

Abstract
JC polyomavirus (JCV) is an important human pathogen that causes the fatal demyelinating disease progressive multifocal leukoencephalopathy (PML). In this study we further delineate the early events of JCV entry in human glial cells and demonstrate that a pentameric subunit of the viral capsid is able to recapitulate early events in viral trafficking. We show that JCV traffics to the endoplasmic reticulum (ER) by 6 h post infection, and that VP1 pentamers arrive at the ER with similar kinetics. Further, this JCV localization to the ER is critical for infection, as treatment of cells with agents that prevent ER trafficking, ER function, or ER quality control reduce JCV infectivity. These pentamers represent a new tool to study polyomavirus entry, and will be particularly useful in studying recently identified polyomaviruses that are difficult to propagate.
Keywords: JC polyomavirus, Endoplasmic reticulum, Trafficking, Endocytosis
Introduction
JC polyomavirus (JCV) is the causative agent of the fatal demyelinating disease progressive multifocal leukoencephalopathy (PML). Infection likely occurs in early to late childhood, with 50–80% of the population seropositive by early adulthood (Egli et al., 2009; Kean et al., 2009; Knowles et al., 2003). JCV infects cells of the kidney, bone marrow and lymphoid tissue (reviewed in (Monaco et al., 1996; Randhawa et al., 2005; Tan et al., 2009;Tan and Koralnik, 2010) resulting in a persistent, lifelong infection. In most healthy individuals, viral replication remains low, resulting in an asymptomatic infection. In immunosuppressed individuals however, especially HIV/AIDS patients, a dramatic increase in viral replication occurs, resulting in lytic infection of oligodendrocytes that is uniformly fatal (Tan and Koralnik, 2010). Importantly, patients undergoing immunomodulatory therapies have also been shown to develop PML as a result of reduced immune function (reviewed in Major, 2010).
JCV is a nonenveloped virus containing a double stranded DNA genome. The viral capsid is approximately 40 nm in diameter and is composed of three structural proteins; VP1, VP2 and VP3. Five VP1 molecules oligomerize to form the viral pentamer, and 72 of these pentamers are linked through elongated C-terminal extensions to form the viral capsid. VP2 and VP3 insert into the axial cavity beneath VP1 and are sequestered from the exterior of the virion (Chen et al., 1998). A truncated form of VP1 that lacks the C-terminal residues can be expressed in bacteria, and produces VP1 pentamers that are unable to assemble into capsids (Neu et al., 2010). These pentamers likely bind cell surface receptors with similar affinity to the intact virions, but bind cells with less avidity due to the lack of valency of the intact capsid (Ewers and Helenius, 2011; Neu et al., 2010; Neu et al., 2008).
JCV binds to cells using the receptor moiety lactoseries tetrasaccaride series c (LSTc) and the serotonin receptor 5HT2A (Elphick et al., 2004; Neu et al., 2010). LSTc contains an α2,6-linked sialic acid, resulting in an “L” shaped conformation. Binding to LSTc is highly specific, as JCV does not bind the closely related carbohydrates LSTb and sialylparagloboside. Unlike most polyomaviruses that bind gangliosides and are endocytosed by non-clathrin dependent mechanisms, JCV is first endocytosed by clathrin-mediated endocytosis before entering the classical endocytic pathway (Pho et al., 2000)(reviewed in Tsai and Qian, 2010). Accordingly, treatment of cells with the clathrin mediated endocytosis inhibitor chlorpromazine but not the caveolae dependent endocytosis inhibitors nystatin or phorbol 12-myristate 13-acetate reduces JCV infection, and cells expressing dominant-negative constructs of the clathrin scaffolding adapter protein EPS15 will reduce JCV infectivity (Pho et al., 2000; Querbes et al., 2004). After endocytosis, JCV enters early endosomes and colocalizes with the early endosomal marker Rab5 (Querbes et al., 2004). Cells expressing dominant negative inhibitors of Rab5 or treated with drugs that prevent endosomal acidification prevents infection (Ashok and Atwood, 2003; Querbes et al., 2006). Conversely, treatment of cells with dominant-negative inhibitors against late endosome (Rab7) or recycling endosome (Rab11)-specific proteins does not affect JCV infection (Ashok and Atwood, 2003; Querbes et al., 2006). JCV also appears to traffic along microtubules and microfilaments, as treatment of host cells with nocodozole and cytochalasin D prevents infection (Ashok and Atwood, 2003). The related polyomaviruses mouse polyomavirus (mPy) and simian virus 40 (SV40) have been shown to traffic to the ER and engage various host-cell chaperones, resulting in destabilization of the viral capsid (Magnuson et al., 2005; Rainey-Barger et al., 2007;Schelhaas et al., 2007). Following localization to the ER, SV40, mPy and BK polyomavirus (BKV) interact with the endoplasmic reticulum associated degradation (ERAD) pathway, resulting in retrotranslocation of the virion into the cytoplasm (Inoue and Tsai, 2011; Jiang et al., 2009; Lilley et al., 2006; Schelhaas et al., 2007). JCV has been shown to colocalize with the ER marker calregulin, and treatment of cells with brefeldin A (BFA), a drug that inhibits COP1 vesicle formation, interferes with the trafficking of early endosomes, and function of the Golgi apparatus, reduces infection (Querbes et al., 2006; Richards et al., 2002). This implies that trafficking to the ER is important for JCV infection. However, it remains unclear whether JCV usurps the same quality control machinery and ERAD pathways as mPy and SV40. In this paper, we further define the infectious pathway of JCV in transformed human glial cells. We show that JCV traffics to the ER by 4–6 h post-infection, similar to SV40 (Pelkmans et al., 2001; Schelhaas et al., 2007). Additionally, siRNA knock down of ER quality control machinery proteins and the ERAD-associated proteins decreases infection, suggesting that these ER components are important for infection. Finally, we show that JCV VP1 pentamers traffic to the ER with similar kinetics to the intact virion, demonstrating the importance of the VP1 pentamer in early events of the polyomavirus replication cycle. This work highlights the importance of JCV trafficking to the ER, despite differences in early entry events from other polyomaviruses. Further, our data show that pentameric VP1, in the absence of the complete virion, is sufficient to recapitulate these trafficking events.
Results
JCV virions and pentamers efficiently enter host cells
Following binding to cellular receptors, JCV enters cells by clathrin-mediated endocytosis, a process that can be influenced by receptor clustering (Liu et al., 2010). We first sought to determine whether the JCV virion, which contains 72 VP1 pentamers for a total of 360 copies of VP1, may be endocytosed much more rapidly into host cells than a less multivalent complex. To analyze the entry of JCV into human glial cells (SVG-A), we labeled JCV virions with either Alexa Fluor 488 (JCV-488) or Alexa Fluor 633 (JCV-633). To test whether intact virions are more efficient at entering cells, we generated purified VP1 pentamers that do not assemble into full virion-like particles, and labeled them with Alexa Fluor 488 (VP1-488) or Alexa Fluor 633 (VP1-633). SDS-PAGE and fluorographic analysis indicate that VP1 is the major labeled protein for both virions and pentamers, while Coomassie staining demonstrates the purity of both labeled and unlabeled pentamers and virions (Fig. 1A). Furthermore, single cycle growth assays reveal that the infectivity of JCV was not adversely affected by labeling, as JCV-488 and JCV-633 were infectious and thus suitable for studies of JCV infectious pathway (Fig. 1B).
Fig. 1.

Characterization of labeled JCV virions and pentamers. (A) Purified virions (JCV) or pentamers (VP1) were labeled with Alexa Fluor 488 or 633 and resolved by SDS-PAGE and imaged by fluorography (top) or subsequently stained with Coomassie G-250 (bottom). Closed arrowhead indicates VP1, open arrowhead indicates a breakdown product of VP1, and asterisk indicates packaged histones. (B) SVG-A cells were infected with equivalent amounts of labeled or unlabeled virions and infected cells were quantitated at 72 hpi based on VP1 expression. Data represent the average number of infected cells per field for triplicate samples. Error bars indicate standard deviations.
Initial events in binding and entry of JCV virions or pentamers were examined in SVG-A cells using confocal microscopy and flow cytometry. Labeled virus or pentamers were bound to cells at 4 °C and allowed to enter at 37 °C for specific time periods before fixation and imaging. At 0 min post binding, both JCV-488 and VP1-488 bound efficiently to cells, with the majority of fluorescence localized at the plasma membrane (Fig. 2A). Two hours after warming, the majority of virions and pentamers are internalized into cells, with little signal remaining on the plasma membrane. To quantify the amount of virus internalization, the plasma membrane was outlined using the bright field image, and the percentage of fluorescent signal on the plasma membrane versus the cytoplasm was determined at each time point. At 0 min post binding, 70–80% of the fluorescent signal is within the plasma membrane region of interest. After 2 h at 37 °C, the percentage of fluorescence from both virus and pentamers at the plasma membrane decreases where approximately 20% of fluorescence remains at the plasma membrane (Fig. 2B).
Fig. 2.

JCV virions and pentamers bind and enter SVG-A cells with similar kinetics. (A) Pre-chilled SVG-A cells were inoculated with JCV-488 or VP1-488 pentamers for 1 h at 4 °C. Cells were washed and fixed at 0 min or incubated at 37 °C for 120 min before fixation. Fixed samples were stained with DAPI before imaging by confocal microscopy. Shown is either the fluorescence image alone (left), or the fluorescence signal overlaid onto the bright field image (right). Scale bars=10 μm. (B) Quantification of A. The plasma membrane of each cell was outlined using the bright field image, and the percentage of virus or pentamer fluorescence intensity on the plasma membrane compared to the entire cell was determined using MetaMorph imaging software for at least ten cells per sample. Error bars indicate standard deviation. (C). Virus or pentamer was bound to cells as in A. Cells were washed and fixed at 0 min or incubated at 37 °C for 120 min before fixation. Cells were imaged by confocal microscopy (left). Extracellular fluorescence was then quenched with trypan blue and the same cells were re-imaged (right). Scale bars=10 μm. (D) Quantification of endocytosis of virions and pentamers. SVG-A cells were detached and chilled prior to binding of JCV-488 or VP1-488 pentamers for 1 h at 4 °C. Cells were then fixed or warmed to 37 °C for the indicated time points before fixation. Cells were then incubated with trypan blue and protected fluorescence was assayed by flow cytometry.
To further quantify the entry kinetics of virions and pentamers, we used a trypan blue based quenching assay and assayed these samples by confocal microscopy or flow cytometry (Engel et al., 2011; Rejman et al., 2004). Labeled pentamers or virions were bound to pre-chilled cells and warmed to 37 °C for the indicated times before fixation. Trypan blue will quench plasma membrane-associated Alexa Flour 488 fluorescence but not internalized fluorophores, resulting in a direct readout of plasma membrane-associated JCV or VP1. In chilled cells, all of the fluorescence is quenched while much of the fluorescence is protected from quenching when cells are warmed to 37 °C for 2 h (Fig. 2C). To further delineate the rate of protection from trypan blue quenching, we used flow cytometry to quantitate virus and pentamer internalization. Entry kinetics using this flow cytometry based assay are similar to results obtained by confocal microscopy, and demonstrate no appreciable difference between virions and pentamers (Lakadamyali et al., 2006) (Fig. 2D).
JCV and VP1 pentamers localize to the ER
We next sought to define the kinetics of JCV virion and pentamer trafficking to the ER. JCV has previously been shown to colocalize with the ER marker calregulin at 12 hpi, and for SV40 and mPy, trafficking to the ER and engagement of ER resident quality control machinery has previously been shown to be critical for infectivity (Querbes et al., 2006; Schelhaas et al., 2007; Walczak and Tsai, 2011). To determine whether VP1 pentamers traffic to the ER with similar kinetics to intact virions we analyzed JCV-633 or VP1-633 colocalization with an ER marker at specific times post-infection and post pentamer treatment. We show that starting at 6 hpi, significant colocalization between both pentamers or virions and the ER was seen (Fig. 3A and B). This colocalization was quantified using Manders’ coefficient of colocalization, using the coefficient from the virus channel to determine colocalization (Bolte and Cordelieres, 2006). Significant increases in colocalization were observed for both virions and pentamers over the time course of infection, reaching a maximum at 8 hpi (Fig. 3B). At 2 hpi, we see significantly more colocalization between VP1 and ER than for the intact virus, suggesting that VP1 may target the ER with more rapid kinetics than the intact virus. At 10 hpi, this colocalization decreased for both virions and pentamers (Fig. 3B). These data show that both JCV and VP1 traffic to the ER with similar kinetics, suggesting localization to this compartment is critical for the infectious life cycle of JCV, and also further highlighting the importance of the VP1 pentamer in directing viral trafficking to the ER.
Fig. 3.

JCV virions pentamers traffic to the ER. SVG-A cells were transfected with the ER marker Heme Oxygenase-CFP (CFP-HO, pseudocolored red). Cells were inoculated with JCV-633 (A) or VP1-633 pentamers (B) (pseudocolored green) at 4 °C for 1 h. Cells were washed and then incubated at 37°C for 6 h before fixation. Confocal micrograph images (left) and enlarged images (right) are shown to demonstrate colocalization. (C) Quantification of virion or VP1 pentamer colocalization with CFP-HO. Virus or pentamer was bound to pre-chilled cells for 1 h at 4 °C. Cells were then washed and fixed, or warmed to 37 °C for the indicated time points prior to fixation. Colocalization was determined using Manders’ coefficient of colocalization. Error bars indicate standard deviation. *p<0.05. (D) ER colocalization after BFA treatment. SVG-A cells on coverslips were left untreated or pretreated for 1 h with BFA. Cells were chilled to 4 °C and inoculated with JCV-633 or VP1-633 in the presence of BFA for 1 h at 4 °C. Cells were washed and either fixed or warmed to 37 °C for 6 h before fixation and imaging. Manders’ coefficient of colocalization was determined for virions and CFP-HO. Error bars indicate standard deviation. *p<0.05. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Chemical inhibitors of ER trafficking and function decrease JCV infection and viral trafficking
To further delineate the importance of the ER in the JCV infectious pathway, we infected SVG-A cells in the presence of pharmacological agents that disrupt components of this trafficking pathway. Cells were pretreated with each drug then inoculated with JCV in the presence of the drug. After 45 h, cells were stained for large T antigen and infected nuclei were scored. To ensure that drug treatment did not have cytotoxic effects on the cells treated, cell viability assays were performed, and drugs were used at concentrations where there was greater than 90% cell viability (data not shown).
Treatment of SVG-A cells with Brefeldin A (BFA) reduced virus infectivity by 50% as compared to an untreated control (Fig. 4A). BFA has been demonstrated to prevent viral trafficking to the ER for other polyomaviruses, and has also been shown to prevent trafficking of viruses through endosomes, suggesting that ER trafficking is important for JCV infectivity (Inoue and Tsai, 2011;Richards et al., 2002). Thus, we sought to determine whether BFA treatment of SVG-A cells affects ER trafficking of JCV virions and pentamers, or whether this inhibition of infection is due to off-target effects. Pretreatment of SVG-A cells with BFA significantly reduces colocalization of either labeled virions or pentamers with the ER marker at 6 hpi (Fig. 3D).
Fig. 4.

Reduction of JCV infectivity by chemical inhibitors. (A) SVG-A cells were pretreated with inhibitors for 1 h, infected with JCV, then incubated in media containing inhibitors for 45 h. Cells were fixed and stained by indirect immunofluorescence using a monoclonal antibody to large T antigen and relative infectivity compared to an untreated control was determined. Error bars denote standard deviation. *p<0.05 as compared to the untreated control. (B) Effect of virion disulfide bond reduction or alkylation on infectivity. JCV virions were pretreated with DTT to reduce disulfide bonds, or AMS to alkylate free disulfide for 2 h at 37 °C. DTT or AMS was then removed from the sample and equivalent amount of virions were then added to SVG-A cells. Results are averaged from three independent experiments. *p<0.05 as compared to the untreated control.
Treatment of SVG-A cells with thapsigargin, a drug that reduces ER calcium ion concentrations and the affects the unfolded protein response (Shmigol et al., 1995), significantly reduced JCV infectivity, providing further evidence that ER trafficking is critical for JCV infectivity (Fig. 4A). Additionally, DTT pretreatment of SVG-A cells also effectively inhibits JCV infection, and likely inhibits cellular enzymes present in the ER (Fig. 4A) (Schelhaas et al., 2007). These data suggest that upon arrival to the ER, JCV is likely engaging enzymes that are inhibited by thapsigargin or DTT treatment. Upon delivery to the ER, interpentmeric disulfide bond isomerization has been shown to be important for mPy and SV40 infectivity. To test whether intact disulfide bonds were necessary for virus infectivity, we incubated JCV with DTT for 2 h at 37 °C, and then removed DTT from the sample. This DTT pretreatment of virions was shown to have no effect on infection, indicating that disruption of disulfide bonds prior to arrival in the ER is not deleterious to infection. To test the need for free sulfhydryl during infection, we incubated JCV with the alkylating agent 4-acetamido-40-maleimidylstilbene-2,20-disulfonic acid (AMS) for 2 h at 37°C, removed the AMS, and infected cells. AMS will alkylate free cysteines, preventing disulfide bond isomerization by the host cell ER isomerases. This pretreatment reduced infection to 20% of our untreated control, demonstrating that free disulfides are needed for productive infection (Fig. 4B). Thus, upon arrival to the ER, JCV interacts with ER resident enzymes, and alkylation of free disulfides in the virion reduces infection
Specific ER enzymes are necessary for productive JCV infection
Upon reaching the ER, productive infection of host cells by SV40 and mPy has been shown to require the ER chaperones PDI, ERP57, ERP72, and additionally ERP29 for mPy, (Gilbert et al., 2006;Magnuson et al., 2005; Schelhaas et al., 2007; Walczak and Tsai, 2011). Since JCV also traffics to the ER, we tested whether these proteins were important for JCV infection using a siRNA based approach (Table 1). Following treatment of SVG-A with at least 2 different siRNAs per gene, cells were infected with JCV for 48 h, before fixation and indirect immunofluorescence staining for large T antigen. Knockdown of the thio-reductase proteins ERp57, PDI, and ERP72 resulted in a significant reduction of JCV infectivity (Fig. 5A). Additionally, knockdown of the disulfide isomerase ERP29 reduced JCV infectivity. siRNA treatment significantly reduced targeted protein levels, as compared to cells treated with a negative control siRNA (Fig. 5B). Additionally, we show that knockdown of the ERAD associated protein Sel1, but not Derlin-1 or Derlin-2 also significantly reduced JCV infectivity. For Derlin-2 and Sel1L, RT-PCR analysis demonstrated that mRNA levels of targeted genes were reduced to levels less than 25% of cells treated with control siRNA (Fig. 5C). These data indicate that after trafficking to the ER, JCV utilizes the ERAD pathway for productive infection.
Table 1.
siRNAs used in this paper.
| siRNA | siRNA #1 product number |
siRNA #2 product number |
Concentration (nM) | Antibody |
|---|---|---|---|---|
| Erp57 | SI02654771 | SI02654778 | 20 | Enzo (ADI-SPA-585) |
| PDI | SI00092414 | SI03109680 | 200 | Enzo (ADI-SPA-890) |
| Erp29 | SI00317702 | SI03153829 | 10 | Abcam (ab11420) |
| Erp72 | SI03078236 | SI00069916 | 10 | Enzo (ADI-SPS-720) |
| Sel1 | SI02664494 | SI03035564 | 100 | ND |
| Derlin1 | SI02778783 | SI00135940 | 50 | Abcam (ab54545) |
| Derlin2 | SI00362789 | SI00362796 | 50 | ND |
| All stars (+) control | SI00135940 | N/A | 50 | ND |
| All stars (−) control | SI00135940 | N/A | 50 | ND |
ND=Not determined.
Fig. 5.

siRNA knockdown of ER proteins reduces JCV infectivity. SVG-A cells were reverse transfected with at least two siRNAs of the indicated gene or scrambled siRNA. At 72 h post-transfection, cells were infected with JCV, fixed and at 45 hpi, and stained with a monoclonal antibody to large T antigen. Results are the average of three independent experiments and error bars denote standard deviations. (B) SVG-A cells were transfected with each siRNA as in A. Cell lysates were collected at 72 hpi and analyzed by SDS-PAGE and immunoblotting with antibodies to each targeted protein or GAPDH. SVG-A cells transfected with scrambled siRNA was used as a negative control for normal protein levels. (C) RT-PCR quantification of targeted mRNA levels after siRNA knockdown. SVG-A cells were transfected with indicated siRNA. 72 h post transfection, cells were harvested and ΔΔCt values of each gene were calculated relative to GAPDH. These levels were then compared to a control which was transfected with negative control siRNA. Results are the average of three independent experiments. Error bars indicate standard deviation.
VP1 pentamers do not localize to the golgi apparatus
The pentameric subunit of JCV shares a high degree of structural similarity with the B subunit of cholera toxin, which undergoes retrograde trafficking to the golgi apparatus prior to trafficking to the ER (reviewed in Lencer and Tsai, 2003; Sandvig and van Deurs, 2005). To investigate whether JCV virus or pentamers also traffic to the golgi, we bound Alexa Fluor 488 labeled cholera toxin B-subunit (CTB-488), VP1-488, or JCV-488 to SVG-A cells and immunostained the golgi apparatus. Using confocal microscopy, colocalization was observed between the Golgi and CTB-488 starting at 0.5 hpi, with significant colocalization of CTB-488 at 6 hpi (Fig. 6). Conversely, JCV and VP1 appear to localize in organelles adjacent to the Golgi apparatus, but at no time points do we see colocalization between virus or pentamers and the golgi apparatus (Fig. 6).
Fig. 6.
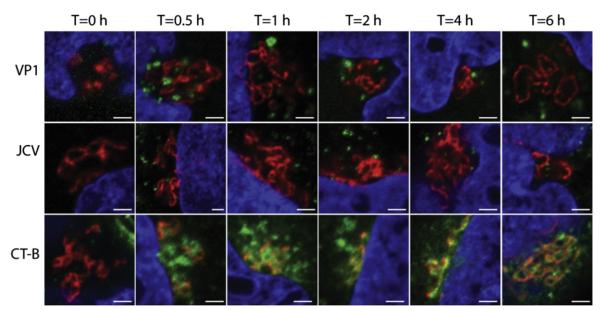
JCV virions or pentamers do not traffic to the golgi apparatus. Cells were chilled to 4 °C inoculated with JCV-488, VP1-488 pentamers, or CT-B-488 (green) for 4 °C for 1 h. Cells were then fixed or warmed to 37 °C for the indicated time points prior to fixation. Cells were stained with an antibody against the golgi marker giantin (red) and coverslips were mounted in mounting media containing DAPI (blue). Scale bars=2 μm. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Discussion
Polyomavirus entry is a complex process that requires viral engagement of numerous host-cell factors in multiple cellular organelles. In this manuscript, we further delineate the infectious pathway of JCV in human glial cells and establish the VP1 pentamer of JC virus as a powerful tool for studying the early events of polyomavirus trafficking. Purified JCV pentamers recapitulate the early trafficking steps of the complete virus, from attachment and entry through localization to the ER. We also show that ER trafficking is critical for infection by disrupting major ER functions using either drug treatment or siRNA knockdown. These results demonstrate VP1 can be used as a model for studying JC virus infection as well as the infectious life cycle of the newly-discovered and difficult to cultivate human polyomaviruses.
The initial interaction between JCV and host cells is mediated by VP1 binding to an α-2,6 linked sialic acid moiety present on LSTc, and the serotonin receptor 5HT2A (Elphick et al., 2004; Neu et al., 2010). Using confocal microscopy, we show entry of both virions and pentamers is rapid, with the majority of VP1 being endocytosed by 30 min (Fig. 2). This rapid entry is consistent with clathrin-mediated endocytosis (Lakadamyali et al., 2006). Although clathrin-mediated endocytosis is a constitutive process, it has been reported that clustering of receptor cargo can increase the rate of endocytosis (Liu et al., 2010). We found no measureable differences in the kinetics of entry between virions and pentamers, indicating that the increased multivalency of the intact virion does not increase endocytosis rates in SVG-A cells (Fig. 2).
VP1 pentamers also traffic to the ER, demonstrating that successful trafficking of JCV to the ER can be orchestrated by this small subunit (Fig. 3A). To quantify colocalization we used Manders’ coefficient of colocalization, which will allow for colocalization measurements to be performed when differences in fluorescence intensities exist between the two acquired channels. This coefficient corresponds to the ratio of summed intensities of pixels in the virus channel for which ER fluorescence is above zero to the total intensity in the virus channel and has a maximum value of 1 (Bolte and Cordelieres, 2006). Since only a small proportion of virions or pentamers are successfully targeted to the ER, we observe coefficients of 0.3, verifying that ER targeting is an inefficient process. Surprisingly, we see more colocalization between VP1 and the ER at 2 hpi than for intact virus, suggesting that the small pentamer targets the ER with increased kinetics (Fig. 3B). The colocalization between the VP1 pentamer and the ER-specific marker CFP-HO suggests that JCV VP1 ER trafficking is strikingly similar to the ER targeting of bacterial toxins including cholera and Shiga toxin. Entry and ER targeting of cholera toxin has been extensively studied, and careful studies have also been performed comparing the ER trafficking of SV40 and mPy to cholera toxin (reviewed in Ewers and Helenius, 2011; Johannes and Popoff, 2008; Johannes and Wunder, 2011; Tsai and Qian, 2010). In the case of mPy and likely other polyomaviruses, ER trafficking is a direct result of ganglioside binding and cholesterol-dependent endocytosis (Qian et al., 2009). As exemplified for cholera toxin, the binding of 5 ganglioside molecules alone is sufficient to direct the toxin to the ER. This is in contrast to the trafficking patterns of antibodies to the ganglioside GD3, which were found to recycle to the plasma membrane rather than traffic to the ER (Crespo et al., 2008). However, JCV has very low affinity for gangliosides, and instead requires serotonin receptor 5HT2A (Elphick et al., 2004) and the sialic acid motif LSTc (Neu et al., 2010) to enter cells by clathrin-mediated endocytosis. It is therefore unclear whether the JCV VP1 pentamer or virus traffics to the ER in a similar fashion to bacterial toxins or other polyomaviruses, especially since Rab7 dominant negative constructs do not inhibit JCV infection, unlike SV40 or mPy (Querbes et al., 2004). Since receptor binding plays a major role in ER targeting of endocytosed cargo and multiple retrograde pathways targeting the ER have been characterized, identifying the proteinaceous or lipid receptor molecule to which LSTc is attached will be instrumental in increasing our understanding of JCV entry (Bonifacino and Hurley, 2008; Johannes and Popoff, 2008;Johannes and Wunder, 2011; Popoff et al., 2007; Rojas et al., 2008).
Confocal microscopy and pharmacological inhibitor studies demonstrate that trafficking to the ER is a critical step for JCV infection, and thus validates a model whereby JC pentamers traffic to this compartment. Previous studies have demonstrated that JCV colocalizes with the ER marker calregulin at 12–16 hpi and treatment of SVG-A cells with BFA reduces infection (Querbes et al., 2004). To verify that JCV ER targeting is critical to infectivity and that BFA is not preventing infection due to off target effects, we undertook a pharmacological and siRNA based approach to reduce ER function. We show that treatment of cells with BFA, which inhibits COP1 vesicle formation, not only reduces infection, but also prevents JCV from trafficking to the ER (Fig. 5A). To further stress the importance of the ER in JCV infection, we treated cells with thapsigargin, a drug that interferes with ER calcium ion homeostasis and the unfolded protein response. This treatment significantly reduced JCV infection (Fig. 4A). Additionally, we show that DTT pretreatment of SVG-A cells, which has been suggested to interfere with the function of resident ER proteins, reduces JCV infectivity (Fig. 5E) (Schelhaas et al., 2007). That DTT pretreatment of cells acts on cellular factors and not virions is consistent with our results demonstrating that DTT pretreatment of virions does not reduce infection (Fig. 5E). Thus, our pharmacological data strongly demonstrates that JCV traffics to the ER, and interference either with ER trafficking, or with ER function, significantly reduces JCV infectivity.
To further delineate what ER factors are utilized by JCV during infection, we employed a siRNA based approach to specifically knock down individual ER chaperones. We show that siRNA mediated knockdown of PDI, ERp29, ERp57, and ERp72 reduces JCV infectivity (Fig. 5A). Similar to mPy but unlike SV40, siRNA mediated knockdown of ERp29 reduces JCV infectivity. ERp29 contains only a single cysteine and therefore can isomerize but not disrupt disulfide bonds, demonstrating that isomerization of JCV disulfide bonds is critical to infection (Hebert and Molinari, 2007). While siRNAs targeting PDI significantly reduced infectivity, appreciable levels of PDI remained after this treatment (Fig. 5b). It is possible that our siRNAs to PDI are resulting in off target effects, however the requirement for PDI in JCV infectivity is consistent with other poylomaviruses. The importance of capsid disulfide bond isomerization during infection is consistent with our results that alkylation of JCV free cysteine residues in the viral capsid reduces infection (Fig. 4E) (Magnuson et al., 2005). In recent studies on mPy, PDI and Erp57 were shown to act in a coordinated fashion to partially disassemble the viral capsid (Walczak and Tsai, 2011). Thus it is likely that upon arrival to the ER, JCV engages a number of these PDI family members in a similar fashion to SV40 and mPy to disrupt the interpentameric disulfide bonds. In SV40, cysteine 9 (C9) and C104 form interpentameric disulfide bonds with adjacent pentamers to link the capsid together, and a disulfide bond at C15 has been shown to be important in mPy (Schelhaas et al., 2007; Walczak and Tsai, 2011). Cysteine residues 104 and 257 are conserved in JCV; however, JCV lacks the C9, and likely engages these ER enzymes in a similar, but not identical fashion. Additionally, knockdown of Sel1 reduces infectivity. Sel1 is a component of the ERAD pathway, suggesting that JCV may also use this retrotranslocation pathway for productive infection. Surprisingly, siRNA knockdown of either Derlin-1 or Derlin-2 fails to significantly reduce JCV infectivity (Fig. 5A). Even with high levels of siRNA, Derlin-1 or Derlin-2 were present at low levels, as determined by western blotting or RT-PCR. Therefore it is possible that the siRNAs used do not reduce targeted proteins to a level where JCV infectivity would be inhibited. Alternatively, JCV may interact with Sel1 independent of Derlin-1 or Derlin-2 for successful retrotranslocation into the cytosol. This interaction has previously been reported for the Sel1 mediated degradation of a mutant of ribosome receptor protein ribophorin (Mueller et al., 2006).
Finally, we investigated whether JCV VP1 pentamers traffic to the golgi apparatus, similar to bacterial toxins. Polyomavirus VP1 pentamers contain a structure similar to AB5 toxins such as cholera toxin (Chen et al., 1998; Sixma et al., 1991; Stehle and Harrison, 1997). Both CTB and polyomaviruses undergo retrograde trafficking to the ER; however, CTB rapidly traffics to the golgi apparatus prior to arrival in the ER. Several possibilities exist for what factors influence golgi localization. JCV pentamers bind the receptor motif LSTc rather than cholera toxin’s receptor GM1, and this interaction may dictate golgi trafficking. Alternatively, low avidity interactions between pentamers or toxins and its receptor may direct traffic to the golgi apparatus, whereas the much higher avidity interaction between the intact virion and its receptor may result in direct ER trafficking. Finally, the larger size of the virion compared to JCV VP1 pentamers or CTB may result in direct ER trafficking. When we analyzed colocalization between CTB and our golgi marker giantin, we saw rapid accumulation of CTB starting at 0.5 hpi, similar to previously published reports (Mallard et al., 1998; Natarajan and Linstedt, 2004). However, we were unable to visualize colocalization between JCV virus or pentamer and the golgi apparatus occurs at time points up to 6 hpi, suggesting that JCV and CTB may target the ER by different trafficking pathways. This result suggests that the specific receptor interactions between VP1 pentamers or cholera toxin and its receptor may dictate sorting to the golgi rather than the avidity of ligand receptor interactions. It is possible that JCV pentamers may be sorted to the golgi apparatus, but any targeting would either be much less efficient than cholera toxin or would be with drastically different kinetics (time points after 6 hpi). Future studies are underway to further delineate the requirements for golgi targeting.
In conclusion, we demonstrate that JCV traffics to the ER with almost identical kinetics to the VP1 pentameric subunit of JC virus alone. Once in the ER, JCV utilizes host-cell chaperones in a similar fashion to mPy and SV40, demonstrating that targeting this organelle is critical for JCV infectivity. This study provides additional insight into the infectious pathway for JCV in human glial cells, and demonstrates that a subunit of the viral capsid can be utilized as a tool to study viral entry and trafficking pathways in viruses that are difficult to cultivate or for higher throughput assays.
Materials and methods
Viruses, cells and plasmids
The MAD1-SVEΔ strain of JCV and the human fetal glial cell line SVG-A maintained in complete growth media (1X MEM, 10%FBS, 0.5% penicillin, 0.5% streptomycin) were used for all experiments (Major et al., 1985; Vacante et al., 1989). The CFP-Heme Oxygenase 2 (CFP-HO) plasmid was a kind gift from Melissa Rolls (Pennsylvania State University). Rab5 fused to monomeric red fluorescent protein (Rab5-mRFP) and Rab7-mRFP were purchased from Addgene (plasmid 14437 and 14436, respectively) (Vonderheit and Helenius, 2005) and the Rab5-GFP and Rab7-GFP were gifts from Stephen Furguson (Robart Research Institute) and Craig Roy (Yale University).
Virus purification and labeling
JCV was purified similar to previously published methods (Shen et al., 2011). Briefly, ten 1700 cm2 roller bottles were seeded with SVG-A cells at 50% confluency and infected with JCV at an MOI of ~0.1 FFU/cell for 14 day, with the cell culture media replaced at 7 day. Viral lysates were harvested by scraping cells in the presence of cell culture media, and this lysate was frozen and thawed 3 times. The lysate was then treated with type V neuraminidase (Sigma) at 37 °C for 1 h to release JCV still bound to cells. Deoxycholate acid (Fisher Scientific) was added to further disrupt cells for 1 h at 37 °C and then sonicated three times on ice (50% amplitude 50% duty cycle, power 4, 1 min). The cellular debris was pelleted by centrifugation, and the viral supernatant was pelleted through a 20% sucrose cushion in a Beckman SW40ti rotor at 150,000g at 4 °C for 3 h. The viral pellet was resuspended into buffer A (10 mM Tris–HCl, 50 mM NaCl, 0.1 mM CaCl2) and sonicated 3 times (30% amplitude 50% duty cycle, power 3, 1 min). The resuspended pellet was loaded onto a CsCl step gradient (1.29–1.35 g/ml) and spun at 115,000g at 4 °C for 18 h in a Beckman SW55ti rotor. The band corresponding to DNA-containing virions was isolated and dialyzed extensively against buffer A. The dialyzed virus was further purified by size exclusion chromatography over an S500HR column previously equilibrated in Buffer A (GE Healthcare). The monomeric virus fraction was collected and concentrated using an Amicon ultra-15 (Millipore) to approximately 0.2 mg/mL. Virion concentrations were determined by Bradford assay. Viral proteins were resolved on a 4–15% gradient SDS-PAGE gel (Bio-Rad) and stained with coomassie G250 (Sigma) to ensure virus purity (Fig. 1). JCV was labeled with Alexa Fluor 488 or Alexa Fluor 633 according to manufacturers’ instructions (Invitrogen). Briefly, purified JCV was desalted in 0.1 M carbonate/bicarbonate buffer pH 8 using a HiTrap 5mL column (GE Healthcare). The virus was then concentrated to ~0.5 mg/ml and dye was added at a molar excess of 200:1. After 1 h of rocking incubation at room temperature, the excess dye was removed over a HiTrap 5mL column. To verify fluorescence labeling, viral proteins were resolved on a 4–15% SDS-PAGE gel and imaged using a Typhoon fluorescence imager (Fig. 1).
Pentamer purification and labeling
JCV VP1 pentamers were purified as described previously (Neu et al., 2010). Briefly, a 2 L bacterial culture transformed with a pET15-b bacterial expression vector containing JCV VP1 residues 22–289 were grown to an OD of ~0.8 at 37 °C with shaking and induced with 0.2 mM IPTG. Cultures were grown at 21 °C for 18 h and the bacteria were pelleted. This pellet was resuspended in 20 mL of loading buffer (50 mM Tris–HCl, 250 mM NaCl, 10 mM imidazole, 5% glycerol) and frozen in liquid nitrogen. After thawing, the pellet was sonicated 5 times on ice and the bacterial lysate was pelleted by low speed centrifugation. The supernatant was filtered through a 0.22 μm filter and purified using a HisTrap 5 mL column using an AKTA Purifier (GE Healthcare). Pentamers were eluted from the column with a linear gradient of 10–500 mM imidazole, and fractions corresponding to pentamers were collected and dialyzed against 20 mM Tris base, 5% glycerol, 250 mM NaCl, 10 mM DTT. The sample was then concentrated and monomeric VP1 pentamers were purified using a Superdex S200 column and eluted in 20 mM Hepes Free acid, 150 mM NaCl, 50 mM DTT (GE Healthcare). These monomeric pentamers were concentrated to 1 mg/ml, aliquoted and stored at 80 °C until use. Concentrations of purified pentamers was determined by spectrophotometric absorbance at 280 nm, using a molar extinction coefficient of 28,0230 M−1 cm−1 per VP1 monomeric subunit. A thawed aliquot of VP1 pentamers was re-purified by size exclusion chromatography to ensure that the VP1 pentamers did not aggregate after freezing and thawing. VP1 pentamers were labeled with Alexa Fluor 488 or Alexa Fluor 633 as described above for JCV virions.
Treatment of cells with inhibitors
To determine the effects of inhibitors on JCV infectivity, SVG-A cells were plated in 96-well plates at a density of 2 × 104 cells/cm2 and incubated at 37 °C 16 h in complete growth media. One hour prior to infection media was removed, and fresh media containing the inhibitor (5 mM DTT, 0.25 μM thapsigargin, 5 ng/mL brefeldin A) was added to cells. After 1 h, cells were infected with JCV at an MOI of 1 FFU/cell in complete growth media containing 2% FBS and allowed to bind for 1 h. Cells were then washed with fresh media containing inhibitors, and incubated in media containing inhibitors for 45 h. Cells were washed in PBS, fixed in ice-cold methanol, and stained with a primary monoclonal antibody specific for JCV large T antigen (Pab962) and a secondary Alexa Fluor 488 goat anti-mouse antibody for 1 h each (Invitrogen). Infected cells were quantitated by counting 3 fields for each well, for triplicate samples from three separate days. The infectivity studies were only performed at concentrations where there was at least 90% viability as compared to the positive control based on a CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega).
Knockdown of ER proteins with siRNA
siRNAs were purchased from Qiagen (Table 1). 3×105 SVG-A cells in 24 well plates were transfected with siRNAs using siRNA max lipofectamine transfection reagent according to manufacturer’s guidelines (Invitrogen). SVG-A cells were reverse transfected by first mixing siRNA and transfection reagent in cell culture media, and then overlaying cells to this mixture. Optimal siRNA concentrations were either determined using fluorescently labeled siRNA (Block-it, Invitrogen), or were based on previously published concentrations (Schelhaas et al., 2007). Two siRNAs per gene were transfected in separate wells to ensure specificity of siRNAs. 72 h post transfection, cells were either infected with JCV or harvested for western blot analysis or realtime PCR (RT-PCR) analysis. Each well was infected with JCV at an MOI of 1 FFU/cell at 37 °C for 1 h. After 1 h, unbound virus was washed, and complete media was added to each well. At 48 hpi, cells were fixed with MeOH and stained with PAB692. Infectivity data were normalized as a percent infectivity of the negative control (Allstars siRNA negative control, Qiagen). A cocktail of siRNAs that are lethal to cells was used a positive control (Allstars siRNA positive control, Qiagen). For western blot analysis, cells were harvested at 72 h post siRNA transfection. Cells were washed with PBS, detached with a cell scraper, pelleted by low speed centrifugation, and the supernatant was aspirated. The remaining cellular pellet was incubated in RIPA buffer with protease inhibitors at 4 °C for 30 min with constant rocking. Cellular debris was then pelleted at 14,000g at 4 °C. The remaining supernatant was then resolved on a 4–15% SDS-PAGE gel and transferred to PVDF membranes. Membranes were blocked for 1 h, and incubated with the appropriate primary antibody for 1 h in blocking buffer according to the manufacturer’s recommended guidelines (Table 1). A primary antibody against GAPDH was also used as a loading control (Sigma). The membrane was washed in PBS-T (0.05% Tween-20), and a secondary antibody conjugated to an infrared dye was added for 1 h. After washing, the membrane was imaged using an Odyssey infrared gel scanner (Li-Cor). For RT-PCR analysis, SVG-A cells were transfected with Derlin-2 or Sel1 siRNAs as above. 72 h post transfection, mRNAs were harvested from cell lysates using the Taqman Cells to CT kit (Life Sciences). Reverse transcriptase and RT-PCR was performed according to manufacturer’s guidelines, using predesigned probes specific for Derlin2, Sel1, or GAPDH (Life Sciences). For PCR analysis, CT threshold levels were set, and the ΔCT values between GAPDH and the gene of interest was determined. RT-PCR was also performed using a negative control siRNA, and negative control ΔCT values were determined, and used to generate ΔΔCT values for each experimental siRNA. Target mRNA levels remaining were determined using the following equation: %mRNA remaining=100–([1–2−ΔΔCT]×100).
Confocal imaging of JCV entry
All microscopy experiments were performed using an LSM-710 laser scanning confocal microscope with a 63 × 1.4NA plan apochromat objective with the pinhole set to one Airy unit (Carl Zeiss). DAPI and CFP-HO were excited using a 405 nm diode laser, Alexa Fluor 488 was excited using a 488 nm argon laser and Alexa Fluor 633 was excited using a 633 nm helium-neon laser. For fixed cell experiments, SVG-A cells were seeded overnight onto #1.5 cover slips (Carl Zeiss). Cells were chilled at 4 °C to prevent endocytosis, and 100 ng of labeled virus or pentamers were bound for 1 h at 4 °C. Excess virus was washed and the samples were returned to 37 °C for the specific length of time. Approximately 2500–5000 particles were bound per cell for these experiments. Cells were fixed with 4% paraformaldehyde (PFA) and mounted in an aqueous media either containing or lacking DAPI.
Viral entry quenching experiments were performed as previously described (Engel et al., 2011). Briefly, SVG-A cells were seeded onto #1.5 coverslips. The following day, cells were pretreated with 15 ng/mL Brefeldin A at 37 °C for 1 h, or left untreated. Cells were then chilled at 4 °C for 30 min and Alexa Fluor 488 labeled JCV or pentamers was bound to cells at 4 °C for 1 h. The cells were then washed and fixed with 4% PFA or warmed to 37 °C for 2 h to allow internalization of virus prior to fixation. Prior to quenching of external fluorescence with trypan blue, a Z-stack of images was taken that imaged the entire height of the cell. After quenching the same Z-series was reimaged, and these images were combined into a single image projecting the maximal fluorescence to demonstrate quenching.
For the golgi colocalization experiments, SVG-A cells were seeded overnight onto #1.5 coverslips. Cells were then chilled at 4 °C for 30 min and 5 μg/ml of JCV-488, 20 μg/ml of VP1-488, or 10 μg/ml of CTB-488 was bound to cells at 4 °C for 1 h. The cells were then washed and fixed with 4% PFA or warmed to 37 °C for the specified prior to fixation. Cells were then permeabalized, with 1% triton X-100 and the golgi was stained with a rabbit polyclonal antibody to the golgi marker giantin (Abcam) and an Alexa Fluor 594 labeled secondary antibody against rabbit. Cells were mounted in aqueous mounting media containing DAPI and imaged by confocal microscopy.
Image analysis of confocal microscopy
For analysis of JCV and viral pentamer entry into cells (Fig. 2B), MetaMorph image analysis software was used (Molecular Devices). The cell boundary of twenty individual cells was traced as a region of interest based on the bright field images. An inclusive threshold was then applied to the fluorescence signal (representing either JCV or pentamer) and average pixel intensity was calculated for that region and the entire cell. The percentage of plasma-membrane associated fluorescence signal was then calculated. Background fluorescence within each cell reduces the percentage of total fluorescence signal on plasma membrane. In order to better define the ER boundaries, images were filtered using the Fast Fourier Transform Band pass filter as described (Qian et al., 2009). Briefly, the settings were specific for filtering structures down to 15 pixels and up to 3 pixels with a tolerance of detection of 5%. Specific colocalized regions are shown as enlarged insets at t=0, t=30 and t=60, and at least 10 cells were analyzed per sample. The images were FFT filtered as described above, and colocalization was assessed using the Manders’ coefficient (Bolte and Cordelieres, 2006). For Manders’ coefficient of colocalization, the coefficient from the virus channel was used to determine colocalization. Error represents standard deviation.
Quantification of virus and pentamer endocytosis by flow cytometry
To quantify endocytosis of virus and pentamers into SVG-A cells, we employed a flow cytometry based trypan blue quenching assay (Engel et al., 2011; Rejman et al., 2004). Trypan blue is a cell impermeant dye that will quench any Alexa Fluor 488 fluorophores attached to extracellular virus or pentamer. This allows the determination of the rate of endocytosis, by measuring the entry of labeled pentamers or virions into cells, which are protected from quenching. SVG-A cells were seeded into six welled plates and were incubated overnight. Cells were detached from each well using cellstripper (Cellgro), and the cells were washed in PBS. Cells were incubated in phenol red free MEM and cooled to 4 °C to prevent endocytosis. Virus or pentamer were then incubated with cells for 1 h on ice to allow binding. Cells were extensively washed, and were then either immediately analyzed by flow cytometry, or heated to 37 °C to allow endocytosis. For each time point, samples were analyzed without trypan blue quenching to determine the total levels of fluorescence per cell. To quench extracellular fluorescence, trypan blue was added to each sample at a final volume of 0.008% (V/V) immediately before analysis by flow cytometry. The level of internalization (i) at each timepoint was determined by comparing the ratio of fluorescence of unquenched (Fq) and unquenched (Fo) according to the following formula: i=(Fq/Fo)*100.
Acknowledgments
We thank all members of the Atwood lab for their helpful comments. The authors declare no conflicts of interest. Work in our laboratory was supported by R01CA071878 (W.J.A.), R01NS043097 (W.J.A.), P01NS065719, and by Ruth L. Kirschstein National Research Service Awards F32NS070687 (C.D.S.N) and F32NS064870 (M.S.M) from the National Institute of Neurological Disorders and Stroke. Confocal microscopy analysis was completed in the Leduc Bioimaging Facility at Brown University. Immunoblot analysis was performed in the Center for Cancer Signaling Networks at Brown University that is supported by P30RR031153 (W.J.A.).
References
- Ashok A, Atwood WJ. Contrasting roles of endosomal pH and the cytoskeleton in infection of human glial cells by JC virus and simian virus 40. J. Virol. 2003;77:1347–1356. doi: 10.1128/JVI.77.2.1347-1356.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Hurley JH. Retromer. Curr. Opin. Cell Biol. 2008;20:427–436. doi: 10.1016/j.ceb.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XS, Stehle T, Harrison SC. Interaction of polyomavirus internal protein VP2 with the major capsid protein VP1 and implications for participation of VP2 in viral entry. EMBO J. 1998;17:3233–3240. doi: 10.1093/emboj/17.12.3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo PM, von Muhlinen N, Iglesias-Bartolome R, Daniotti JL. Complex gangliosides are apically sorted in polarized MDCK cells and internalized by clathrin-independent endocytosis. FEBS J. 2008;275:6043–6056. doi: 10.1111/j.1742-4658.2008.06732.x. [DOI] [PubMed] [Google Scholar]
- Egli A, Infanti L, Dumoulin A, Buser A., Samaridis, J., Stebler C, Gosert R, Hirsch HH. Prevalence of polyomavirus BK and JC infection and replication in 400 healthy blood donors. J. Infect. Dis. 2009;199:837–846. doi: 10.1086/597126. [DOI] [PubMed] [Google Scholar]
- Elphick GF, Querbes W, Jordan JA, Gee GV, Eash S, Manley K, Dugan A, Stanifer M, Bhatnagar A, Kroeze WK, Roth BL, Atwood WJ. The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science. 2004;306:1380–1383. doi: 10.1126/science.1103492. [DOI] [PubMed] [Google Scholar]
- Engel S, Heger T, Mancini R, Herzog F, Kartenbeck J, Hayer A, Helenius A. The role of endosomes in SV40 entry and infection. J. Virol. 2011 doi: 10.1128/JVI.02179-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewers H, Helenius A. Lipid-mediated endocytosis. Cold Spring Harb. Perspect. Biol. 2011 doi: 10.1101/cshperspect.a004721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert J, Ou W, Silver J, Benjamin T. Downregulation of protein disulfide isomerase inhibits infection by the mouse polyomavirus. J. Virol. 2006;80:10868–10870. doi: 10.1128/JVI.01117-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert DN, Molinari M. In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 2007;87:1377–1408. doi: 10.1152/physrev.00050.2006. [DOI] [PubMed] [Google Scholar]
- Inoue T, Tsai B. A large and intact viral particle penetrates the endoplasmic reticulum membrane to reach the cytosol. PLoS Pathog. 2011;7:e1002037. doi: 10.1371/journal.ppat.1002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Abend JR, Tsai B, Imperiale MJ. Early events during BK virus entry and disassembly. J. Virol. 2009;83:1350–1358. doi: 10.1128/JVI.02169-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannes L, Popoff V. Tracing the retrograde route in protein trafficking. Cell. 2008;135:1175–1187. doi: 10.1016/j.cell.2008.12.009. [DOI] [PubMed] [Google Scholar]
- Johannes L, Wunder C. Retrograde transport: two (or more) roads diverged in an endosomal tree? Traffic. 2011;12:956–962. doi: 10.1111/j.1600-0854.2011.01200.x. [DOI] [PubMed] [Google Scholar]
- Kean JM, Rao S, Wang M, Garcea RL. Seroepidemiology of human polyomaviruses. PLoS Pathog. 2009;5:e1000363. doi: 10.1371/journal.ppat.1000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles WA, Pipkin P, Andrews N, Vyse A, Minor P, Brown DW, Miller E. Population-based study of antibody to the human polyomaviruses BKV and JCV and the simian polyomavirus SV40. J. Med. Virol. 2003;71:115–123. doi: 10.1002/jmv.10450. [DOI] [PubMed] [Google Scholar]
- Lakadamyali M, Rust MJ, Zhuang X. Ligands for clathrin-mediated endocytosis are differentially sorted into distinct populations of early endosomes. Cell. 2006;124:997–1009. doi: 10.1016/j.cell.2005.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lencer WI, Tsai B. The intracellular voyage of cholera toxin: going retro. Trends Biochem. Sci. 2003;28:639–645. doi: 10.1016/j.tibs.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Lilley BN, Gilbert JM, Ploegh HL, Benjamin TL. Murine polyomavirus requires the endoplasmic reticulum protein Derlin-2 to initiate infection. J. Virol. 2006;80:8739–8744. doi: 10.1128/JVI.00791-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu AP, Aguet F, Danuser G, Schmid SL. Local clustering of transferrin receptors promotes clathrin-coated pit initiation. J. Cell Biol. 2010;191:1381–1393. doi: 10.1083/jcb.201008117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnuson B, Rainey EK, Benjamin T, Baryshev M, Mkrtchian S, Tsai B. ERp29 triggers a conformational change in polyomavirus to stimulate membrane binding. Mol. Cell. 2005;20:289–300. doi: 10.1016/j.molcel.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Major EO. Progressive multifocal leukoencephalopathy in patients on immunomodulatory therapies. Annu Rev. Med. 2010;61:35–47. doi: 10.1146/annurev.med.080708.082655. [DOI] [PubMed] [Google Scholar]
- Major EO, Miller AE, Mourrain P, Traub RG, de Widt E, Sever J. Establishment of a line of human fetal glial cells that supports JC virus multiplication. Proc. Natl. Acad. Sci. USA. 1985;82:1257–1261. doi: 10.1073/pnas.82.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallard F, Antony C, Tenza D, Salamero J, Goud B, Johannes L. Direct pathway from early/recycling endosomes to the Golgi apparatus revealed through the study of shiga toxin B-fragment transport. J. Cell Biol. 1998;143:973–990. doi: 10.1083/jcb.143.4.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco MC, Atwood WJ, Gravell M, Tornatore CS, Major EO. JC virus infection of hematopoietic progenitor cells, primary B lymphocytes, and tonsillar stromal cells: implications for viral latency. J. Virol. 1996;70:7004–7012. doi: 10.1128/jvi.70.10.7004-7012.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller B, Lilley BN, Ploegh HL. SEL1L, the homologue of yeast Hrd3p, is involved in protein dislocation from the mammalian ER. J. Cell Biol. 2006;175:261–270. doi: 10.1083/jcb.200605196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan R, Linstedt AD. A cycling cis-Golgi protein mediates endosome-to-Golgi traffic. Mol. Biol. Cell. 2004;15:4798–4806. doi: 10.1091/mbc.E04-05-0366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu U, Maginnis MS, Palma AS, Stroh LJ, Nelson CD, Feizi T, Atwood WJ, Stehle T. Structure-function analysis of the human JC polyomavirus establishes the LSTc pentasaccharide as a functional receptor motif. Cell Host Microbe. 2010;8:309–319. doi: 10.1016/j.chom.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu U, Woellner K, Gauglitz G, Stehle T. Structural basis of GM1 ganglioside recognition by simian virus 40. Proc. Natl. Acad. Sci. USA. 2008;105:5219–5224. doi: 10.1073/pnas.0710301105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkmans L, Kartenbeck J, Helenius A. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat. Cell Biol. 2001;3:473–483. doi: 10.1038/35074539. [DOI] [PubMed] [Google Scholar]
- Pho MT, Ashok A, Atwood WJ. JC virus enters human glial cells by clathrin-dependent receptor-mediated endocytosis. J. Virol. 2000;74:2288–2292. doi: 10.1128/jvi.74.5.2288-2292.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popoff V, Mardones GA, Tenza D, Rojas R, Lamaze C, Bonifacino JS, Raposo G, Johannes L. The retromer complex and clathrin define an early endosomal retrograde exit site. J. Cell Sci. 2007;120:2022–2031. doi: 10.1242/jcs.003020. [DOI] [PubMed] [Google Scholar]
- Qian M, Cai D, Verhey KJ, Tsai B. A lipid receptor sorts polyomavirus from the endolysosome to the endoplasmic reticulum to cause infection. PLoS Pathog. 2009;5:e1000465. doi: 10.1371/journal.ppat.1000465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querbes W, Benmerah A, Tosoni D, Di Fiore PP, Atwood WJ. A JC virus-induced signal is required for infection of glial cells by a clathrin- and eps15-dependent pathway. J. Virol. 2004;78:250–256. doi: 10.1128/JVI.78.1.250-256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querbes W, O’Hara BA, Williams G, Atwood WJ. Invasion of host cells by JC virus identifies a novel role for caveolae in endosomal sorting of noncaveolar ligands. J. Virol. 2006;80:9402–9413. doi: 10.1128/JVI.01086-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainey-Barger EK, Magnuson B, Tsai B. A chaperone-activated nonenveloped virus perforates the physiologically relevant endoplasmic reticulum membrane. J. Virol. 2007;81:12996–13004. doi: 10.1128/JVI.01037-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randhawa P, Shapiro R, Vats A. Quantitation of DNA of polyomaviruses BK and JC in human kidneys. J. Infect. Dis. 2005;192:504–509. doi: 10.1086/431522. [DOI] [PubMed] [Google Scholar]
- Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem J. 2004;377:159–169. doi: 10.1042/BJ20031253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards AA, Stang E, Pepperkok R, Parton RG. Inhibitors of COP-mediated transport and cholera toxin action inhibit simian virus 40 infection. Mol. Biol. Cell. 2002;13:1750–1764. doi: 10.1091/mbc.01-12-0592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas R, van Vlijmen T, Mardones GA, Prabhu Y, Rojas AL, Mohammed S, Heck AJ, Raposo G, van der Sluijs P, Bonifacino JS. Regulation of retromer recruitment to endosomes by sequential action of Rab5 and Rab7. J. Cell Biol. 2008;183:513–526. doi: 10.1083/jcb.200804048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandvig K, van Deurs B. Delivery into cells: lessons learned from plant and bacterial toxins. Gene Therapy. 2005;12:865–872. doi: 10.1038/sj.gt.3302525. [DOI] [PubMed] [Google Scholar]
- Schelhaas M, Malmstrom J, Pelkmans L, Haugstetter J, Ellgaard L, Grunewald K, Helenius A. Simian Virus 40 depends on ER protein folding and quality control factors for entry into host cells. Cell. 2007;131:516–529. doi: 10.1016/j.cell.2007.09.038. [DOI] [PubMed] [Google Scholar]
- Shen PS, Enderlein D, Nelson CD, Carter WS, Kawano M, Xing L, Swenson RD, Olson NH, Baker TS, Cheng RH, Atwood WJ, Johne R, Belnap DM. The structure of avian polyomavirus reveals variably sized capsids, non-conserved inter-capsomere interactions, and a possible location of the minor capsid protein VP4. Virology. 2011;411:142–152. doi: 10.1016/j.virol.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmigol A, Kostyuk P, Verkhratsky A. Dual action of thapsigargin on calcium mobilization in sensory neurons: inhibition of Ca2+ uptake by caffeine-sensitive pools and blockade of plasmalemmal Ca2+ channels. Neuroscience. 1995;65:1109–1118. doi: 10.1016/0306-4522(94)00553-h. [DOI] [PubMed] [Google Scholar]
- Sixma TK, Pronk SE, Kalk KH, Wartna ES, van Zanten BA, Witholt B, Hol WG. Crystal structure of a cholera toxin-related heat-labile enterotoxin from E. coli. Nature. 1991;351:371–377. doi: 10.1038/351371a0. [DOI] [PubMed] [Google Scholar]
- Stehle T, Harrison SC. High-resolution structure of a polyomavirus VP1-oligosaccharide complex: implications for assembly and receptor binding. EMBO J. 1997;16:5139–5148. doi: 10.1093/emboj/16.16.5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CS, Dezube BJ, Bhargava P, Autissier P, Wuthrich C, Miller J, Koralnik IJ. Detection of JC virus DNA and proteins in the bone marrow of HIV-positiveand HIV-negative patients: implications for viral latency and neurotropic transformation. J. Infect. Dis. 2009;199:881–888. doi: 10.1086/597117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CS, Koralnik IJ. Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: clinical features and pathogenesis. Lancet Neurol. 2010;9:425–437. doi: 10.1016/S1474-4422(10)70040-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai B, Qian M. Cellular entry of polyomaviruses. Curr. Top. Microbiol. Immunol. 2010;343:177–194. doi: 10.1007/82_2010_38. [DOI] [PubMed] [Google Scholar]
- Vacante DA, Traub R, Major EO. Extension of JC virus host range to monkey cells by insertion of a simian virus 40 enhancer into the JC virus regulatory region. Virology. 1989;170:353–361. doi: 10.1016/0042-6822(89)90425-x. [DOI] [PubMed] [Google Scholar]
- Vonderheit A, Helenius A. Rab7 associates with early endosomes to mediate sorting and transport of Semliki forest virus to late endosomes. PLoS Biol. 2005;3:e233. doi: 10.1371/journal.pbio.0030233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walczak CP, Tsai B. A PDI family network acts distinctly and coordinately with ERp29 to facilitate polyomavirus infection. J. Virol. 2011;85:2386–2396. doi: 10.1128/JVI.01855-10. [DOI] [PMC free article] [PubMed] [Google Scholar]