

Abstract
Since leucine-rich repeat kinase 2 (LRRK2) was linked to Parkinson's disease in 2004, kinase activity of LRRK2 has been believed to play a critical role in the pathogenesis of Parkinson's disease. As a result, identification of LRRK2 inhibitors has been a focus for drug discovery. However, most LRRK2 mutations do not simply increase kinase activity. In this review we summarize the potential mechanisms that regulate the kinase activity of LRRK2. We outline some currently available kinase inhibitors, including the identification of a DFG-out (type-II) inhibitor. Finally, we discuss the relationship of LRRK2 with tau and α-synuclein. The fact that all three proteins are autophapgy-related provides a future strategy for the identification of LRRK2 physiological substrate(s).
Since mutations of leucine-rich repeat kinase 2 (LRRK2) were identified in Parkinson's disease (PD) patients in 2004, LRRK2 has emerged as the most relevant gene to PD pathogenesis [1,2]. More than 40 mutations of LRRK2 have been found in both familial and sporadic forms of PD [3–5]. LRRK2 is ubiquitously expressed in the substantia nigra of the brain, the region where degeneration of dopaminergic neurons starts in PD patients [6,7]. LRRK2 has also been reported to be a prominent part of Lewy body deposits in PD [8]. Currently, the cellular functions of LRRK2 are poorly understood due to its unknown physiological substrate(s), although several proteins have been reported to be phosphorylated by LRRK2, including ezrin/radixin/moesin, 4E-BP, MKKs, β-tubulin, α-synuclein, peroxiredoxin 3, Akt1 and ArfGAP1 [9–18] Recently, accumulated evidence has suggested roles of LRRK2 in autophagy [19–21] and neuro-inflammation [22–25], indicating various functions of LRRK2. Reviews regarding the biological functions of LRRK2 and animal models have been recently published [26–28]. In this review, we focus on the possible functions associated with the different domains of LRRK2, the mechanisms of kinase regulation, inhibitors of kinase activity, and the relationship that LRRK2 may have with α-synuclein and tau.
Structural biology of LRRK2
LRRK2 is a large gene whose transcript encodes a 2527 amino acid protein (286 kDa) that is comprised of 51 exons. Sequence analysis predicts that LRRK2 contains multiple domains, including an ankyrin-like (ANK) domain, leucine-rich repeat (LRR) domain, a ROC domain followed by its associated C-terminal of Roc (COR) domain, a mitogen-activated protein kinase (MAPK) domain and a C-terminal WD40 domain (Figure 1A). The presence of both protein interaction domains (ANK, LRR and WD40) and the enzymatic domains (ROC and MAPK) within LRRK2 suggests that this protein may serve as a scaffold for assembly of a multiprotein complex and act as a central integrator of multiple signaling pathways.
Figure 1. Position of Parkinson's disease-linked mutations of LRRK2 indicated on linear domain structure and homology models.

(A) A linear representation of LRRK2 sequence and the domain organization with some of the most commonly occurring Parkinson's disease mutations annotated on these domains. The two mutations in the kinase activation loop G2019S and I2020T are indicated in italics. (B) Ribbon representation of the x-ray structure of the GTPase domain of LRRK2 (2ZEJ) showing the positions of Parkinson's disease-linked mutations. (C) Ribbon representation of the kinase domain of LRRK2 showing the positions of various Parkinson's disease-linked mutations. The mutations in the activation loop G2019S and I2020T are indicated in italics.
With its domain structure, LRRK2 is considered a member of the ROCO family. The ROCO protein family has a conserved core, consisting of a Ras-like GTPase called Roc (Ras of complex proteins) and a COR domain, often with a C-terminal kinase domain and several N-terminal LRR. The first ROCO family member to be identified was Dictyostelium cGMP-binding protein GbpC [29]. This marked a new research area for cell biologists and biochemists when the genome sequence of this model organism became available. Although the initial description of the ROCO family of proteins did not draw much attention in the field, this rapidly changed when dominant mutations of LRRK2, a member of the human ROCO family, were found to be linked to PD. The most common genetic PD-associated mutations are found throughout the functional domains of LRRK2 (Figure 1A), and therefore have the potential to impact both its enzymatic properties and protein interactions. No published x-ray crystal structure is yet available of LRRK2, and therefore structural analysis is confined to homology modeling. An examination and modeling of LRRK2 domain organization are important for understanding the underlying mechanisms.
Understanding the function of each domain is important for drug design since each domain provides drug interaction sites additional to the ones on the kinase domain.
Protein–protein interaction domains
ANK, LRR & WD40 domains
The ANK, LRR and WD40 domains are found in a number of different proteins and are suggested to provide a versatile framework for recruiting a number of different substrates to the kinase domain for phosphorylation. The ANK domain is generally believed to work as attachment for integral membrane proteins and cytoskeleton. Indeed, recent biochemical data suggested increased dimerization and kinase activity of LRRK2 upon membrane localization [30].
The LRR domain is a well-studied domain present in many protein structures. The folded form of this domain forms a crescent-shaped structure creating a large solvent-exposed concave surface made up of parallel β-strands that acts as a docking point for other proteins to bind and interact. Some of the well-known functions of the LRR domain include hormone–receptor interactions, cell adhesion, enzyme inhibition, cellular trafficking and regulation of cytoskeletal dynamics [31]. Based on a homology modeling of the LRR domain, many of the LRRK2 amino acid substitutions in the LRR domain are predicted to lie on the surface of this domain, such as S1226T, and therefore, it is conceivable that these mutations either disrupt or weaken protein–protein interactions [32]. However, at least one PD-linked mutant I1122V appears to be a mutation in the protein core that might lead to loss of thermodynamic stability and unfolding [32].
The C-terminal WD40 domain, which forms a β-propeller-like structure, is found fused to the kinase domain and this feature is also observed in other members of the ROCO family. The WD40 domain has been associated with a diverse cellular function including cytoskeletal assembly and vesicle trafficking, but its exact function in LRRK2 is undetermined. A mutation in this domain (M2397T) of LRRK2 has been associated with Crohn's disease [33].
ROC & COR domains
The ROC and COR domains are fused to each other, a feature that is conserved throughout evolution and common to all members of the ROCO protein family. The ROC domain has a fold that resembles the Ras-related superfamily of small GTPases [29]. Three PD-associated mutations have been linked to residue R1441 in this domain (R1441C, R1441G and R1441H) [32]. The COR domain contains the mutations, Y1699C and R1628P. Recent crystallographic structural determinations of human LRRK2 ROC domain and bacterial ROC–COR domains suggest at least two different mechanisms for dimerization. Cookson and co-workers have reported that the ROC domain undergoes extensive domain swapping with the C-terminal portion of the domain exchanged between the two monomers (Figure 1B) [34], whereas Wittinghofer and co-workers have suggested that majority of the dimerization interactions come from the COR [35]. Evolutionarily, domain swapping in proteins occurs only in the termini, therefore it is highly unlikely that the observed domain swapping in the LRRK2 ROC domain crystal structure is physiologically relevant [36].
In a predicted model based on the Wittinghoffer structure, residue R1441 appears to be exposed on the surface and away from the residues responsible for GTPase activity [35]. It is likely that this residue is involved in protein–protein interactions with other domains of LRRK2 or with protein substrates. Substitution of arginine by small side-chain residues glycine or cystine may weaken protein–protein interactions if a charge pair is involved in the binding [32]. Since R1441C has been associated with both α-synuclein Lewy body and tau pathology [2], it is conceivable that LRRK2 may lie in the upstream part of PD pathogenesis pathway. Recent biochemical studies from our laboratory and others have shown that the ROC domain is indeed a functional GTPase [37,38].
The kinase domain
The kinase domain is attractive because kinases have been very amenable to therapeutic intervention. Some PD-linked LRRK2 mutations in the kinase domain are shown in Figure 1C. Based on sequence similarity, the MAPK domain of LRRK2 belongs to the tyrosine kinase-like subfamily of human protein kinases, whose members show sequence similarity to both serine/threonine and tyrosine kinases [39,40]. The MAPK domain of LRRK2 most resembles receptor-interacting protein kinases, which are crucial sensors of cellular stress and can activate MAPK pathways [41].
Molecular modeling studies on this domain have been carried out by ourselves and others. There is agreement that the most common PD-linked mutations G2019S and I2020T occur within the conserved region of the activation loop (ADYGIAQYCC) in the MAPK domain (Figure 2). A major conformational change common to all kinases in the tyrosine kinase-like family is the repositioning of the activation loop bearing a conserved DFG motif (DYG motif in LRRK2).
Figure 2. Structural details of the ATP-binding site of LRRK2.

The DYG regulatory motif is shown in the `DYG-in' state. D2017 participates in a hydrogen bond with Mg2+ ion and ATP molecule in the binding pocket. The CPK representations of the Parkinson's disease-associated mutations G2019S (orange) and I2020T (purple) are shown on the activation loop.
The exchange of Asp with Phe (Tyr in LRRK2) switches the kinase from an active form (DYG-in) to an inactive form (DYG-out) of the kinase (Figure 3). In LRRK2, D2017 and G2019 are part of the DYG motif. The modeled structure of the LRRK2 kinase domain (Figure 2) is constructed on the `DYG-in' active form. The figure shows ATP and Mg2+ ion docked into this structure. The LRRK2 kinase model shows all the expected subdomains of a Ser/Thr protein kinase [42]. The ATP binding cleft shows a glycine-rich loop (residues 1885–1982) facilitating backbone interactions with the β-phosphate of ATP [42]. D2017 makes a stabilizing interaction with ATP via Mg2+ ion. In addition, the catalytic loop shows H1998, K1996 and D1994 correctly positioned for catalysis in close proximity to the γ-phosphate of ATP. The LRRK2 kinase domain shows spatial conservation of residues in the regulatory spine (L1924, L1935, Y1992 and Y2018) as with other kinases in this subfamily [43]. The catalytic spine comprises V1893, L1955, L2001, L2062 and I2066. The adenine group of ATP fits the catalytic spine, positioning itself between V1893 and L2001. In addition, the model shows C2024 and C2025, which are both on the activation loop and exposed to solvent. The modeling undoubtedly reveals that the PD-linked mutation G2019S is part of the DYG-motif while I2020T lies adjacent to the critical motif that is responsible for regulation of the kinase activation/deactivation. The DYG conserved region in the activation loop tends to create a flexible conformation as a result of the presence of a small glycine residue in the enzyme. This allows the kinase to switch easily between active (DYG-in) and inactive (DYG-out) forms. Modeling suggests that the serine substitution in the G2019S mutant results in a backbone hydrogen-bond interaction with D1994 from the adjacent helix when in the active conformation. The G2019S mutant appears to be locked in a more active state, which leads to an increase in the kinase activity. We and others have consistently shown that the G2019S mutant increases the kinase activity by two- to three-fold compared with wild-type (wt) LRRK2 [42,44,45] without influencing ATP or LRRKtide binding. The enhanced kinase activity is correlated with increased cytotoxicity, suggesting a gain-of-function mechanism for this mutation [45,46]. In addition, molecular dynamic simulations carried out in our laboratory indicate that the threonine residue side chain in the I2020T mutation makes a hydrogen bond with the backbone of D2017, which is involved in binding Mg2+ and ATP. This would explain the sixfold higher binding affinity for ATP observed for this mutant [47].
Figure 3. Ribbon representation of the active and inactive forms of LRRK2.

D2017 and Y2018 act as bipositional switches that exchange positions as the enzyme switches between the active and inactive form (DYG-in: green; DYG-out: red). The mutations in the activation loop G2019S and I2020T are indicated by CPK representation (G2019S: orange; I2020T: pink).
Mechanisms of LRRK2 kinase regulation
Some PD-linked LRRK2 mutations have increased kinase activity, which correlates with increased neuronal cytotoxicity [48–53]. However, most of the LRRK2 mutations do not manifest their effects by simply increasing kinase activity. Even for the most common mutant G2019S, only two- to three-fold increases in kinase activity were observed in vitro [42,44]. It is difficult to reconcile these observations with the hypothesis that LRRK2 kinase plays a key role in the pathogenesis of PD. Therefore a more comprehensive understanding of LRRK2 mutations must take into account possible complex mechanisms that regulate LRRK2 kinase activity. The potential regulatory mechanisms include intramolecular regulation, post-translational modifications, dimerization and interaction with regulatory proteins.
Intramolecular regulation
The presence of both a ROC domain (GTPase domain) and a kinase domain in LRRK2 suggests that the two domains may be acting a manner analogous to that seen with small GTPases and associated kinases, exemplified by Ras and Raf [54,55]. This would suggest that the ROC domain of LRRK2 acts as a molecular switch for the kinase domain, cycling between GTP- and GDP-bound states. With GTP bound, the switch region on the outside of the domain is in its active state and leads to an increase in kinase activity. With GDP bound, the tertiary structure of the switch domain is in an inactive state and leads to decrease in kinase activity. Structural models have proposed that the COR domain folds back on itself allowing the two enzymatic domains to interact [53]. In addition, several lines of experimental evidence supporting this intramolecular interaction have been reported. The mutants R1441C, R1441G and Y1699C located in the ROC and COR domains reduce GTP hydrolysis or enhance GTP binding and lead to an increased kinase activity [37,53,56]. The T1348N mutant in the ROC domain disrupts GTP binding and leads to inactive kinase [57]. The combination of R1441C with the G2019S (a kinase domain mutant) increases the kinase activity by sevenfold compared with wt LRRK2 [58]. Binding of nonhydrolyzable analogues of GTP (e.g., GTPγS) results in an increased kinase activity of LRRK2 [51]. On the other hand, there is experimental evidence that does not support this intramolecular interaction. Downregulation of GTPase activity caused by R1441C and R1441G mutations has been observed with the kinase activity unaffected [56,59]. In addition, recently two independent groups reported that GTP or GDP binding has no effect on the kinase activity of LRRK2 when tested in vitro using recombinant LRRK2 protein [38,60]. One possibility for this observation as suggested by Taymans et al. is that LRRK2 GTPase regulates other targets rather than its own kinase domain. It is clear that the LRRK2 GTPase can function independently, whereas the kinase activity strictly requires the GTPase domain. It is very likely that LRRK2 GTPase has additional targets regardless of whether it regulates its own kinase domain. This possibility highlights the need to identify the potential downstream effector(s) of the LRRK2 GTPase domain. The other possibility is the lack of required GTPase-activating protein (GAP) or guanine exchange factor of LRRK2 when tested in an in vitro biochemical assay. Recently, ArfGAP1 was reported as a GAP of LRRK2 by two groups [9,10]. It is directly phosphorylated by LRRK2 and markedly enhances GTPase activity of LRRK2. Both groups reported that interaction of ArfGAP1 with LRRK2 was not changed by LRRK2 pathogenic mutations. However, the effect of ArfGAP1 on LRRK2 kinase activity is inconsistent – one group reported increased LRRK2 autophosphorylation and kinase activity in its presence, while the other group reported reduced LRRK2 kinase activity. Despite this controversy, it is clear that LRRK2 and ArfGAP1 interact in a reciprocal manner and GTP hydrolysis of LRRK2 plays a critical role in regulating its kinase domain in the presence of ArfGAP1.
Regulation by phosphorylation
Phosphopeptide analysis has revealed 17 autophosphorylation sites of LRRK2 mainly located in the GTPase domain [61], supporting the hypothesis that LRRK2 kinase modulates its GTPase function. In addition to the autophosphorylation sites, 20 constitutive phosphorylation sites in a region upstream of the LRR domain have also been identified. These findings suggest that the discrete phosphorylation states of LRRK2 play critical roles in regulating interactions with others proteins as well as its own enzymatic activities. This hypothesis is further supported by the recent discovery that LRRK2 interacts with proteins 14–3–3 [44,62]. The interaction is regulated by two constitutive phosphorylation states at Ser-910 and -935 of LRRK2. Reduced phosphorylation of Ser-910/-935 and the subsequent disrupted interaction with proteins 14–3–3 were observed for the mutant R1441C along with nine other PD-linked LRRK2 mutations [44]. But it was not observed for the most common mutant G2019S or other LRRK2 mutations, suggesting a mutant-specific regulating mechanism. A selective LRRK2 kinase inhibitor LRRK2-In-1 leads to dephosphorylation of Ser-910/-935, which in turn disrupts 14–3–3 binding [63]. This observation suggests a potential regulatory feedback mechanism – a downstream kinase whose activity is regulated by LRRK2 is required for phosphorylating Ser-910/-935. However, the kinase(s) responsible for phosphorylation of these critical sites was unknown at the time of writing this manuscript. The identification of this kinase(s) in the future will be critical for understanding LRRK2 function and revealing the signaling pathway of LRRK2.
Other mechanisms of regulation
Other mechanisms of LRRK2 regulation include dimerization and interaction with chaperon proteins. LRRK2 dimer is more active than monomer and is enriched in cell membranes [30]. It has also been reported that the chaperone protein Hsp90 forms a complex with LRRK2 and stabilizes LRRK2 [64,65]. Inhibition of Hsp90 leads to LRRK2 degradation in a proteasome-dependent manner by promoting Hsp70 expression. Clearly, the mechanisms of LRRK2 regulation are very complex. Elucidation of its regulating mechanisms in the future will provide potential therapeutic strategies for the treatment of LRRK2-associated PD.
LRRK2 kinase activity
Inhibitors
Since LRRK2 was linked to PD in 2004, development of LRRK2 inhibitors has become a focus in drug discovery for the treatment of PD. A review of LRRK2 inhibitors and their in vitro and in vivo inhibition has recently been published [66]. The common feature of these published inhibitors is the lack of brain–blood barrier (BBB) penetration, which limits their therapeutic application in PD. Recently, Zhang et al. reported TAE684 as a potent LRRK2 inhibitor with BBB penetration. But TEA684 shows no evidence of LRRK2 inhibition in brain despite high levels measured in a pharmaco-kinetic study [67]. In addition, the majority of LRRK2 inhibitors are ATP-competitive, which may have selectivity issues. Although a few inhibitors of LRRK2 show good selectivity, such as LRRK2-In-1 [63], they may display loss of potency in some LRRK2 mutations due to a dramatic difference on KATP compared with wt LRRK2. For example, I2012T has been reported to exhibit tenfold loss of potency to ATP competitive inhibitors than wt LRRK2 due to its small KATP [47]. The IC50 values of ATP competitive inhibitors at any given ATP concentration directly depends on the KATP of kinase targets as described by Cheng–Prusoff equation [68]:
Inhibitors with similar binding affinity to two targets are much weaker against the target with lower KATP. This effect of KATP on inhibitor potency requires different dosing strategy for different mutant carriers and results in personalized medicine.
One strategy to develop inhibitors that would be equally effective on wt and mutant LRRK2 is to develop DFG-out inhibitors or type-II inhibitors. DFG-out inhibitors target the inactive/DFG-out (DYG-out in LRRK2) kinase conformation. They exploit an allosteric site, which is immediately adjacent to the ATP binding pocket and is only available in the inactive/DFG-out conformation (Figure 3). Currently, there are over 30 known DFG-out inhibitors targeting a variety of kinases. We have reported the identification of an ATP noncompetitive inhibitor through high-throughput screening [69]. However, the compound is not structurally attractive. Through docking of known DFG-out inhibitors into a LRRK2 model, we have identified four DFG-out inhibitors as summarized in Table 1. Bosutinib and imatinib made hydrogen bonds mainly in the hinge region and very few interactions in the DYG-out pocket and were equally potent against wt LRRK2 and the G2019S mutant. On the other hand sorafenib, which made almost no interactions in the hinge region (Figure 4) was effective against wt LRRK2, but was poorly effective against the G2019S and I2020T mutants. Among all the four DFG-out inhibitors, the most promising inhibitor is ponatinib, which is potent toward wt LRRK2 and has low polar surface area (PSA) value, predicting likelihood for BBB penetration. Ponatinib was capable of making one hydrogen bond in the hinge region and a number of interactions in the DFG-out pocket of wt LRRK2. This made ponatinib exceptionally potent against wt LRRK2, but it also retained some potency against the mutants due to hinge interactions. As expected for a DFG-out inhibitor, ponatinib inhibits wt LRRK2 in an ATP noncompetitive manner. However, for the two mutations G2019S and I2020T that reside in the activation loop, ponatinib inhibits them in an ATP competitive mechanism. Similar inhibition patterns were also observed for imatinib and sorafenib as shown in Table 1. These results suggest that the conventional view of DFG-out inhibitors cannot be applied to the two mutants G2019S and I2020T. However, for LRRK2 mutations not located in the activation loop, the identification of ponatinib provides a new avenue for developing more selective and non-ATP competitive inhibitors.
Table 1.
IC50 values of DYG-out inhibitors of LRRK2.
| Compound | Dominant binding mode to LRRK2 | IC50, μM† |
||
|---|---|---|---|---|
| wt LRRK2 | G2019S | I2020T | ||
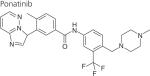
|
DFG-out and hinge | 0.031 ± 0.004‡ | 0.2 ± 0.03§ | 0.6 ± 0.07§ |
|
| ||||
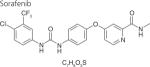
|
DFG-out | 0.7 ± 0.3‡ | 9.7 ± 3.2§ | >10¶ |
|
| ||||
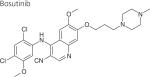
|
Hinge | 0.3 ± 0.02§ | 0.2 ± 0.02§ | 2.6 ± 0.8¶ |
|
| ||||
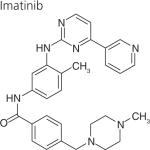
|
Hinge | 5.1 ± 1.4‡ | 5.6 ± 0.4§ | >10¶ |
IC50 was determined at 50 μM ATP.
ATP noncompetitive inhibiton.
ATP competitive inhibition.
Inhibition mechanism is not determined.
Figure 4. Homology model of LRRK2 bound to the DFG-out inhibitor sorafenib.

(A) Ribbon diagram with residues that interact with sorafenib in stick repesentation is shown in this figure. Sorafenib is shown to bind in the DYG-out conformation making interactions with both the ATP-binding hinge region of the kinase as well as the DYG-out allosteric pocket. (B) A ligand interaction diagram for the model of LRRK2 bound to sorafenib is shown here. All residues with 4.0A distance are highlighted. Hydrophobic residues are shown in green, red indicates negatively charged residues, blue indicates positively charged residues. The arrows indicate hydrogen bonds.
The fact that the G2019S mutant stabilizes the DYG-in active conformation requires a new strategy to successfully inhibit it if noncompetitive ATP inhibitors are sought. It is conceivable that a molecule that can force a DYG-flip to an inactive conformation of the mutant would end up `locking' the mutant enzymes in a permanent inactive form.
Biochemical & cell-based kinase assays
With the lack of known LRRK2 substrates, kinase assays monitoring LRRK2 kinase activity have relied on autophosphorylation or phosphorylation of nonphysiological substrates, such as LRRKtide (a peptide derived from moesin with a sequence of RLGRDKYKTLRQIRQ), PLK-peptide (a PLK-derived peptide with a motif of RRRSLLE), nictide (a peptide of RLGWWRFYTLRRARQGNTKQR), myelin basic protein (MBP) and moesin [7,69–71]. Among them, LRRKtide and MBP are the two commonly used generic substrates of LRRK2 for inhibitor characterization. Peptide substrates have often been chosen as phosphoryl acceptors, motivated by ease of assay design or by the lack of unknown physiological substrate. Some kinases, such as cdk5, behave kinetically the same toward its natural substrate tau and peptide substrate and respond to inhibitors in the same manner [72]. However, in some cases, peptide substrates may not truly represent the natural protein substrates. For example, we recently observed thar b-Raf, a structurally related kinase of LRRK2, can also phosphorylate LRRKtide. The kinetics between natural substrate MEK1 and LRRKtide phosphorylation are quite different: KATP for LRRKtide phosphorylation is 600-fold higher than that of MEK1 phosphorylation (180 μM in LRRKtide phosphorylation versus 0.3 μM in MEK1 phosphorylation). Even more surprising, the known potent b-Raf inhibitor GDC-0879 with a reported IC50 of 0.1 nM [73] does not inhibit b-Raf at concentration up to 100 μM when LRRKtide is the substrate. We have constructed a model of the kinase domain of LRRK2 using the crystal structure of b-Raf and suggested that b-Raf and LRRK2 are structurally related [42]. The structural similarity between b-Raf and LRRK2 raises the likelihood that LRRK2 might behave differently toward its real substrate and peptide substrate. It raises the concern whether inhibitors of LRRK2 currently being assessed using peptide substrate truly represent the efficacy with physiological substrates. At present, the cell-based assays for LRRK2 are very limited due to its unidentified natural substrate(s). Currently, cellular activity is measured by LRRK2 aggregation, interaction with 14–3–3 and phosphorylation at Ser-910/-935 [63,67,74]. So far, the inhibitors show dose-dependent inhibition of Ser-910/-935 phosphorylation and disruption of 14–3–3 binding in cells consistent with their biochemical activity. These results have eased some of our concerns, but still it is worth noting that LRRK2 and its inhibitors should be recharacterized using its natural substrate(s) when it is available.
The choice of peptide as substrate for compound screening or inhibitor characterization should be judged based on a thorough kinetic study.
LRRK2, α-synuclein & tau: three risk factors of PD
As with most neurodegenerative diseases, PD is characterized by the formation of proteinaceous inclusions. Most PD patients with LRRK2 mutations have α-synuclein-positive Lewy bodies [2]. LRRK2 mutant carriers without Lewy bodies show tau-positive lesions [2]. In certain cases, TAR DNA-binding protein 43 (TDP43) inclusions were also observed [75]. The association of LRRK2, α-synuclein and tau with PD was consistent with recent genome-wide association studies that identified the genes coding for α-synuclein, LRRK2 and tau as increased risk factors of PD [76,77]. The mechanism that LRRK2 contributes to α-synuclein and tau aggregation is controversial. There has been evidence that LRRK2 directly phosphorylates α-synuclein at Ser-129 [78]. However, the slow kinetics of α-synuclein phosphorylation by LRRK2 makes it unlikely that LRRK2 contributes α-synuclein via direct interaction as the enzyme and the substrate [28,79]. Phosphorylation of tubulin-associated tau by LRRK2 at Thr181 was also observed [80]. But the question remains unaddressed of how good LRRK2 is compared with cdk5, a kinase which has been widely believed to phosphorylate tau in vivo. It is likely that LRRK2 contributes to α-synuclein and tau aggregation mainly through an indirect mechanism. What is unclear at present is where the functional links exist among these three proteins?
Since the first report of neurite shortening associated with autophagy abnormalities in differentiated SH-SY5Y cells expressing the G2019S mutant [81], more and more evidence from animal model and cells generated from PD patients suggests a role of LRRK2 in autophagy. Transgenic mice expressing G2019S mutation show autophagic and mitochondrial abnormalities in the brain [82]. In a LRRK2-knockout mouse model, the loss of LRRK2 impairs the autophagy pathway in kidney in an age-dependent manner: the autophagic activity increases at a young age and decreases with aging [19]. Inducing autophagy in Caenorhabditis elegans coexpressing LRRK2 mutants and V337M tau increases neuronal function to the same level as nontransgenic nematodes [83]. Recently, over long-term culture of induced pluripotent stem (iPS) cells generated from PD patients carrying G2019S mutant show accumulated autophagic vacuoles, which were not present in control iPS cells [84]. In addition to the implication of LRRK2 in autophagy, there is evidence that the process of α-synuclein and tau degradation is autophagy dependent. wt α-synuclein can be efficiently degraded by chaperone-mediated autophagy, whereas degradation of pathogenic α-synuclein mutant was impaired by autophagy [85]. Inhibition of autophagy by compound 3-methylamphetamine or by inhibiting PLD1 activity results in tau aggregation [86,87], while tau-overexpressing Drosophila treated with rapamycin, an autophagy inducer, led to reduced tau aggregation and tau-induced neurotoxicity [88]. Collectively, it becomes more evident that LRRK2 mutations misregulate autophagy and lead to α-synuclein and tau aggregation and cell death. The future direction for identifying LRRK2 physiological substrates should be focused on autophagy related genes.
In summary, recent findings in LRRK2 research have provided a wealth of approaches facilitating LRRK2 inhibitor design, its substrate identification, and understanding upstream and downstream players in LRRK2 biology.
Future perspective
The critical role of LRRK2 in PD pathogenesis and the increasing understanding of its various functions will ensure that LRRK2 remains as attractive opportunity for therapeutic intervention in the next decade. The focus will remain in elucidating its function and identifying its physiological substrates by cell culture, animal models and tool compounds, obtaining x-ray crystallographic structure, and developing selective and BBB penetrating compounds. How rapid all these goals can achieve is difficult to assess, but will depend on the efficient strategies developed by biologists and chemists.
It will not be long before the x-ray structure of LRRK2 kinase domain is determined. However, given the fact that LRRK2 has several protein-interaction domains, one should expect that the substrate-binding site of LRRK2 would exhibit a high degree of conformational flexibility in order to accommodate different substrates. While the x-ray structure would intrinsically be of high value for structure-based drug design, the true potential of the crystal structure will be exploited by combining it with molecular dynamic simulations to understand the subtle difference in LRRK2 active site conformations resulting from various substrates. Considering the involvement of LRRK2 in multiple inflammatory diseases, including Crohn's disease, in various organs, such studies would help develop more organ selective and targeted therapeutics.
Given the intensive interest and effort put into drug discovery, targeting LRRK2 kinase activity, more research and development are urgently required to develop functional cell-based assays in order to evaluate inhibitors of LRRK2 in more reliable assays. The design of the assay should be focused on the known LRRK2 functions, such as mitochondrial abnormalities and autophagic dysfunction. Given the difficulty of designing selective and potent kinase inhibitors, especially for the two mutants G2019S and I2020T, where DFG-out inhibitors do not have any advantage over type-I inhibitors, the focus of drug discovery should be broadened to compounds that inhibit LRRK2 through blocking its interaction with other proteins or interfering the GTPase function. The availability of above-mentioned cell-based assays will not only facilitate the identification of such compounds, but also be of great help in compound evaluation in cells from PD patients with different LRRK2 mutations. LRRK2 are expressed in different organs in the body, such as brain, heart, liver, kidney and spleen. The specific functions of each organ make it very likely that the physiological substrate(s) of LRRK2 may vary in different organs, especially brain versus other organs. The best patient cells for accurate inhibitor evaluation will be the iPS cells, from which dopaminergic neurons will be derived over long-term culture.
The association of LRRK2 and tau suggests that LRRK2 might play a role in the pathogenesis of Alzheimer's disease (AD) in addition to its role in PD. AD animal models will be good tools in the future to elucidate the implication of LRRK2 in AD and to understand the linkage between these two most common neurodegenerative diseases.
Key Terms.
LRRK2: Leucine-rich repeat kinase 2 is a risk factor of Parkinson's disease.
Parkinson's disease: Disease characterized by tremor, rigidity, bradykinesia and postural instability. It is the second most common neurodegenerative disorder after Alzheimer's disease.
Kinase inhibitor: Type of enzyme inhibitor that specifically inhibits the activity of protein kinases.
Executive summary
-
■
Considering the complexity of leucine-rich repeat kinase 2 (LRRK2) domain structures, the focus of drug design for LRRK2 should be broadened to compounds that block LRRK2 interaction with other proteins or interfere with its GTPase activity.
-
■
Given the fact that the G2019S and I2020T mutants are located in the activation loop and regulate the activation state of LRRK2, designing DFG-out inhibitors to obtain selectivity is only a good strategy for mutations not involved in the activation loop.
-
■
Peptide substrates have often been chosen as phosphoryl acceptors in drug discovery, a choice motivated by ease of assay design, data analysis, or the lack of unknown physiological substrate. The choice of peptide as substrate for compound screening or inhibitor characterization should be judged based on a thorough kinetic study.
Acknowledgements
The authors thank M Glicksman for valuable comments.
This work is sponsored by NIH R21 grant NS072519.
No writing assistance was utilized in the production of this manuscript.
Footnotes
Financial & competing interests disclosure The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
Papers of special note have been highlighted as:
■ of interest
- 1.Paisan-Ruiz C, Jain S, Evans EW, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 2.Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 3.Berg D, Schweitzer KJ, Leitner P, et al. Type and frequency of mutations in the LRRK2 gene in familial and sporadic Parkinson's disease. Brain. 2005;128:3000–3011. doi: 10.1093/brain/awh666. [DOI] [PubMed] [Google Scholar]
- 4.Farrer M, Stone J, Mata IF, et al. LRRK2 mutations in Parkinson disease. Neurology. 2005;65:738–740. doi: 10.1212/01.wnl.0000169023.51764.b0. [DOI] [PubMed] [Google Scholar]
- 5.Khan NL, Jain S, Lynch JM, et al. Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson's disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain. 2005;128:2786–2796. doi: 10.1093/brain/awh667. [DOI] [PubMed] [Google Scholar]
- 6.Higashi S, Moore DJ, Colebrooke RE, et al. Expression and localization of Parkinson's disease-associated leucine-rich repeat kinase 2 in the mouse brain. J. Neurochem. 2007;100:368–381. doi: 10.1111/j.1471-4159.2006.04246.x. [DOI] [PubMed] [Google Scholar]
- 7.Anand VS, Reichling LJ, Lipinski K, et al. Investigation of leucine-rich repeat kinase 2: enzymological properties and novel assays. FEBS J. 2009;276:466–478. doi: 10.1111/j.1742-4658.2008.06789.x. [DOI] [PubMed] [Google Scholar]
- 8.Miklossy J, Arai T, Guo JP, et al. LRRK2 expression in normal and pathologic human brain and in human cell lines. J. Neuropathol. Exp. Neurol. 2006;65:953–963. doi: 10.1097/01.jnen.0000235121.98052.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stafa K, Trancikova A, Webber PJ, et al. GTPase activity and neuronal toxicity of Parkinson's disease-associated LRRK2 is regulated by ArfGAP1. PLoS Genet. 2012;8:e1002526. doi: 10.1371/journal.pgen.1002526. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■ Reports the identification of the first GTPase-activating protein of LRRK2. The results elucidate the functional link between the GTPase domain and the kinase domain of leucine-rich repeat kinase 2 (LRRK2). They provide strong support for the intramolecular regulating mechanims of LRRK2, which has been a long-term debate.
- 10.Xiong Y, Yuan C, Chen R, Dawson TM, Dawson VL. ArfGAP1 is a GTPase activating protein for LRRK2: reciprocal regulation of ArfGAP1 by LRRK2. J. Neurosci. 2012;32:3877–3886. doi: 10.1523/JNEUROSCI.4566-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■ Reports the identification of the first GTPase-activating protein of LRRK2. The results elucidate the functional link between the GTPase domain and the kinase domain of LRRK2. They provide strong support for the intramolecular regaluting mechanims of LRRK2, which has been a long-term debate.
- 11.Parisiadou L, Xie C, Cho HJ, et al. Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J. Neurosci. 2009;29:13971–13980. doi: 10.1523/JNEUROSCI.3799-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gillardon F. Leucine-rich repeat kinase 2 phosphorylates brain tubulin-beta isoforms and modulates microtubule stability – a point of convergence in parkinsonian neurodegeneration? J. Neurochem. 2009;110:1514–1522. doi: 10.1111/j.1471-4159.2009.06235.x. [DOI] [PubMed] [Google Scholar]
- 13.Imai Y, Gehrke S, Wang HQ, et al. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 2008;27:2432–2443. doi: 10.1038/emboj.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gloeckner CJ, Schumacher A, Boldt K, Ueffing M. The Parkinson disease-associated protein kinase LRRK2 exhibits MAPKKK activity and phosphorylates MKK3/6 and MKK4/7, in vitro. J. Neurochem. 2009;109:959–968. doi: 10.1111/j.1471-4159.2009.06024.x. [DOI] [PubMed] [Google Scholar]
- 15.Hsu CH, Chan D, Greggio E, et al. MKK6 binds and regulates expression of Parkinson's disease-related protein LRRK2. J. Neurochem. 2010;112:1593–1604. doi: 10.1111/j.1471-4159.2010.06568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qing H, Wong W, McGeer EG, McGeer PL. Lrrk2 phosphorylates alpha synuclein at serine 129: Parkinson disease implications. Biochem. Biophys. Res. Commun. 2009;387:149–152. doi: 10.1016/j.bbrc.2009.06.142. [DOI] [PubMed] [Google Scholar]
- 17.Angeles DC, Gan BH, Onstead L, et al. Mutations in LRRK2 increase phosphorylation of peroxiredoxin 3 exacerbating oxidative stress-induced neuronal death. Hum. Mutat. 2011;32:1390–1397. doi: 10.1002/humu.21582. [DOI] [PubMed] [Google Scholar]
- 18.Ohta E, Kawakami F, Kubo M, Obata F. LRRK2 directly phosphorylates Akt1 as a possible physiological substrate: impairment of the kinase activity by Parkinson's disease-associated mutations. FEBS Lett. 2011;585:2165–2170. doi: 10.1016/j.febslet.2011.05.044. [DOI] [PubMed] [Google Scholar]
- 19.Tong Y, Giaime E, Yamaguchi H, et al. Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. Mol. Neurodegener. 2012;7:2. doi: 10.1186/1750-1326-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanchez-Danes A, Richaud-Patin Y, Carballo-Carbajal I, et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson's disease. EMBO Mol. Med. 2012;4:380–395. doi: 10.1002/emmm.201200215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferree A, Guillily M, Li H, et al. Regulation of physiologic actions of LRRK2: focus on autophagy. Neurodegener. Dis. 2012;10:238–241. doi: 10.1159/000332599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim B, Yang MS, Choi D, et al. Impaired inflammatory responses in murine Lrrk2-knockdown brain microglia. PLoS ONE. 2012;7:e34693. doi: 10.1371/journal.pone.0034693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gillardon F, Schmid R, Draheim H. Parkinson's disease-linked leucine-rich repeat kinase 2(R1441G) mutation increases proinflammatory cytokine release from activated primary microglial cells and resultant neurotoxicity. Neuroscience. 2012;208:41–48. doi: 10.1016/j.neuroscience.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 24.Botta-Orfila T, Sanchez-Pla A, Fernandez M, et al. Brain transcriptomic profiling in idiopathic and LRRK2-associated Parkinson's disease. Brain Res. 2012;1466:152–157. doi: 10.1016/j.brainres.2012.05.036. [DOI] [PubMed] [Google Scholar]
- 25.Moehle MS, Webber PJ, Tse T, et al. LRRK2 inhibition attenuates microglial inflammatory responses. J. Neurosci. 2012;32:1602–1611. doi: 10.1523/JNEUROSCI.5601-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seol W. Biochemical and molecular features of LRRK2 and its pathophysiological roles in Parkinson's disease. BMB Rep. 2010;43:233–244. doi: 10.5483/bmbrep.2010.43.4.233. [DOI] [PubMed] [Google Scholar]
- 27.Yue Z. Genetic mouse models for understanding LRRK2 biology, pathology and pre-clinical application. Parkinsonism Relat. Disord. 2012;18(Suppl. 1):S180–S182. doi: 10.1016/S1353-8020(11)70056-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cookson MR. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson's disease. Nat. Rev. Neurosci. 2010;11:791–797. doi: 10.1038/nrn2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marin I, van Egmond WN, van Haastert PJ. The Roco protein family: a functional perspective. FASEB J. 2008;22:3103–3110. doi: 10.1096/fj.08-111310. [DOI] [PubMed] [Google Scholar]
- 30.Berger Z, Smith KA, Lavoie MJ. Membrane localization of LRRK2 is associated with increased formation of the highly active LRRK2 dimer and changes in its phosphorylation. Biochemistry. 2010;49:5511–5523. doi: 10.1021/bi100157u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Vet E, Gebhardt WA, Sinnige J, et al. Implementation intentions for buying, carrying, discussing and using condoms: the role of the quality of plans. Health Educ. Res. 2011;26:443–455. doi: 10.1093/her/cyr006. [DOI] [PubMed] [Google Scholar]
- 32.Mata IF, Wedemeyer WJ, Farrer MJ, Taylor JP, Gallo KA. LRRK2 in Parkinson's disease: protein domains and functional insights. Trends Neurosci. 2006;29:286–293. doi: 10.1016/j.tins.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 33.Lewis PA, Manzoni C. LRRK2 and human disease: a complicated question or a question of complexes? Sci. Signal. 2012;5(207):pe2. doi: 10.1126/scisignal.2002680. [DOI] [PubMed] [Google Scholar]
- 34.Greggio E, Zambrano I, Kaganovich A, et al. The Parkinson disease-associated leucine-rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. J. Biol. Chem. 2008;283:16906–16914. doi: 10.1074/jbc.M708718200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gotthardt K, Weyand M, Kortholt A, Van Haastert PJ, Wittinghofer A. Structure of the Roc-COR domain tandem of C. tepidum, a prokaryotic homologue of the human LRRK2 Parkinson kinase. EMBO J. 2008;27:2239–2249. doi: 10.1038/emboj.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bennett MJ, Schlunegger MP, Eisenberg D. 3D domain swapping: a mechanism for oligomer assembly. Protein Sci. 1995;4:2455–2468. doi: 10.1002/pro.5560041202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo L, Gandhi PN, Wang W, et al. The Parkinson's disease-associated protein, leucine-rich repeat kinase 2 (LRRK2), is an authentic GTPase that stimulates kinase activity. Exp. Cell Res. 2007;313:3658–3670. doi: 10.1016/j.yexcr.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu M, Dobson B, Glicksman MA, Yue Z, Stein RL. Kinetic mechanistic studies of wild-type leucine-rich repeat kinase 2: characterization of the kinase and GTPase activities. Biochemistry. 2010;49:2008–2017. doi: 10.1021/bi901851y. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■ Reported for the first time that GTP binding has no effect on the kinase activity of LRRK2 in the biochemical assay.
- 39.Braconi Quintaje S, Orchard S. The annotation of both human and mouse kinomes in UniProtKB/Swiss-Prot: one small step in manual annotation, one giant leap for full comprehension of genomes. Mol. Cell. Proteomics. 2008;7:1409–1419. doi: 10.1074/mcp.R700001-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manning G, Plowman GD, Hunter T, Sudarsanam S. Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci. 2002;27:514–520. doi: 10.1016/s0968-0004(02)02179-5. [DOI] [PubMed] [Google Scholar]
- 41.Meylan E, Tschopp J. The RIP kinases: crucial integrators of cellular stress. Trends Biochem. Sci. 2005;30:151–159. doi: 10.1016/j.tibs.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 42.Liu M, Kang S, Ray S, et al. Kinetic, mechanistic, and structural modeling studies of truncated wild-type leucine-rich repeat kinase 2 and the G2019S mutant. Biochemistry. 2011;50:9399–9408. doi: 10.1021/bi201173d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taylor SS, Kornev AP. Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem. Sci. 2011;36:65–77. doi: 10.1016/j.tibs.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nichols RJ, Dzamko N, Morrice NA, et al. 14–13–3 binding to LRRK2 is disrupted by multiple Parkinson's disease-associated mutations and regulates cytoplasmic localization. Biochem. J. 2010;430:393–404. doi: 10.1042/BJ20100483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.West AB, Moore DJ, Biskup S, et al. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl Acad. Sci. USA. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winner B, Melrose HL, Zhao C, et al. Adult neurogenesis and neurite outgrowth are impaired in LRRK2 G2019S mice. Neurobiol. Dis. 2011;41:706–716. doi: 10.1016/j.nbd.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reichling LJ, Riddle SM. Leucine-rich repeat kinase 2 mutants I2020T and G2019S exhibit altered kinase inhibitor sensitivity. Biochem. Biophys. Res. Commun. 2009;384:255–258. doi: 10.1016/j.bbrc.2009.04.098. [DOI] [PubMed] [Google Scholar]
- 48.Gloeckner CJ, Kinkl N, Schumacher A, et al. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum. Mol. Genet. 2006;15:223–232. doi: 10.1093/hmg/ddi439. [DOI] [PubMed] [Google Scholar]
- 49.Greggio E, Jain S, Kingsbury A, et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 50.Luzon-Toro B, Rubio de la Torre E, Delgado A, Perez-Tur J, Hilfiker S. Mechanistic insight into the dominant mode of the Parkinson's disease-associated G2019S LRRK2 mutation. Hum. Mol. Genet. 2007;16:2031–2039. doi: 10.1093/hmg/ddm151. [DOI] [PubMed] [Google Scholar]
- 51.Smith WW, Pei Z, Jiang H, et al. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat. Neurosci. 2006;9:1231–1233. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- 52.Smith WW, Pei Z, Jiang H, et al. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc. Natl Acad. Sci. USA. 2005;102:18676–18681. doi: 10.1073/pnas.0508052102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.West AB, Moore DJ, Choi C, et al. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum. Mol. Genet. 2007;16:223–232. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]
- 54.Avruch J, Zhang XF, Kyriakis JM. Raf meets Ras: completing the framework of a signal transduction pathway. Trends Biochem. Sci. 1994;19:279–283. doi: 10.1016/0968-0004(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 55.Kolch W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem. J. 2000;351(Pt 2):289–305. [PMC free article] [PubMed] [Google Scholar]
- 56.Lewis PA, Greggio E, Beilina A, et al. The R1441C mutation of LRRK2 disrupts GTP hydrolysis. Biochem. Biophys. Res. Commun. 2007;357:668–671. doi: 10.1016/j.bbrc.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ito G, Okai T, Fujino G, et al. GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial Parkinson's disease. Biochemistry. 2007;46:1380–1388. doi: 10.1021/bi061960m. [DOI] [PubMed] [Google Scholar]
- 58.Webber PJ, Smith AD, Sen S, et al. Autophosphorylation in the leucine-rich repeat kinase 2 (LRRK2) GTPase domain modifies kinase and GTP-binding activities. J. Mol. Biol. 2011;412:94–110. doi: 10.1016/j.jmb.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li X, Tan YC, Poulose S, et al. Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson's disease R1441C/G mutants. J. Neurochem. 2007;103:238–247. doi: 10.1111/j.1471-4159.2007.04743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taymans JM, Vancraenenbroeck R, Ollikainen P, et al. LRRK2 kinase activity is dependent on LRRK2 GTP binding capacity but independent of LRRK2 GTP binding. PLoS ONE. 2011;6:e23207. doi: 10.1371/journal.pone.0023207. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■ Reproduced the same observation as [38] and highlighted the possibility that LRRK2 GTPase has additional targets other than its own kinase domain.
- 61.Gloeckner CJ, Boldt K, von Zweydorf F, et al. Phosphopeptide analysis reveals two discrete clusters of phosphorylation in the N-terminus and the Roc domain of the Parkinson-disease associated protein kinase LRRK2. J. Proteome Res. 2010;9:1738–1745. doi: 10.1021/pr9008578. [DOI] [PubMed] [Google Scholar]
- 62.Li X, Wang QJ, Pan N, et al. Phosphorylation-dependent 14–13–3 binding to LRRK2 is impaired by common mutations of familial Parkinson's disease. PLoS ONE. 2011;6:e17153. doi: 10.1371/journal.pone.0017153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Deng X, Dzamko N, Prescott A, et al. Characterization of a selective inhibitor of the Parkinson's disease kinase LRRK2. Nat. Chem. Biol. 2011;7:203–205. doi: 10.1038/nchembio.538. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■ Reported the most selective and potent LRRK2 inhibitor so far, LRRK2-In-1. The inhibitor is currently being widely used as a tool compound in order to elucidate LRRK2 function.
- 64.Wang L, Xie C, Greggio E, et al. The chaperone activity of heat shock protein 90 is critical for maintaining the stability of leucine-rich repeat kinase 2. J. Neurosci. 2008;28:3384–3391. doi: 10.1523/JNEUROSCI.0185-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ko HS, Bailey R, Smith WW, et al. CHIP regulates leucine-rich repeat kinase-2 ubiquitination, degradation, and toxicity. Proc. Natl Acad. Sci. USA. 2009;106:2897–2902. doi: 10.1073/pnas.0810123106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kramer T, Lo Monte F, Amombo GMO, Schmidt B. Small molecule kinase inhibitors for LRRK2 and their application to Parkinson's disease models. ACS Chem. Neurosci. 2012;3:151–160. doi: 10.1021/cn200117j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang J, Deng X, Choi HG, Alessi DR, Gray NS. Characterization of TAE684 as a potent LRRK2 kinase inhibitor. Bioorg. Med. Chem. Lett. 2012;22:1864–1869. doi: 10.1016/j.bmcl.2012.01.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 69.Lovitt B, Vanderporten EC, Sheng Z, et al. Differential effects of divalent manganese and magnesium on the kinase activity of leucine-rich repeat kinase 2 (LRRK2) Biochemistry. 2010;49:3092–3100. doi: 10.1021/bi901726c. [DOI] [PubMed] [Google Scholar]
- 70.Pedro L, Padros J, Beaudet L, et al. Development of a high-throughput AlphaScreen assay measuring full-length LRRK2(G2019S) kinase activity using moesin protein substrate. Anal. Biochem. 2010;404:45–51. doi: 10.1016/j.ab.2010.04.028. [DOI] [PubMed] [Google Scholar]
- 71.Covy JP, Giasson BI. Identification of compounds that inhibit the kinase activity of leucine-rich repeat kinase 2. Biochem. Biophys. Res. Commun. 2009;378:473–477. doi: 10.1016/j.bbrc.2008.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu M, Choi S, Cuny GD, et al. Kinetic studies of Cdk5/p25 kinase: phosphorylation of tau and complex inhibition by two prototype inhibitors. Biochemistry. 2008;47:8367–8377. doi: 10.1021/bi800732v. [DOI] [PubMed] [Google Scholar]
- 73.Wong H, Belvin M, Herter S, et al. Pharmacodynamics of 2-[4-[(1E)-1-(hydroxyimino)-2,3-dihydro-1H-inden-5-yl]-3-(pyridine-4-yl)-1H-pyraz ol-1-yl]ethan-1-ol (GDC-0879), a potent and selective B-Raf kinase inhibitor: understanding relationships between systemic concentrations, phosphorylated mitogen-activated protein kinase kinase 1 inhibition, and efficacy. J. Pharmacol. Exp. Ther. 2009;329:360–367. doi: 10.1124/jpet.108.148189. [DOI] [PubMed] [Google Scholar]
- 74.Dzamko N, Deak M, Hentati F, et al. Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser(910)/Ser(935), disruption of 14–13–3 binding and altered cytoplasmic localization. Biochem. J. 2010;430:405–413. doi: 10.1042/BJ20100784. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■ Along with [44], reports that the interaction between 14–13–3 proteins with LRRK2 is regulated by the phosphorylation state of Ser-910/-935, two constitutive phosphorylation sites of LRRK2. They support a regulatory feedback mechanism – a downstream kinase of LRRK2 is responsible for phosphorylating Ser-910/-935. The discovery has become the basis for the curently widely used cell-based assay to evaluate LRRK2 inhibitors in a cellular context of patients with LRRK2 mutations.
- 75.Covy JP, Yuan W, Waxman EA, et al. Clinical and pathological characteristics of patients with leucine-rich repeat kinase-2 mutations. Mov. Disord. 2009;24:32–39. doi: 10.1002/mds.22096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Satake W, Nakabayashi Y, Mizuta I, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat. Genet. 2009;41:1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]; ■ Identified tau, α-synuclein and LRRK2 as the risk factors of LRRK2. Provides strong evidence of the association between tau and LRRK2 and has significant impact on our understanding of Alzheimer's disease and Parkinson's disease.
- 77.Simon-Sanchez J, Schulte C, Bras JM, et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat. Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■ Identified tau, α-synuclein and LRRK2 as the risk factors of LRRK2. Provides strong evidence of the association between tau and LRRK2 and has significant impact on our understanding of Alzheimer's disease and Parkinson's disease.
- 78.Qing H, Zhang Y, Deng Y, McGeer EG, McGeer PL. LRRK2 interaction with alpha-synuclein in diffuse Lewy body disease. Biochem. Biophys. Res. Commun. 2009;390:1229–1234. doi: 10.1016/j.bbrc.2009.10.126. [DOI] [PubMed] [Google Scholar]
- 79.Devine MJ, Lewis PA. Emerging pathways in genetic Parkinson's disease: tangles, Lewy bodies and LRRK2. FEBS J. 2008;275:5748–5757. doi: 10.1111/j.1742-4658.2008.06707.x. [DOI] [PubMed] [Google Scholar]
- 80.Kawakami F, Yabata T, Ohta E, et al. LRRK2 phosphorylates tubulin-associated tau but not the free molecule: LRRK2-mediated regulation of the tau-tubulin association and neurite outgrowth. PLoS ONE. 2012;7:e30834. doi: 10.1371/journal.pone.0030834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Plowey ED, Cherra SJ, 3rd, Liu YJ, Chu CT. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J. Neurochem. 2008;105:1048–1056. doi: 10.1111/j.1471-4159.2008.05217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ramonet D, Daher JP, Lin BM, et al. Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS ONE. 2011;6:e18568. doi: 10.1371/journal.pone.0018568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ferree A, Guillily M, Li H, et al. Regulation of physiologic actions of LRRK2: focus on autophagy. Neurodegener. Dis. 2011;10:238–241. doi: 10.1159/000332599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sanchez-Danes A, Richaud-Patin Y, Carballo-Carbajal I, et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson's disease. EMBO Mol. Med. 2012;4:380–395. doi: 10.1002/emmm.201200215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 86.Wang Y, Martinez-Vicente M, Kruger U, et al. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum. Mol. Genet. 2009;18:4153–4170. doi: 10.1093/hmg/ddp367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dall'Armi C, Hurtado-Lorenzo A, Tian H, et al. The phospholipase D1 pathway modulates macroautophagy. Nat. Commun. 2010;1:142. doi: 10.1038/ncomms1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Berger Z, Ravikumar B, Menzies FM, et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 2006;15:433–442. doi: 10.1093/hmg/ddi458. [DOI] [PubMed] [Google Scholar]