signaling. J Cell Physiol. 2009;221(3):707–715. doi: 10.1002/jcp.21910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WD. Lung mucus: a clinician’s view. Eur Respir J. 1997;10(8):1914–1917. doi: 10.1183/09031936.97.10081914. [DOI] [PubMed] [Google Scholar]
- King P, Holdsworth S, Freezer N, Holmes P. Bronchiectasis. Intern Med J. 2006;36(11):729–737. doi: 10.1111/j.1445-5994.2006.01219.x. [DOI] [PubMed] [Google Scholar]
- Kirkham S, Sheehan JK, Knight D, Richardson PS, Thornton DJ. Heterogeneity of airways mucus: variations in the amounts and glycoforms of the major oligomeric mucins MUC5AC and MUC5B. Biochem J. 2002;361(3):537–546. doi: 10.1042/0264-6021:3610537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozul CD, Ely KH, Enelow RI, Hamilton JW. Low-dose arsenic compromises the immune response to influenza A infection in vivo. Environ Health Perspect. 2009;117:1441–1447. doi: 10.1289/ehp.0900911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozul CD, Nomikos AP, Hampton T, Warnke L, Gosse JA, Davey JC, et al. Laboratory diet profoundly alters gene expression and confounds genomic analysis in mouse liver and lung. Chem Biol Interact. 2008;173:129–140. doi: 10.1016/j.cbi.2008.02.008. [DOI] [PubMed] [Google Scholar]
- Kozul-Horvath CD, Zandbergen F, Jackson BP, Enelow RI, Hamilton JW.2012Effects of low-dose drinking water arsenic on mouse fetal and postnatal growth and development. PLoS One 75e38249; doi: 10.1371/journal.pone.0038249[Online 31 May 2012] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuperman DA, Lewis CC, Woodruff PG, Rodriguez MW, Yang YH, Dolganov GM, et al. Dissecting asthma using focused transgenic modeling and functional genomics. J Allergy Clin Immunol. 2005;116(2):305–311. doi: 10.1016/j.jaci.2005.03.024. [DOI] [PubMed] [Google Scholar]
- Larcombe A, Foong R, Berry L, Zosky G, Sly P. In utero cigarette smoke exposure impairs somatic and lung growth in BALB/c mice. Eur Respir J. 2011;38(4):932–938. doi: 10.1183/09031936.00156910. [DOI] [PubMed] [Google Scholar]
- Letterio J, Roberts A. Regulation of immune responses by TGF-β. Annu Rev Immunol. 1998;16:137–161. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- Leverkoehne I, Gruber AD. The murine mCLCA3 (alias gob-5) protein is located in the mucin granule membranes of intestinal, respiratory, and uterine goblet cells. J Histochem Cytochem. 2002;50(6):829–838. doi: 10.1177/002215540205000609. [DOI] [PubMed] [Google Scholar]
- Mandal BK, Suzuki KT. Arsenic round the world: a review. Talanta. 2002;58(1):201–235. [PubMed] [Google Scholar]
- McAleer J, Choi S, Davila E, Ouyang W, Qin S, Reinhart T, et al. Elucidating the role of Reg3 family members in pulmonary host defense. Am J Respir Crit Care Med. 2011;183:A1071. [Abstract] [Google Scholar]
- McCall MG, Acheson ED. Respiratory disease in infancy. J Chronic Dis. 1968;21(5):349–359. doi: 10.1016/0021-9681(68)90043-x. [DOI] [PubMed] [Google Scholar]
- Nakanishi A, Morita S, Iwashita H, Sagiya Y, Ashida Y, Shirafuji H, et al. Role of gob-5 in mucus overproduction and airway hyperresponsiveness in asthma. Proc Natl Acad Sci USA. 2001;98(9):5175–5180. doi: 10.1073/pnas.081510898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngalame N, Micciche A, Feil M, States JC.2012Delayed temporal increase of hepatic Hsp70 in ApoE knockout mice after prenatal arsenic exposure. Toxicol Sci; doi: 10.1093/toxsci/kfs264[Online 5 September 2012] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaidis NM, Wang TC, Hogan SP, Rothenberg ME. Allergen induced TFF2 is expressed by mucus-producing airway epithelial cells but is not a major regulator of inflammatory responses in the murine lung. Exp Lung Res. 2006;32(10):483–497. doi: 10.1080/01902140601059547. [DOI] [PubMed] [Google Scholar]
- Nikolaidis NM, Zimmermann N, King NE, Mishra A, Pope SM, Finkelman FD, et al. Trefoil factor-2 is an allergen-induced gene regulated by Th2 cytokines and STAT6 in the lung. Am J Respir Cell Mol Biol. 2003;29(4):458–464. doi: 10.1165/rcmb.2002-0309OC. [DOI] [PubMed] [Google Scholar]
- Petrick J, Blachere F, Selmin O, Lantz R. Inorganic arsenic as a developmental toxicant: In utero exposure and alterations in the developing rat lungs. Mol Nutr Food Res. 2009;53(5):583–591. doi: 10.1002/mnfr.200800019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podolsky DK. Mechanisms of regulatory peptide action in the gastrointestinal tract: trefoil peptides. J Gastroenterol. 2000;35(suppl 12):69–74. [PubMed] [Google Scholar]
- Rahman A, Vahter M, Ekström EC, Persson LÅ. Arsenic exposure in pregnancy increases the risk of lower respiratory tract infection and diarrhea during infancy in Bangladesh. Environ Health Perspect. 2011;119:719–724. doi: 10.1289/ehp.1002265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raqib R, Ahmed S, Sultana R, Wagatsuma Y, Mondal D, Hoque AW, et al. Effects of in utero arsenic exposure on child immunity and morbidity in rural Bangladesh. Toxicol Lett. 2009;185:197–202. doi: 10.1016/j.toxlet.2009.01.001. [DOI] [PubMed] [Google Scholar]
- Rogers DF. The airway goblet cell. Int J Biochem Cell Biol. 2003;35(1):1–6. doi: 10.1016/s1357-2725(02)00083-3. [DOI] [PubMed] [Google Scholar]
- Rose MC, Nickola TJ, Voynow JA. Airway mucus obstruction: mucin glycoproteins, MUC gene regulation and goblet cell hyperplasia. Am J Respir Cell Mol Biol. 2001;25(5):533–537. doi: 10.1165/ajrcmb.25.5.f218. [DOI] [PubMed] [Google Scholar]
- Roy MG, Rahmani M, Hernandez JR, Alexander SN, Ehre C, Ho SB, et al. Mucin production during prenatal and postnatal murine lung development. Am J Respir Cell Mol Biol. 2011;44(6):755–760. doi: 10.1165/rcmb.2010-0020OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo-Attwood T, Ramos-Nino M, Bond J, Butnor K, Heintz N, Gruber A, et al. Gene expression profiles reveal increased mClca3 (Gob5) expression and mucin production in a murine model of asbestos-induced fibrogenesis. Am J Pathol. 2005;167(5):1243–1256. doi: 10.1016/S0002-9440(10)61212-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shale DJ, Ionescu AA. Mucus hypersecretion: a common symptom, a common mechanism? Eur Respir J. 2004;23(6):797–798. doi: 10.1183/09031936.0.00018404. [DOI] [PubMed] [Google Scholar]
- Simoes EA. Respiratory syncytial virus infection. Lancet. 1999;354(9181):847–852. doi: 10.1016/S0140-6736(99)80040-3. [DOI] [PubMed] [Google Scholar]
- Sly P, Collins R, Thamrin C, Turner D, Hantos Z. Volume dependence of airway and tissue impedance in mice. J Appl Physiol. 2003;94:1460–1466. doi: 10.1152/japplphysiol.00596.2002. [DOI] [PubMed] [Google Scholar]
- Smith AH, Marshall G, Yan Y, Ferreccio C, Liaw J, von Ehrenstein O, et al. Increased mortality from lung cancer and bronchiectasis in young adults after exposure to arsenic in utero and in early childhood. Environ Health Perspect. 2006;114:1293–1296. doi: 10.1289/ehp.8832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. 2004. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3:Article 3.
- Vahter ME. Interactions between arsenic-induced toxicity and nutrition in early life. J Nutr. 2007;137(12):2798–2804. doi: 10.1093/jn/137.12.2798. [DOI] [PubMed] [Google Scholar]
- WHO (World Health Organization) 2011a. Guidelines for Drinking-water Quality 4ED. Geneva: WHO. Available: http://whqlibdoc.who.int/publications/2011/9789241548151_eng.pdf [accessed 6 January 2013]
- WHO (World Health Organization) World Health Statistics 2011. Geneva:WHO. 2011b. Available: http://www.who.int/whosis/whostat/EN_WHS2011_Full.pdf [accessed 2 January 2013]
- Yao H, Edirisinghe I, Rajendrasozhan S, Yang SR, Caito S, Adenuga D, et al. Cigarette smoke-mediated inflammatory and oxidative responses are strain-dependent in mice. Am J Physiol Lung Cell Mol Physiol. 2008;294(6):L1174–L1186. doi: 10.1152/ajplung.00439.2007. [DOI] [PubMed] [Google Scholar]
- Zhang X, Firestein S. The olfactory receptor gene superfamily of the mouse. Nat Neurosci. 2002;5(2):124–133. doi: 10.1038/nn800. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14(3):282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Shapiro M, Dong Q, Louahed J, Weiss C, Wan S, et al. A calcium-activated chloride channel blocker inhibits goblet cell metaplasia and mucus overproduction. Novartis Found Symp. 2002;248:150–165. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.