

Abstract
Dysregulation of gene expression is a frequent occurrence in oral squamous cell carcinoma (OSCC). However, accumulating evidence suggests that in contrast to genetics, epigenetic modifications consisting of aberrant DNA methylation, histone modifications and altered expression of miRNAs induce OSCC tumorigenesis and perhaps play a more central role in the evolution and progression of this disease. The unifying theme among these three epigenetic mechanisms remains the same, which is aberrant regulation of gene expression. In this article, we provide a comprehensive review of the impact of epigenetics on oral tumorigenesis with a systematic report on aberrant DNA methylation, histone modifications and miRNA regulation in the pathogenesis of OSCC. We provide insights into recent studies on the prospect of biomarkers for early detection and indication of disease recurrence, and novel treatment modalities.
Keywords: DNA methylation, epigenetics, genomic instability, histone modification, miRNA, oral cancer
Each year more than 100,000 patients die of oral squamous cell carcinoma (OSCC) worldwide [1]. OSCC is a multifactorial disease in which chronic tobacco and alcohol use constitute two major risk factors, while chronic inflammation, viral infections (human papillomavirus), betel quid chewing and genetic predisposition are supplementary factors that contribute towards its pathogenesis. In order to improve survival by means of enhanced prevention and therapeutic options, it is of the essence to understand the basic molecular mechanisms driving oral tumorigenesis. Besides the limited and somewhat poorly understood role of genetic alterations in the pathogenesis of OSCC, accumulating evidence over the last decade highlights that epigenetic alterations, consisting of aberrant DNA methylation, histone modifications and altered expression of miRNAs, may actually play a much larger role in the initiation and progression of OSCC. An accumulating body of data suggests that the two major risk factors, smoking and alcohol consumption, have a direct impact on the dynamic regulation of gene expression orchestrated by epigenetic mechanisms.
Epigenetic mechanisms may influence dysregulation of gene expression in a number of different ways. Aberrant promoter hypermethylation can interfere with the binding of transcription factors to the DNA of tumor suppressor genes, resulting in transcriptional silencing of key genes involved in protection of cells from the process of unregulated growth. By contrast, global hypomethylation may cause reactivation of methylation-silenced proto-oncogenes as well as leading to enhanced genomic instability. Post-translational histone modifications (e.g., acetylation or methylation of histone residues) cause conformational changes of the DNA tertiary structure, thereby either enhancing or blocking the binding of transcription factors to promoter DNA. Finally, miRNAs regulate gene expression on a post-transcriptional level by inhibiting protein translation through degradation or repression of the mRNA transcript.
In the last decade, epigenetics has become the major focus of numerous investigations, as it provides previously unrecognized mechanistic insights into the etiology of OSCC genesis. This review article provides a comprehensive and an up-to-date description of the current state of epigenetic research, with full reporting of aberrant DNA methylation, histone modifications and effects on miRNA expression in OSCC tumors. This includes all pertinent articles that were accessible through a PubMed search that was performed at the time of compilation of this article in May 2012. The keywords used for literature search included ‘oral cancer methylation’, ‘oral cancer histone acetylation OR oral cancer histone deacetylation OR oral cancer histone modification’ and ‘oral cancer miRNA’.
DNA methylation in oral cancer
Both genome-wide hypomethylation and promoter hypermethylation play a critical role in cancer development. Figure 1 illustrates the process of OSCC tumorigenesis and highlights the molecular alterations that accrue during the progression of this malignant disease. There is an increased recognition that global DNA hypomethylation contributes to tumorigenesis by multiple potential mechanisms. First, through the reduction of methylation at DNA repetitive elements (e.g., LINE-1, Alu sequences), which are distributed throughout the human genome. Demethylation at these methylated repeat elements results in enhanced chromosomal instability. Second, by accidental demethylation of some of the evolutionarily conserved and methylation-silenced promoter regions of various endoparasitic elements and proto-oncogenes, which can lead to their reactivation and contribute towards accelerated carcinogenesis. Third, DNA methylation alterations may possibly contribute to oral carcinogenesis by loss of imprinting. In such a scenario, hypomethylation may result in reactivation of naturally methylated silent, imprinted bialleles and alter gene expression. This mechanism has been discussed in numerous different tumor entities including head and neck squamous cell carcinoma; however, it has not been investigated in OSCC as yet.
Figure 1. Progression of oral squamous cell carcinoma.

While oral squamous cell carcinoma progresses from one stage to the next, the oral tissue is marked by distinct molecular alterations during each step. Risk factors for oral cancers, such as alcohol consumption or tobacco abuse, lead to a series of genetic and epigenetic alterations in the oral tissues, which eventually may progress to advanced oral cancer. This figure highlights some of the specific genetic and aberrant methylation alterations that occur during oral squamous cell carcinoma progression.
On the other hand, aberrant promoter hypermethylation is another key player in OSCC tumorigenesis. Aberrant methylation in this instance occurs at the CpG-rich sequences (or CpG islands) present within the promoter regions of many tumor suppressor genes. Hypermethylation of promoter CpG dinucleotides results in a closed chromatin configuration (or heterochromatin), which blocks access for transcription factors to bind to the promoter region of tumor suppressor genes, resulting in their transcriptional silencing. This process of aberrant hypermethylation of 5-methyl-cytosines at CpG dinucleotides is catalyzed by DNA methyltransferases (DNMTs). Given the growing awareness for the aberrant methylation in human cancer, DNMT inhibitors, aimed at reactivating potentially methylation-silenced tumor suppressor genes, are now being explored as potential treatment choices for OSCC.
Unequivocal evidence indicates that DNA methylation is closely related to OSCC tumorigenesis. Analysis of DNA extracted from OSCC tissues and oral premalignant lesions (OPLs) found they exhibit more frequent and higher levels of DNA methylation compared with healthy or corresponding normal tissue from neoplastic tissues [2]. The use of tobacco, which is a major risk factor for the development of OSCC, has been linked to nonspecific global hypomethylation [3,4]. In contrast to smokers, patients who drink heavily have an increased risk for CpG hypermethylation of multiple OSCC-related genes. Chronic inflammation of the oral mucosa is another risk factor that can potentially modify the methylation status of various genes in OSCC tumors [5]. The occurrence of multiple CpG methylation sites in a panel of tumor-related genes in OSCC was highly associated with cancer stage and may correlate with lymph node metastasis [6]. Abundant investigations have focused on gene-specific aberrant DNA methylation in OSCC tumors. This systematic review covers all investigations on DNA methylation in oral cancer as displayed in Table 1 (for a more comprehensive list of aberrantly methylated genes, please refer to Supplementary Table 1; see online at www.futuremedicine.com/doi/full/10.2217/fon.12.138). To date, most oral cancer-related publications focused on CpG methylation of APC, Survivin, E-cadherin, MGMT, MLH1, p14ARF, p15INK4B, p16INK4A, RARβ and RASSF genes. Hence, these genes were selected for a more detailed discussion below.
Table 1.
Key genes frequently hypermethylated in oral cancer†.
| Gene | Locus | Function | Methylation frequency (%), n/N | Ref. | ||
|---|---|---|---|---|---|---|
| Normal | Adjacent tumor tissue | OSCC | ||||
| APC | 5q21 | WNT signaling | 6 (347) | 13 (6/47) | [6] | |
| BIRC5/SURVIVIN | 17q25 | Cell cycle progression | 0 | [9] | ||
| CACNA1G | 17q22 | Voltage-gated ion channel activity | 0 (0/2) | 0 (0/96) | [107] | |
| CCNA1/CYCLIN A | 13q12.3-q13 | Cell cycle regulation | 0 (0/9) | 53 (42/78) | [17] | |
| CDH1/E-CADHERIN | 16q22.1 | Cell adhesion | 13 (6/47) | 42 (20/47) | [6] | |
| CHFR | 12q24.33 | Cell cycle regulation | 0 (0/18) | 8 (1/13) | 46 (6/13) | [108] |
| DAPK | 9q34.1 | Apoptosis | 0 (0/20) | 60 (36/60) | 68 (41/60) | [109] |
| DCC | 18q21.1 | Transcription coactivator | 0 (0/30) | 10 (5/48) | 59 (54/92) | [36] |
| EDNRB | 13q22 | Receptor activity | 0 (0/30) | 10 (5/48) | 73 (67/92) | [36] |
| EPCAM | 2p21 | Calcium-independent cell adhesion molecule | 51 (37/72) | [110] | ||
| FHIT | 3p14.2 | Cell cycle progression | 0 (0/13) | 28 (8/29) | [111] | |
| GSTP1 | 11q13 | Glutathione transferase activity | 0 (0/20) | 0 (0/60) | 0 (0/60) | [109] |
| KIF1A | 2q37.3 | Axon transport | 0 (0/30) | 10 (5/48) | 72 (66/92) | [36] |
| MGMT | 10q26.3 | DNA damage response | 0 (0/20) | 27 (16/60) | 52 (31/60) | [109] |
| MINT1 | 9q13-q21 | Movement, exocytosis and adhesion | 0 (0/2) | 23 (22/96) | [107] | |
| MINT2 | 15q11-q12 | Protein binding and exocytosis | 0 (0/2) | 8 (8/96) | [107] | |
| MINT27 | 1p36 | Transcriptional regulator | 0 (0/2) | 16 (15/96) | [107] | |
| MINT31 | 1p36 | Transcriptional regulator | 0 (0/2) | 15 (14/96) | [107] | |
| MLH1 | 3p21.3 | DNA damage response | 0 (0/200) | 76 (38/50) | [25] | |
| MSH2 | 2p21 | DNA damage response and mismatch repair | 50 (14/28) | [24] | ||
| Notch1 | 9q34.3 | Transcription factor and receptor activity | 6 (2/34) | [112] | ||
| p14ARF | 9p21 | Cell cycle regulation | 0 (0/2) | 14 (13/96) | [107] | |
| p15INK4B/CDKN2B | 9p21 | Cell cycle regulation | 0 (0/2) | 0 (0/25) | 23 (12/51) | [26] |
| p16INK4A/CDKN2A | 9p21 | Cell cycle regulation | 0 (0/20) | 50 (30/60) | 67 (40/60) | [109] |
| p53 | 17p13.1 | Cell cycle regulation | 4 (2/48) | [30] | ||
| RARβ | 3p24.2 | Transcription factor and receptor activity | 62 (13/21) | 73 (58/80) | [17] | |
| RASSF1 | 3p21.3 | Cell cycle progression | 13 (6/47) | 38 (18/47) | [6] | |
| RASSF2 | 20p13 | Cell cycle progression | 28 (134/482) | [44] | ||
| RECK | 9p13.3 | Protein binding and peptidase inhibitor activity | 0 (0/12) | 30 (6/20) | 55 (11/20) | [113] |
| RUNX3 | 1p36 | WNT signaling | 0 (0/10) | 53 (16/30) | 70 (21/30) | [114] |
| SFRP1 | 8p11.21 | Protein binding | 24 (10/42) | [115] | ||
| SFRP2 | 4q31.3 | Protein binding | 36 (16/44) | [115] | ||
| SFRP5 | 10q24 | Protein binding | 16 (7/43) | [115] | ||
| VHL | 3p25.3 | Transcription factor binding | 0 (0/48) | [30] | ||
| WIF1 | 12q14.3 | WNT signaling | 9 (4/47) | 42 (20/47) | [6] | |
For a more comprehensive list, please refer to Supplementary Table 1.
OSCC: Oral squamous cell carcinoma.
Adenomatous polyposis coli
APC (adenomatous polyposis coli) is a tumor suppressor gene that indirectly inhibits cell proliferation through the WNT-1 (wingless) signaling pathway in human cancer cells. APC gene methylation and its associated gene silencing occur early in the development of oral carcinogenesis, by allowing increased β-catenin target gene expression, which results in increased cell division [7]. Hypermethylation of the APC promoter was detected in 15% of OSCC tumor tissues and 25% of OSCC cell lines [7,8]. Thus, hypermethylation of the APC promoter may be an important step in the dysregulation of the WNT signaling pathway, leading to an increase in cell proliferation during carcinogenesis.
Survivin/BIRC5
The function of Survivin, or BIRC5 (baculo-viral inhibitor of apoptosis repeat-containing protein 5), is to increase tumor cell numbers by promoting cell proliferation and preventing cell apoptosis. There is no evidence for hypermethylation of Survivin in OSCC tissues [9]. This oncogene is ordinarily methylated (silenced) in normal tissues, but is frequently upregulated in OSCC due to its hypomethylation. Thus, over-expression of Survivin due to hypomethylation leads to an increase in cell proliferation with a concomitant decrease in cell death promoting carcinogenesis. In one study, increased Survivin expression was associated with a more aggressive and invasive tumor phenotype [10]. Using the hamster buccal pouch mucosa as an experimental model for oral carcinogenesis, Chen et al. observed that mineral oil-treated animals had normal Survivin methylated alleles, while the carcinogen 7,12-dimethylbenz[a]anthracene (DMBA)-treated animals had buccal pouch carcinomas with a hypomethylated Survivin allele [11,12]. The results of these studies indicate that hypomethylation of the Survivin gene appears to be an important step in the process of OSCC carcinogenesis.
CDH1/E-cadherin
Hypermethylation of the CDH1 (cadherin 1) gene, which encodes the adhesion protein E-cadherin, is highly associated with OSCC and may be an early event in oral carcinogenesis [13]. CDH1 promoter hypermethylation in OSCC tumors is associated with invasive tumor behavior and a worse prognosis [14,15]. Interestingly, hypermethylation and loss of heterozygosity of the APC gene have also been linked with a change in the cytoplasmic expression pattern of E-cadherin [8]. However, results of CDH1 methylation studies have been inconsistent. Several studies reported the frequency of CDH1 methylation in OSCC tissues as ranging from as little as 17% to as high as 85% [6,14,16,17]. Basaloid cells of dysplastic lesions were reported to exhibit increased CDH1 hypermethylation [18], while two studies did not detect any significant difference in methylation status between OSCC and healthy control tissues [16,17]. The variability in results of these studies indicates that CDH1 methylation may not serve as a good marker for OSCC.
O-6-methylguanine-DNA methyltransferase
The DNA repair gene MGMT (O-6-methylguanine-DNA methyltransferase) is important in preventing carcinogenesis, since it removes mutagenic adducts from O(6)-alkyl-guanine in DNA. Silencing MGMT via methylation is thought to be an early event in carcinogenesis [19], as 75% of OSCC tissues demonstrate MGMT gene silencing via hypermethylation of its promoter region [20]. It has been postulated that CpG methylation may be only one way to silence MGMT expression, as additional factors may also modulate its gene expression [21]. For example, betel quid chewing is a known risk factor for development of OSCC, and patients who had chewed betel quid demonstrated a lack of MGMT expression [20]. Hypermethylation of the MGMT promoter is associated with poorer survival in patients with OSCC [22]. Multiple independent studies evaluated OSCC tissues and found that 12–74% had MGMT methylation [6,16,19,22,23]. Based upon these studies, it is apparent that methylation of the MGMT gene in OSCC tumors may be useful both as a diagnostic tool, and possibly as a predictor of patient survival.
mutL homolog 1
Owing to the significant role of the DNA mismatch repair gene MLH1 (mutL homolog 1) in prevention of the accumulation of mutations in DNA, it is not surprising that it is a frequent target of epigenetic modifications in many human cancers. Hypermethylation of its 5′-promoter region is believed to silence MLH1 gene expression and, hence, downregulate protein expression. MLH1 methylation may be associated with a higher risk for development of oral malignancies [24], and in a study of MLH1 methylation status and OSCC, 76% of OSCC tissues showed MLH1 promoter methylation compared with 0% of the control tissues [25]. Interestingly, most of the MLH1-methylated promoters in OSCC samples corresponded to early clinical stages, with only a few late-stage OSCCs demonstrating MLH1 promoter methylation [25]. This observation of a lack of CpG methylation of MLH1 in healthy tissue or tumor-adjacent normal tissue has been subsequently validated in other studies [25,26]. Overall, methylation of MLH1 in OSCC tissue is observed in up to 76% of the examined cases [24–26], indicating it may serve as a biomarker for the early presence of OSCC.
p14ARF
p14ARF is a tumor-suppressor gene that not only works to control cell proliferation and division, but was also recently discovered to regulate tumor-induced angiogenesis [27]. Hypermethylation of p14ARFresults in loss of p53 function and deactivation of p21-induced cell proliferation. p14ARF hypermethylation, a rather late event in carcinogenesis, is associated with increased tumor size and tumor stage, and nodal metastasis [23]. It has been shown that p14ARF hypermethylation in late-stage tumors is associated with a lower recurrence rate and a better clinical outcome compared with patients with tumors that were not p14ARF-hypermethylated [28]. While neither healthy tissue nor tumor-adjacent tissues exhibited any signs of p14ARF methylation, a small proportion (4%) of the OPLs demonstrated p14ARF hypermethylation [29]. In studies of OSCC tumors, 14 –44% of the tumors had hypermethylation of the p14ARF promoter [23,28]. In a study focusing on betel quid-related OSCC, p14ARF hypermethylation was frequently detected in OPLs, suggesting it could serve as a prognostic marker for early detection of betel quid-associated OSCC [29]. The fact that the majority of the hypermethylation of p14ARF promoters was observed in dysplasia or early stages of OSCC may denote it is a transient early event in the process of carcinogenesis in OSCC.
p15INK4B
p15INK4B is a tumor suppressor gene that acts to inhibit cell growth by preventing cell cycle progression during the G1 phase. Hypermethylation and gene inactivation of the p15INK4B checkpoint control gene are commonly observed in OPLs and OSCC [29]. While healthy and tumor-adjacent tissues did not typically exhibit signs of methylation for p15INK4B [29], 9–28% of the examined OSCC tissues revealed promoter hypermethylation of this gene [30]. Taken together, these reports indicate that aberrant methylation of the p15INK4B gene may serve as a marker for OSCC.
p16INK4A
The product of the p16INK4A gene is another cell cycle inhibitor, and the p16INK4A gene was one of the first genes investigated for the role of methylation in oral cancer. p16INK4A gene expression is highly regulated by hypermethylation of its promoter, as well as heterozygous deletion of its chromosomal locus [31]. Hypermethylation of the p16INK4A gene is frequently detected in corresponding normal mucosa of OSCC tissues, which is thought to contribute to the risk of the ‘field defect’ concept during the early stages of OSCC [19]. In line with these results, three individual studies showed that 20–58% of the OPLs have methylated p16INK4A promoters [32–34], and the risk for progression to OSCC was generally greater in patients with p16INK4A-methylated OPLs [32]. p16INK4A hypermethylation is associated with increased tumor size and stage (stages III and IV), nodal metastasis, increased risk for disease recurrence and a poor prognosis [13,23,28]. When OSCC tissues were tested for the presence of p16INK4A methylation, 12–88% of the OSCC cases revealed evidence for aberrant methylation of this tumor suppressor gene [6,19,22,23,28,35–37]. Besides the biological variation in p16INK4A methylation among various studies, these results are also dependent upon samples and methodology used in different laboratories [38,39]. Two animal studies examined methylation of p16INK4A in OSCC versus normal tissues. In the first study, a hamster buccal pouch mucosa experiment demonstrated aberrant p16INK4A promoter methylation in 26% of all DMBA-treated animals [40]. In another study using the rat tongue model to study dysplasia induced by 4-nitroquinoline 1-oxide, it was noted that p16INK4A methylation increased with the degree of dysplasia, and this observation was inversely correlated with the expression of this gene in dysplastic tissues [41]. These results suggest that p16INK4A promoter methylation may serve as a good prognostic indicator for the development of OSCC.
Retinoic acid receptor β
RARβ (retinoic acid receptor β) is a member of the thyroid-steroid hormone receptor superfamily of nuclear transcription factors associated with cell growth and differentiation. There has been much interest in the role of RARβ promoter methylation due to the use of retinoids as chemopreventive agents in OSCC; however, the role of RARβ in OSCC remains unclear. A high frequency of RARβ promoter methylation (73%) has been observed in an OSCC tissue pyrosequencing study, although 62% of aberrant methylation has been noted in adjacent normal tissues [17]. In an examination of OPLs, over half (53%) of the cases had methylated RARβ promoters [42]. Based upon these results, it is unclear whether methylation of RARβ is a very early step in OSCC carcinogenesis, or whether it simply represents a generalized aberration across all oral mucosal tissues.
Ras-association domain family 2
There are ten RASSF (Ras-association domain family) gene members, which have a variety of important cell functions. RASSF proteins function as tumor suppressors and regulators of the cell cycle, apoptosis, and microtubule formation. In recent years, it has been recognized that gene expression of several members of this family can be controlled by their aberrant methylation. In a study of RASSF2, 39% of OSCC tissues were methylated in at least one RASSF2 gene [43], while another investigation of OSCC tissues reported 22% methylation of the RASSF1A gene and 28% for the RASSF2A gene [44]. Interestingly, simultaneous methylation of both the RASSF1A and RASSF2A genes was associated with a poor disease-free survival [44]. Other trials have reproduced these results and demonstrated up to 12–38% methylation of the RASSF1 gene in OSCC tissues [6,22,44]. In one study of betel quid-associated OSCC, 93% of the OSCC tissues demonstrated methylation of the RASSF1 gene, suggesting a high association of RASSF1 methylation in tumors due to betel nut chewing [45]. Thus, epigenetic silencing of the RASSF family of genes via methylation may play an important role, not only in the induction, but also in the outcome, of patients with OSCC.
DNA methylation alterations as diagnostic biomarkers in OSCC
Accumulating evidence suggests that early detection of OSCC is perhaps one of the most promising approaches for the treatment of this deadly malignancy. Tumor tissues certainly have a very high diagnostic value; however, patient’s compliance for additional biopsies is rather low and the surgical method is quite invasive for frequent screening. By contrast, saliva provides an unlimited, easily available specimen resource for the early detection of OSCC in high-risk patients, and is suitable for monitoring tumor recurrence. Thus, screening a set of methylated cancer-related genes in the saliva or oral rinses of patients can provide an attractive panel of biomarkers for the early detection of OSCC. Accordingly, in a quest to identify and develop robust and preferably noninvasive biomarkers, Viet and Schmidt performed a methylation array analysis for detection of 807 cancer-associated genes comparing the saliva of healthy subjects with that of OSCC patients, preoperative and postoperative saliva and OSCC tissue of cancer patients [46]. This elegant study identified 34 genes that were highly methylated in tumor tissues and preoperative saliva samples, while there was no evidence for methylation of these CpG loci in the postoperative saliva of OSCC patients and healthy subjects [46]. With the help of an array-based analysis interrogating 27,578 CpG sites, Guerrero-Preston et al. tested OSCC saliva samples for their methylation status. Compared with saliva of healthy subjects, the saliva of OSCC patients showed significant differences in HOXA9 and NID2 methylation. The genes HOXA9, NID2 or a combination of HOXA9 and NID2 have therefore been suggested as biomarkers due to high sensitivity of 75, 87 and 50% and high specificity of 53, 21 and 90%, respectively [47]. Another clinical oral rinse study by Nagata et al. investigated the methylation status of a panel of tumor-related genes to develop a noninvasive method of detection for OSCC. In a comparison of oral rinse samples from OSCC and control patients, aberrant methylation was detected in a total of 13 genes; eight of these genes demonstrated higher levels of methylation in OSCC versus control washings. Upon further evaluation, a combination of ECAD, TMEFF2 and MGMT methylation offered a very high sensitivity (97%) and specificity (92%) for detecting OSCC[48].
The LINE-1 (long interspersed nuclear element) gene is used as a surrogate marker for global methylation levels in human cancers. Even though the sequence of each LINE element is homologous, LINE-1 methylation levels may differ at various loci. In OSCC cell lines, 28–39% of CpG sites occur within LINE-1 repeat elements [5]. Analyses of oral rinses and the corresponding OSCC tissues of cancer patients both revealed LINE-1 hypomethylation [49]. Compared with oral tissue from healthy subjects, OSCC tissues demonstrated LINE-1 hypomethylation independent of tumor stage, tumor site, histological grading or risk factors. In addition to hypermethylated gene loci, LINE-1 hypomethylation may therefore be an excellent biomarker for use in noninvasive early OSCC detection [49].
Several studies have suggested p16INK4A may be used as a biomarker to predict malignant transformation, as hypermethylation of this gene is associated with metastasis and overall poor survival in OSCC patients [37]. While healthy normal tissues do not show any signs of p16INK4A hypermethylation, normal tissues from smokers without cancer often demonstrate methylation of this gene [50], suggesting that p16INK4A hypermethylation may be a very early event in OSCC development [50]. In patients with leukoplakia, an oral precancerous condition, methylation of p16INK4A was observed in the oral rinses of 44% of patients, and hypermethylation increased with stage of the disease [51]. In one longitudinal clinical study investigating the prognostic usefulness of methylation biomarkers in the malignant transformation of OPL, hypermethylation of the p16INK4A gene correlated with malignant progression [52]. In addition to predicting malignant transformation, p16INK4A hypermethylation was shown to predict local OSCC recurrence [53]. To identify a patient’s individual risk for OSCC recurrence, it may be useful to evaluate more than just the p16INK4A methylation status of their tumor tissue. One study assessed the effectiveness of testing for p16INK4A promoter hypermethylation in the tumor tissues and in the surgical margins. Promoter hypermethylation of p16INK4A was observed in 87% of the tongue squamous cell carcinoma (TSCC) tissues. But, most importantly, patients with histologically tumor-free and p16INK4A-hypermethylated surgical margins had a sixfold increased risk for local recurrence compared with patients with histologically tumor-free and negative margins [54]. In another report, p16INK4A promoter hypermethylation was detected in 65% of the OSCC tissues, 55% of which also revealed evidence for its methylation in the matching serum DNA. Interestingly, 75% of the patients with p16INK4A-hypermethylation both in tumor tissue and serum were affected by tumor recurrence [55]. Thus, monitoring patients with precancerous lesions or patients after OSCC treatment via p16INK4A hypermethylation may be a valuable tool in identifying malignancy or malignant recurrence in OSCC patients.
In addition to histological examination of the surgical specimens, the detection of promoter hypermethylation in cancer-free surgical margins or in oral rinses/scrapings may be a simple and noninvasive approach for predicting future OSCC recurrence or malignant transformation. For instance, hypermethylation of genes such as p16INK4A and MGMT is significantly associated with poor overall survival, which can facilitate establishment of individualized treatment plans in OSCC patients [6]. In summary, the discovery and development of epigenetic biomarkers provides us with not only more attractive and practically feasible cancer screening modalities, but also equips us with molecular tools that can guide personalized treatment decision-making in the clinics in the not so distant future.
DNA methylation inhibitors as therapeutic agents in OSCC
As mentioned previously, CpG methylation alterations may also provide an avenue for establishing epigenetic therapeutic regimens for this malignancy. For instance, treatment of OSCC cells (HSC-3) with the DNMT inhibitor zebularine alone resulted in tumor growth inhibition, as evidenced by decreased cell growth and reduction in the number of cells accumulated in the G2/M cell cycle phase [56]. While the molecular mechanisms for these observations remain unclear at this time, it was intriguing to observe that when zebularine was used as an adjuvant to cisplatin or 5-fluoro-uracil chemotherapy, it greatly enhanced the apoptotic activity of the cisplatin treatment, but it decreased the efficacy of treatment with 5-fluorouracil[57].
MGMT is another gene involved in OSCC whose expression is modulated via abnormal methylation. OSCC cell lines with low levels of MGMT methylation responded with a greater antiproliferative response to 5-fluorouracil chemotherapy, compared with cell lines with high levels of MGMT methylation [58]. However, when cells were pretreated with O6-benzylguanine (O6-BG), a potent inhibitor of MGMT, the anti-proliferative effect of 5-fluorouracil was much greater in these nonresponsive cells in comparison with mock-treated cell lines [58]. Therefore, one must be cautious when considering combination chemotherapies and inhibitors for use in the treatment of OSCC.
Green tea extracts are thought to prevent malignant transformation of precancerous lesions to OSCC. Overall, the clinical response rate tended to be better in patients treated with green tea extracts versus placebo, and there was a significantly greater achievable response rate with the higher doses of extracts (750 or 1000 mg/m2) compared with placebo groups [59]. In studies with OSCC cell lines, green tea extracts suppressed cancer cell invasion by reversing hypermethylation of the RECK gene, resulting in its enhanced mRNA expression [60]. In summary, data from these preliminary experiments clearly favor the enthusiasm and excitement for the development of safe and effective DNMT inhibitors that can be used alone or in conjunction with other chemotherapeutic regimens for better management of patients with oral cancer.
Histone modifications in oral cancer
Post-translational modifications of histones are frequently observed in oral cancer. These epigenetic alterations occur primarily at the N-terminal tails within each of the four histone complexes (H3, H4, H2A and H2B). Various modifications include methylation, acetylation, ADP-ribosylation, phosphorylation, ubiquitination and sumoylation of specific residues within these histone tails. These processes are generally reversible and modify the tertiary DNA structure. Alterations at histone tails have a direct impact on chromatin condensation, which can exist either in a heterochromatin or euchromatin configuration. The heterochromatin form is frequently observed in neoplastic cells, during which the DNA is coiled up into a closed, compacted chromatin configuration, making it impossible for transcription factors to access DNA and allow active gene transcription. By contrast, euchromatin configuration is a theme in healthy, normal cells, in which DNA is present in an open configuration, allowing transcription factors easier access to DNA. Histone modifications are tightly associated with DNA methylation, particularly in the context of tumor suppressor gene silencing, leading to an enhanced rate of tumorigenesis.
Methylation of specific lysine residues within histone tails is associated with either activation or transcriptional repression of gene transcription. However, this effect is primarily dependent upon the position of the specific lysine residue being methylated, as well as the number of sites methylated [61]. Histones can be methylated at only one or all of the six lysine residues within the histone tails H3 (K4, K9, K27, K36 and K79) and H4 (K20). Histone methylation at H3K9, H3K27 and/or H4K20 is generally associated with repressive histone marks that lead to transcriptional silencing of the corresponding gene [62], while methylation of H3K4, H3K36 and H3K79 is associated with activating histone marks that allow active transcription of the involved gene [63]. Histone methylation can occur in a mono- (me1), di- (me2) or tri-methylated (me3) form on various lysine residues.
The processes of DNA methylation and histone methylation are tightly coregulated and collectively play an important role in carcinogenesis. In one study, the patterns of DNA and histone methylation were positively correlated in normal, OPL and OSCC tissues [64]. In another study, the pattern of H3K4 histone methylation was associated with OSCC malignancy. The frequency of transcriptionally inactive, H3K4me2 histones was greater in OSCC compared with normal tissues, while the converse was true for activating H3K4me3 modifications [65]. A similar pattern of histone methylation was found in OPLs, such as leukoplakia, compared with normal tissues. Leukoplakia tissues presented themselves more like OSCC tissues with regards to their pattern of histone methylation. The results of these investigations help reinforce the notion that in many instances, leukoplakia should be considered a premalignant condition for the development of OSCC and treated accordingly. Thus, changes in H3K4 histone methylation may occur as an early event during OSCC carcinogenesis [65].
Hypomethylation of H3K9, rather than hypermethylation, is associated with transcriptional activation due to the establishment of loosely configured euchromatin. When an OSCC cell line was treated with ornithine decarboxylase antizyme-1, it induced hypomethylation of H3K9me2, as well as changes in the levels of a number of DMNTs. These results indicate that hypomethylation of histones may be a potent means for accelerating OSCC tumorigenesis, which causes enhanced DNA instability, loss of cell cycle control and decreased ability for DNA repair [62].
Similar to histone methylation, histone deacetylation, catalyzed by various histone deacetylases (HDACs), plays a significant role during oral carcinogenesis. It has been demonstrated that HDAC2 improves the stability of the HIF-1α protein, which may lead to enhanced invasion and migration in OSCC [66]. Likewise, expression of HDAC6 was upregulated in OSCC and was found to be stage-specific; the higher the stage, the greater the activity [67]. The authors concluded that HDAC6 expression may be important in determining tumor aggressiveness in oral cancers. In addition to deacetylating histones, HDAC6 was revealed to be capable of deacetylating α-tubulin as well, thus promoting microtubule-dependent cell motility, a process that may be important to the development of metastasis [68].
Interestingly, Arif et al. discovered that histone H3, primarily H3K14, is hyperacetylated in OSCC [69]. These investigators discovered that in the KB oral cancer cell line, increased H3 acetylation was nitric oxide-dependent. Since nitric oxide is produced during the inflammatory process, which has been linked to the initiation and development of OSCC, this is a particularly important finding[69]. Furthermore, hydrazinocurcumin, a potential therapeutic drug, was able to inhibit histone acetyltransferase activity and reduce oral tumor growth in a xenograft mouse model [69]. The results of these experiments demonstrate an important mechanistic role for hyperacetylation of H3K14 in OSCC pathogenesis.
The poly(ADP-ribose) polymerase (PARP) family of enzymes is responsible for the post-translational covalent transfer of ADP-ribose to proteins, as well as formation of polymers of poly(ADP-ribose). The activity of PARP-1, DNA synthesis rates and the degree of ADP-ribosylation is greatest in actively proliferating OSCC [70], a finding that is ascribed to histones rather than nonhistone chromosomal proteins. The addition of poly(ADP-ribose) to histones loosens the chromatin structure. This nucleosome modification facilitates DNA repair through large protein complexes such as chromatin assembly factor (CAF)-1. CAF-1 is a molecular chaperone consisting of three subunits (p48, p60 and p150). Its role is the integration of H3K56-acetylated histones into the chromatin. The nuclear expression of CAF-1/p60 is upregulated in multiple tumor entities including OSCC. Worst prognosis in terms of survival and metastasizing behavior may be predicted in PARP-1high and CAF-1/p60high OSCC tissues [71]. Concurrent CAF-1-mediated deregulation of cell proliferation as well as DNA repair also occurs in aggressive TSCC. An immunohistochemical evaluation reported that CAF-1/p60 is commonly expressed in TSCC tissues, whereas CAF-1/p150 may be downregulated. Both parameters are associated with poor clinical outcome and worse prognosis [72]. Taken together, DNA methylation, acetylation and/or poly(ADP)-ribosylation may all be important factors in the development of OSCC.
Histone modifications as diagnostic biomarkers in OSCC
In contrast with advancements made with regards to identification of DNA methylation alterations in OSCC, noninvasive measurement of histone modifications still remains a concept in its infancy. Overexpression of HDAC2 in OSCC tissue was identified to be associated with advanced tumor stage, tumor size, metastasis and significantly shorter overall survival, and may therefore make a good biomarker [73]. A very recent pilot study determined the feasibility of using DNA collected from oral rinse samples to perform chromatin immunoprecipitation assays to analyze the interaction of DNA methylation, histone modifications and gene expression. The authors provided very encouraging data showing the epigenetic control of p16INK4A/CDKN2A gene could be determined in the DNA from oral rinses. The positive results obtained in this study have provided a springboard to embark upon future studies using oral rinse samples to investigate patterns of histone modifications in OSCC[74]. In 1993, Das investigated ADP-ribosylation in OSCC tissues from two different tumor stages, along with the analysis of normal adjacent oral tissues. The activity of poly(ADP-ribose) synthetase was reported to increase during oral carcinogenesis. With increasing malignancy, a progressive increase in ADP-ribosylation of histones was noticed in purified nuclei of OSCC tissue, suggesting that ADP-ribosylation may be a potential marker for OSCC [70].
HDAC inhibitors as therapeutic agents in OSCC
Based upon the recognition for the role of histone modifications in human cancer, there is growing interest in treating OSCC patients with a variety of epigenetic inhibitors, more specifically using a panel of different inhibitors of HDAC activity. There are 18 assorted HDACs that can be divided into four distinct classes (Class I–IV) based on their sequence identity and function. The various classes of HDAC inhibitors currently available are summarized in Table 2. Interestingly, they have varied affinities for the different classes of HDACs. HDAC inhibitors represent a novel, promising compilation of antineoplastic compounds that inhibit deacetylation of histones and thereby act to promote the uncoiling of chromatin allowing activation of a variety of genes associated with the regulation of cell survival, proliferation, differentiation and apoptosis. HDAC inhibitors may enable the re-expression of silenced tumor suppressor genes in OSCC and thereby reverse the malignant phenotype. When combined with well-established chemotherapeutics, HDAC inhibitors synergistically enhance the efficacy of conventional chemotherapy [75]. HDAC inhibitors fall into the category of prospective therapeutics for a variety of diseases, perhaps most importantly for their application in cancer therapy. In 2006, the US FDA approved the HDAC inhibitor suberoylanilide hydroxamic acid for use as a therapeutic agent for advanced cutaneous T-cell lymphoma. At present, several novel HDAC inhibitors are undergoing clinical trials and may soon be launched, in combination with more traditional chemotherapies, as therapy to treat cancer [76].
Table 2.
Classification of histone deacetylase inhibitors.
| Histone deacetylase inhibitor group | Representative example | Chemical structure |
|---|---|---|
| Hydroxamic acids | Trichostatin A, Suberoylanilidehydroxamic acid |
|
| Cyclic tetrapeptides and depsipeptides | FR901228 |
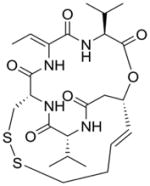
|
| Benzamides | MS-275 |
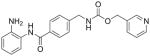
|
| Electrophilic ketones | Trifluoromethyl ketone |
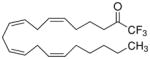
|
| Aliphatic acid compounds | Phenylbutyric acid |
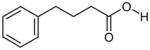
|
Trichostatin A
The hydroxamic acid trichostatin A (TSA) was one of the earliest HDAC inhibitors to be discovered. TSA and other hydroxymates are the only known inhibitors of the classical HDACs (Class I, II and IV). TSA owes its pharmacological effect to zinc ion complexation [77]. TSA was shown to inhibit proliferation in oral cancer cells (YD-10B) by causing cell cycle arrest at the G2/M phase [78]. Additionally, TSA induced apoptosis in these cells by enhancing p21WAF1 expression, decreasing Cyclin B1 and blocking the phosphorylation of Cdc2 [78]. In another study examining OSCC cell lines (HSC-4, Ho-1-N-1 and Ho-1-U-1), similar results were obtained with TSA with regards to inhibiting cell growth and inducing apoptosis in all three OSCC cell lines and inducing expression of p21WAF1 protein expression [79]. Additionally, TSA enhanced the protein expression of p21, cyclin E, cyclin A, Bak and Bax, while it simultaneously inhibited the expression of HDAC1, p53, E2F-1, E2F-4 and hyperphosphorylated Rb [79]. Analogous results were found using TSCC cells (TCA8113), in which TSA inhibited cell growth by inducing a G2/M arrest and induced apoptosis through upregulation of the apoptosis-inducing protein Bax [80]. This study also showed that the NF-κB pathway is involved in these effects, as TSA blocked NF-κB activation, which in turn caused a decrease in the levels of the anti-apoptotic proteins Bcl-2 and Bcl-xL [80]. Even though TSA has these many effects in blocking the proliferation of cancer cells, its use as an anticancer agent is limited due to its high toxicity.
Butyric acid derivatives
The butyric acid derivatives represent another well-investigated category of HDAC inhibitors. Phenylbutyrate, a prominent butyric acid derivative, promotes DNA repair and survival in healthy cells. Phenylbutyrate treatment has been shown to reduce oxidative stress, TNF-α expression and the incidence of severe oral mucositis [81]. As an adjuvant to radiotherapy, it lowered the risk of OSCC and tumor progression in hamster cheek pouches [81]. Sodium butyrate, another butyric acid derivative, inhibited OSCC proliferation and induced G1 and G2/M cell cycle arrest in vitro in multiple human OSCC cell lines, including HSC-3, HSC-4, SCC-1 and SCC-9 [82,83]. In concert with inhibiting cell cycle progression, sodium butyrate induced expression of G1 phase cell cycle regulatory proteins CDK6, p21WAF-1 and p27, while decreasing the expression of the S/G2 phase protein CDK2 as well as phosphorylation of Rb. The HDAC inhibitor sodium butyrate caused Cyclin D1 upregulation and Cyclin B1 and Cyclin E downregulation [83]. However, even though phenylbutyrates and sodium butyrates are generally viewed as rather weak HDAC inhibitors [77], they have been entered into clinical trials for study, particularly for their effects on myelodysplastic disorders.
In addition, other novel HDAC inhibitors have been described in the field of oral cancer. (S)-HDAC42 is a phenylbutyrate-based inhibitor that is potent in suppressing tumor growth in a number of cancers. It acts through histone acetylase-dependent and -independent pathways. Using OSCC tumor cell lines, it was observed to have a more pronounced antiproliferative effect compared with suberoylanilide hydroxamic acid [84].
Romidepsin (FR901228/FK228)
Romidepsin is a naturally occurring depsipeptide that is a potent inhibitor of Class I HDACs [77]. Analogous to butyric acid derivatives, romidepsin induced cell cycle arrest at G1 and G2/M [85]. In vitro studies targeting the role of mapsin, a proposed tumor suppressor gene, discovered that addition of romidepsin to OSCC cells caused a time-dependent re-expression of mapsin transcripts [86,87]. These results suggested that reinduction of mapsin expression after romidepsin treatment may help to explain the recovery of normal biological functions such as blockage of cell invasion and tumor angiogenesis. In another study using OSCC cell lines, it was shown that expression of the cell senescence regulating gene hTERT (telomerase reverse transcriptase) increased after treatment with romidepsin [86,87]. Since mapsin and hTERT both may be important factors in regulating cell mortality and invasion, targeted treatment with the HDAC inhibitor romidepsin to induce their expression may be an important addition to cancer therapy. Besides these, there are currently a whole host of HDAC inhibitors being developed and tested for their antitumor activities [88].
HDAC inhibitors as adjuvant chemotherapeutic agents
Numerous studies have revealed the synergistic behavior of the HDAC inhibitors with well-established chemotherapeutics such as cisplatin and 5-fluorouracil; hence, these agents have been suggested as adjuvants to chemotherapy as well as radiotherapy [75]. The synthetic benzamide HDAC inhibitor MS-275 enhanced the cytotoxic effectiveness as adjuvant to cisplatin in the treatment of OSCC [75,77], while the combination of low-dose cisplatin and suberoylanilide hydroxamic acid displayed a synergistic effect in inducing greater cytotoxicity and apoptosis than cisplatin treatment alone. As an adjuvant to chemotherapy, suberoylanilide hydroxamic acid may enhance OSCC cell sensitivity, demonstrating a much greater effect in the cell cycle fractions from the G0/G1 phase and G1/S phase [75,89,90]. In addition, suberoylanilide hydroxamic acid enhances treatment efficiency in combination with 5-fluorouracil [56]. The results of all of these experiments indicate that HDAC inhibitors have an important use in sensitizing oral cancer cells to the effects of chemo- and radiotherapy.
miRNAs in oral cancer
miRNAs are the most recent entrants into the category of epigenetic gene expression regulators. miRNAs represent small RNA molecules with important regulatory functions, wherein each miRNA has the potential to target multiple mRNAs or gene targets. miRNAs regulate gene expression on a post-transcriptional level, inhibiting protein formation by degradation or repression of translation of the mRNA transcript. As the function and role of miRNAs are still a fairly new field of research, the studies associated with miRNA regulation of OSCC are recent, but increasing rapidly. To date, 1898 unique mature human miRNAs have been identified. An overview of the known OSCC-associated miRNAs is depicted in Figure 2 and Table 3 (for a more detailed list of individual studies on miRNA expression patterns in OSCC please refer to Supplementary Table 2).
Figure 2. Role of miRNAs in the development and progression of oral squamous cell carcinoma.

Two important epigenetic mechanisms support OSCC tumorigenesis: overexpression of oncogenic miRNAs (or oncoMiRs) and underexpression of tumor suppressor miRNAs (or tsMiRs). Listed in this figure are some of the most important miRNAs and their gene targets that have emerged as key regulators of gene expression in oral cancer.
OSCC: Oral squamous cell carcinoma.
Table 3.
Important miRNAs frequently altered in oral cancer†.
| miRNA | Regulation | Malignancy | Sample type | Method | Ref. |
|---|---|---|---|---|---|
| let-7a/7b | Down | OSCC | Cell lines | qRT-PCR | [102] |
| miR-16 | Down | OSCC | Hamster cheek pouch | Microarray, qRT-PCR | [98] |
| miR-17-5p | Up | TSCC | Tissue | qRT-PCR (TaqMan® miRNA assay, 156miRNAs) | [91] |
| miR-21 | Up | Leukoplakia | Tissue | TaqMan® low density arrays, qRT-PCR | [96] |
| miR-24 | Up | TSCC | Tissue | qRT-PCR | [93] |
| miR-26a/b | Down | OSCC | Hamster cheek pouch | Microarray, qRT-PCR | [91,98] |
| miR-29a | Down | OSCC | Hamster cheek pouch | Microarray, qRT-PCR | [98] |
| miR-30a-3p | Up | TSCC | Tissue | qRT-PCR (TaqMan® miRNA assay, 156miRNAs) | [91] |
| miR-31 | Up | OSCC | Plasma | qRT-PCR | [103] |
| miR-34b/c | Up | TSCC | Tissue | qRT-PCR (TaqMan® miRNA assay, 156miRNAs) | [91] |
| miR-99a | Down | TSCC | Tissue | qRT-PCR (TaqMan® miRNA assay, 156miRNAs) | [91] |
| miR-104 | Up | TSCC | Tissue | qRT-PCR (TaqMan® miRNA assay, 156miRNAs) | [91] |
| miR-107 | Down | TSCC | Tissue | qRT-PCR (TaqMan® miRNA assay, 156miRNAs) | [91] |
| miR-124a | Down | OSCC | Hamster cheek pouch | Microarray, qRT-PCR | [98] |
| miR-124b | Up | TSCC | Tissue | qRT-PCR (TaqMan® miRNA assay, 156miRNAs) | [91] |
| miR-125a/b | Down | OSCC | Saliva | qRT-PCR | [98,105] |
| miR-128a | Up | TSCC | Tissue | qRT-PCR (TaqMan® miRNA assay, 156miRNAs) | [91] |
| miR-132 | Up | TSCC | Tissue | qRT-PCR (TaqMan® miRNA assay, 156miRNAs) | [91] |
| miR-133a/b | Down | TSCC | Tissue | qRT-PCR (TaqMan® miRNA assay, 156miRNAs) | [91] |
| miR-134 | Up | TSCC | Tissue | qRT-PCR (TaqMan® miRNA assay, 156miRNAs) | [91] |
| miR-137 | Up | TSCC | Tissue | qRT-PCR (TaqMan® miRNA assay, 156miRNAs) | [91] |
| miR-143 | Down | OSCC | Hamster cheek pouch | Microarray, qRT-PCR | [98] |
| miR-145 | Down | OSCC | Hamster cheek pouch | Microarray, qRT-PCR | [98] |
| miR-148b | Down | OSCC | Hamster cheek pouch | Microarray, qRT-PCR | [98] |
| miR-149 | Down | TSCC | Tissue | qRT-PCR (TaqMan® miRNA assay, 156miRNAs) | [91] |
| miR-155 | Down | OSCC | Hamster cheek pouch | Microarray, qRT-PCR | [98] |
| miR-181b | Up | Leukoplakia | Tissue | TaqMan® low density arrays, qRT-PCR | [96] |
| miR-181c | Up | TSCC | Tissue | qRT-PCR (TaqMan® miRNA assay, 156miRNAs) | [91] |
| miR-194 | Down | TSCC | Tissue | qRT-PCR (TaqMan® miRNA assay, 156miRNAs) | [91] |
| miR-195 | Down | TSCC | Tissue | qRT-PCR (TaqMan® miRNA assay, 156miRNAs) | [91] |
| miR-198 | Up | TSCC | Tissue | qRT-PCR (TaqMan® miRNA assay, 156miRNAs) | [91] |
| miR-200a | Down | OSCC | Saliva | qRT-PCR | [105] |
| miR-200b | Up | OSCC | Hamster cheek pouch | Microarray, qRT-PCR | [98] |
| miR-203 | Down | OSCC | Hamster cheek pouch | Microarray, qRT-PCR | [98] |
| miR-219 | Down | TSCC | Tissue | qRT-PCR (TaqMan® miRNA assay, 156miRNAs) | [91] |
| miR-221 | Up | OSCC | Hamster cheek pouch | Microarray, qRT-PCR | [98] |
| miR-345 | Up | Leukoplakia | Tissue | TaqMan® low density arrays, qRT-PCR | [96] |
| miR-375 | Down | OSCC | Tissue | qRT-PCR (TaqMan® miRNA assay) | [106] |
| miR-762 | Up | OSCC | Hamster cheek pouch | Microarray, qRT-PCR | [98] |
For a more comprehensive list, please refer to Supplementary Table 2.
OSCC: Oral squamous cell carcinoma; qRT-PCR: Quantitative reverse transcriptase PCR; TSCC: Tongue squamous cell carcinoma.
miRNA expression patterns in TSCC
In studies of oral cancer, researchers have looked for specific miRNA signatures or profiles in oral cancer tumors. Wong et al. performed miRNA profiling by examining the expression level of 156 miRNAs in TSCC and compared the expression with normal tissues. Using a threefold expression difference as a cutoff threshold, this group identified 24 miRNAs that were overexpressed in TSCC (miR-17-5p, miR-21, miR-30a-3p, miR-31, miR-34b, miR-34c, miR-104, miR-124a, miR-124b, miR-128a, miR-132, miR-134, miR-137, miR-147, miR-154, miR-155, miR-181c, miR-184, miR-197, miR-198, miR-213, miR-325, miR-338 and miR-372) while 13 miRNAs were underexpressed (miR-26b, miR-99a, miR-100, miR-107, miR-125b, miR-133a, miR-133b, miR-138, miR-139, miR-149, miR-194, miR-195 and miR-219) [91]. The number of miRNAs modulated in TSCC may signify the importance of dysregulated expression of these specific miRNAs in this disease.
A microarray-based miRNA profiling analysis identified that miR-21 is frequently overexpressed in TSCC compared with adjacent normal mucosa, and high miR-21 expression is associated with poor prognosis in TSCC by virtue of inhibiting apoptosis [92]. Likewise, upregulation of miR-24 [93] and miR-184 [94] in TSCC is associated with enhanced proliferation and reduced apoptosis [93]. By contrast, reduced miR-138 expression correlates with enhanced risk for metastasis in TSCC, potentially through regulation of RhoC and ROCK2 Rho GTPases [95]. Our understanding of the functions of these miRNAs may help us better understand their role in TSCC.
miRNA expression patterns in OSCC
Identification of a miRNA signature that could determine the risk for malignant transformation of OPL would be an extremely useful tool for the early detection of OSCC. In a comparison of miRNA profiles from leuoplakia with and without progression, 109 miRNAs were found to be differentially expressed only in progressive leukoplakia and invasive OSCC [96]. However, most interestingly was that increased expression of miR-21, miR-181b and miR-345 was associated with an increased severity of OPL, suggesting these miR-NAs may serve as biomarkers for the early identification of progressive leukoplakias that are at risk for progressing into malignant lesions [96].
While examining the miRNA profiles in 18 OSCC cell lines compared with the immortalized oral keratinocyte cell line RT7, a panel of 148 miRNAs were analyzed [97]. A total of 54 miRNAs (37%) were underexpressed in OSCC cell lines compared with control cells. Interestingly, it was also noted that the down-regulated expression of miR-34b, miR-137, miR-193a and miR-203 was a consequence of CpG hypermethylation; a finding that further reiterated the intimate association between different epigenetic mechanisms in human carcinogenesis. These in vitro results were validated in primary OSCC tissues, wherein downregulation of the tumor suppressors miR-137 and miR-193a via epigenetic silencing was a more frequent occurrence than the reduced expression of miR-34b and miR-203 [97]. Thus, the interrelationship between miRNA expression and other epigenetic mechanisms have the potential to be extremely important in the process of carcinogenesis.
In an OSCC animal model, six Syrian hamsters were treated with the carcinogen DMBA, followed by miRNA microarray analysis to investigate the expression profiles of miRNAs during the development of oral carcinogenesis. While five miRNAs (miR-21, miR-200b, miR-221, miR-338 and miR-762) were over-expressed, 12 miR-NAs (miR-16, miR-26a, miR-29a, miR-124a, miR-125b, miR-126-5p, miR-143, miR-145, miR-148b, miR-155, miR-199a and miR-203) were downregulated due to this oncogenic stimulus [98]. Overexpression of miR-21 has subsequently been validated in other studies, and it has been proposed that miR-21 downregulates the expression of the tumor suppressor gene PDCD4, which is associated with nodal metastasis and invasive potential in OSCC [99].
While the list of miRNAs that are differentially expressed in oral cancers is continuously evolving, it has been shown that miR-211, which is mapped to 15q13, is frequently overexpressed, and is associated with tumor progression, nodal metastasis, vascular invasion and poor prognosis of OSCC [100]. On the other hand, miR-133a and miR-133b are downregulated in OSCC. This promotes oral carcinogenesis through the pathway of inducing proliferation and inhibition of apoptosis, by permitting overexpression of pyruvate kinase type M2, a potential oncogene in solid tumors [101]. The mechanistic details underlying differential regulation of miRNAs remain poorly understood. In a recent study, it was shown that dicer, an RNase III endo-nuclease responsible for miRNA maturation, exhibited aberrantly high protein expression in OSCC cell lines and tissues. Other reports indicate the let-7 family of miRNAs might regulate the expression of dicer post-transcriptionally in oral carcinogenesis [102].
These data clearly support the idea that aberrant expression of miRNAs may have a causal link with OSCC pathogenesis and, once these expression patterns are more firmly established, some of these may serve as important diagnostic, prognostic and predictive biomarkers in oral carcinogenesis.
miRNA alterations as diagnostic biomarkers in OSCC
Selected studies have thus far explored the potential of miRNAs as biomarkers for the early detection of oral neoplasias. Given the abundance of miRNA alterations in human cancer and their stability in a wide variety of bodily fluids and tissues, it is not surprising that miRNA detection may become a valuable tool for OSCC diagnostics in tumor tissues, plasma and/or saliva obtained from OSCC patients.
Plasma levels of miR-31 were suggested to serve as tumor biomarkers for early OSCC detection and indicators for recurrence [103]. Plasma miR-31 was not only increased in OSCC patients compared with healthy controls, but its levels also significantly dropped in patients following surgical removal of the tumor [103]. On similar lines, miR-184 was frequently overexpressed in pre-operative plasma samples from TSCC patients, while the postoperative samples demonstrated a significant drop after successful removal of the primary tumor [91,94]; a concept highlighting the usefulness of this miRNA as an indicator of tumor recurrence. Furthermore, increased expression of miR-10b was observed in plasma samples from patients with OSCC and premalignant OPLs compared with normal subjects, suggesting its usefulness as an early diagnostic tool for identification of OSCC[104].
While analyzing saliva samples from OSCC patients, Park et al. found that miR-125a and miR-200a are significantly underexpressed in OSCC patients compared with healthy controls [105]. Oral rinses obtained from OSCC patients and healthy subjects displayed aberrant miR-200c and miR-141 methylation and aberrant miR-375 and miR-200a expression [106]. Collectively, there is accumulating evidence that highlights not merely the feasibility, but potential superiority of miRNA-based biomarkers for the earlier diagnosis and prognosis of patients with oral cancers.
Conclusion & future perspective
Epigenetic mechanisms have clearly emerged as important contributors in the pathogenesis of various human cancers. More importantly, since these alterations occur frequently in the process, and are potentially reversible, this makes them ideal for exploitation as disease biomarkers as well as therapeutic targets in human cancer. In terms of oral cancer, the data thus far have been from somewhat preliminary experiments; nonetheless, the results are exciting, highlighting the involvement of epigenetic mechanisms in every stage of oral carcinogenesis. Given the promising results, future research should concentrate on a more focused approach that can reap the benefits of existing data for the earlier diagnosis of OSCC. Additionally, it can better appreciate the possible role of epigenetic drugs as possible adjuvants to chemo- and radiotherapy. It is just a matter of time before most of these approaches are put into everyday clinical practice, which will undoubtedly help reduce OSCC morbidity and mortality in the not-so-distant future.
Supplementary Material
Executive summary.
Epigenetics of oral cancer
Regulation of gene expression may be modified by three epigenetic mechanisms: aberrant DNA methylation, histone modifications and expression of miRNAs.
DNA methylation in oral cancer
Aberrant promoter hypermethylation occurs at CpG sites within gene promoters leading to impeded access for transcription factors binding to the DNA, and eventually resulting in transcriptional silencing of key growth regulatory tumor suppressor genes.
Global hypomethylation of CpG dinucleotides within the body of the gene may invoke enhanced genomic instability by permitting reactivation of evolutionarily methylation-silenced proto-oncogenes and retrotransposons.
Histone modifications in oral cancer
Post-translational histone modifications (e.g., acetylation or methylation of histone residues) cause conformational changes of the tertiary DNA structure in such a manner that it inhibits transcription factor binding to DNA and results in transcriptional gene silencing.
miRNAs in oral cancer
miRNAs regulate gene expression on a post-transcriptional level, either by inhibiting protein translation through degradation or by repression of translation of the mRNA transcript.
Future perspective
Detection of aberrant gene expression is possible from tissue, plasma or saliva providing a rationale for the development of noninvasive screening biomarkers for oral squamous cell carcinoma.
Epigenetic therapy, including the use of DNA methyltransferases and histone deacetylase inhibitors, is gaining attention. Specific and safe epigenetic drugs are identified that have the potential for a more effective and personalized medicine for oral malignancies.
Acknowledgments
The authors would like to thank M Hinshelwood for her critical suggestions and editing to further improve the quality of this article.
Footnotes
For reprint orders, please contact: reprints@futuremedicine.com
Financial & competing interests disclosure
The present work was supported by grants R01 CA129286 from the National Cancer Institute, NIH and funds from the Baylor Research Institute. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
▪ ▪ of considerable interest
- 1.Parkin DM, Pisani P, Ferlay J. Estimates of the worldwide incidence of 25 major cancers in 1990. Int J Cancer. 1999;80(6):827–841. doi: 10.1002/(sici)1097-0215(19990315)80:6<827::aid-ijc6>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 2.Piyathilake CJ, Bell WC, Jones J, et al. Pattern of nonspecific (or global) DNA methylation in oral carcinogenesis. Head Neck. 2005;27(12):1061–1067. doi: 10.1002/hed.20288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guerrero-Preston R, Baez A, Blanco A, Berdasco M, Fraga M, Esteller M. Global DNA methylation: a common early event in oral cancer cases with exposure to environmental carcinogens or viral agents. P R Health Sci J. 2009;28(1):24–29. [PubMed] [Google Scholar]
- 4.Baba S, Yamada Y, Hatano Y, et al. Global DNA hypomethylation suppresses squamous carcinogenesis in the tongue and esophagus. Cancer Sci. 2009;100(7):1186–1191. doi: 10.1111/j.1349-7006.2009.01171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gasche JA, Hoffmann J, Boland CR, Goel A. Interleukin-6 promotes tumorigenesis by altering DNA methylation in oral cancer cells. Int J Cancer. 2011;129(5):1053–1063. doi: 10.1002/ijc.25764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6▪ ▪.Supic G, Kozomara R, Jovic N, Zeljic K, Magic Z. Prognostic significance of tumor-related genes hypermethylation detected in cancer-free surgical margins of oral squamous cell carcinomas. Oral Oncol. 2011;47(8):702–708. doi: 10.1016/j.oraloncology.2011.05.014. This translational study introduces experimental knowledge into clinics. The identified prognostic markers may help to create individual treatment plans depending on the risk of recurrence after tumor resection. [DOI] [PubMed] [Google Scholar]
- 7.Uesugi H, Uzawa K, Kawasaki K, et al. Status of reduced expression and hypermethylation of the APC tumor suppressor gene in human oral squamous cell carcinoma. Int J Mol Med. 2005;15(4):597–602. [PubMed] [Google Scholar]
- 8.Gao S, Eiberg H, Krogdahl A, Liu CJ, Sorensen JA. Cytoplasmic expression of E-cadherin and β-Catenin correlated with LOH and hypermethylation of the APC gene in oral squamous cell carcinomas. J Oral Pathol Med. 2005;34(2):116–119. doi: 10.1111/j.1600-0714.2004.00275.x. [DOI] [PubMed] [Google Scholar]
- 9.Tanaka C, Uzawa K, Shibahara T, Yokoe H, Noma H, Tanzawa H. Expression of an inhibitor of apoptosis, survivin, in oral carcinogenesis. J Dent Res. 2003;82(8):607–611. doi: 10.1177/154405910308200807. [DOI] [PubMed] [Google Scholar]
- 10.Lo ML, Staibano S, Pannone G, et al. Expression of the apoptosis inhibitor survivin in aggressive squamous cell carcinoma. Exp Mol Pathol. 2001;70(3):249–254. doi: 10.1006/exmp.2001.2367. [DOI] [PubMed] [Google Scholar]
- 11.Chen YK, Hsue SS, Lin LM. Survivin expression is regulated by an epigenetic mechanism for DMBA-induced hamster buccal-pouch squamous-cell carcinomas. Arch Oral Biol. 2005;50(6):593–598. doi: 10.1016/j.archoralbio.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 12.Hsue SS, Wang WC, Chen YK, Lin LM. Expression of inhibitors of apoptosis family protein in 7,12-dimethylbenz[a]anthracene-induced hamster buccal-pouch squamous-cell carcinogenesis is associated with mutant p53 accumulation and epigenetic changes. Int J Exp Pathol. 2008;89(5):309–320. doi: 10.1111/j.1365-2613.2008.00583.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Su PF, Huang WL, Wu HT, Wu CH, Liu TY, Kao SY. p16(INK4A) promoter hypermethylation is associated with invasiveness and prognosis of oral squamous cell carcinoma in an age-dependent manner. Oral Oncol. 2010;46(10):734–739. doi: 10.1016/j.oraloncology.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 14.Chang HW, Chow V, Lam KY, Wei WI, Yuen A. Loss of E-cadherin expression resulting from promoter hypermethylation in oral tongue carcinoma and its prognostic significance. Cancer. 2002;94(2):386–392. doi: 10.1002/cncr.10211. [DOI] [PubMed] [Google Scholar]
- 15.Kudo Y, Kitajima S, Ogawa I, et al. Invasion and metastasis of oral cancer cells require methylation of E-cadherin and/or degradation of membranous β-catenin. Clin Cancer Res. 2004;10(16):5455–5463. doi: 10.1158/1078-0432.CCR-04-0372. [DOI] [PubMed] [Google Scholar]
- 16.Kordi-Tamandani DM, Moazeni-Roodi AK, Rigi-Ladiz MA, Hashemi M, Birjandian E, Torkamanzehi A. Promoter hypermethylation and expression profile of MGMT and CDH1 genes in oral cavity cancer. Arch Oral Biol. 2010;55(10):809–814. doi: 10.1016/j.archoralbio.2010.06.017. [DOI] [PubMed] [Google Scholar]
- 17.Shaw RJ, Liloglou T, Rogers SN, et al. Promoter methylation of P16, RARβ, E-cadherin, cyclin A1 and cytoglobin in oral cancer: quantitative evaluation using pyrosequencing. Br J Cancer. 2006;94(4):561–568. doi: 10.1038/sj.bjc.6602972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alvarado CG, Maruyama S, Cheng J, et al. Nuclear translocation of β-catenin synchronized with loss of E-cadherin in oral epithelial dysplasia with a characteristic two-phase appearance. Histopathology. 2011;59(2):283–291. doi: 10.1111/j.1365-2559.2011.03929.x. [DOI] [PubMed] [Google Scholar]
- 19.Kato K, Hara A, Kuno T, et al. Aberrant promoter hypermethylation of p16 and MGMT genes in oral squamous cell carcinomas and the surrounding normal mucosa. J Cancer Res Clin Oncol. 2006;132(11):735–743. doi: 10.1007/s00432-006-0122-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang SH, Lee HS, Mar K, Ji DD, Huang MS, Hsia KT. Loss expression of O6-methylguanine DNA methyltransferase by promoter hypermethylation and its relationship to betel quid chewing in oral squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2010;109(6):883–889. doi: 10.1016/j.tripleo.2009.12.019. [DOI] [PubMed] [Google Scholar]
- 21.Murakami J, Asaumi J, Maki Y, et al. Influence of CpG island methylation status in O6-methylguanine-DNA methyltransferase expression of oral cancer cell lines. Oncol Rep. 2004;12(2):339–345. [PubMed] [Google Scholar]
- 22.Taioli E, Ragin C, Wang XH, et al. Recurrence in oral and pharyngeal cancer is associated with quantitative MGMT promoter methylation. BMC Cancer. 2009;9:354. doi: 10.1186/1471-2407-9-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ishida E, Nakamura M, Ikuta M, et al. Promotor hypermethylation of p14ARF is a key alteration for progression of oral squamous cell carcinoma. Oral Oncol. 2005;41(6):614–622. doi: 10.1016/j.oraloncology.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 24.Czerninski R, Krichevsky S, Ashhab Y, Gazit D, Patel V, Ben-Yehuda D. Promoter hypermethylation of mismatch repair genes, hMLH1 and hMSH2 in oral squamous cell carcinoma. Oral Dis. 2009;15(3):206–213. doi: 10.1111/j.1601-0825.2008.01510.x. [DOI] [PubMed] [Google Scholar]
- 25.Gonzalez-Ramirez I, Ramirez-Amador V, Irigoyen-Camacho ME, et al. hMLH1 promoter methylation is an early event in oral cancer. Oral Oncol. 2011;47(1):22–26. doi: 10.1016/j.oraloncology.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 26.Viswanathan M, Tsuchida N, Shanmugam G. Promoter hypermethylation profile of tumor-associated genes p16, p15, hMLH1, MGMT and E-cadherin in oral squamous cell carcinoma. Int J Cancer. 2003;105(1):41–46. doi: 10.1002/ijc.11028. [DOI] [PubMed] [Google Scholar]
- 27.Zerrouqi A, Pyrzynska B, Febbraio M, Brat DJ, Van Meir EG. P14ARF inhibits human glioblastoma-induced angiogenesis by upregulating the expression of TIMP3. J Clin Invest. 2012;122(4):1283–1295. doi: 10.1172/JCI38596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sailasree R, Abhilash A, Sathyan KM, Nalinakumari KR, Thomas S, Kannan S. Differential roles of p16INK4A and p14ARF genes in prognosis of oral carcinoma. Cancer Epidemiol Biomarkers Prev. 2008;17(2):414–420. doi: 10.1158/1055-9965.EPI-07-0284. [DOI] [PubMed] [Google Scholar]
- 29.Takeshima M, Saitoh M, Kusano K, et al. High frequency of hypermethylation of p14, p15 and p16 in oral pre-cancerous lesions associated with betel-quid chewing in Sri Lanka. J Oral Pathol Med. 2008;37(8):475–479. doi: 10.1111/j.1600-0714.2008.00644.x. [DOI] [PubMed] [Google Scholar]
- 30.Yeh KT, Chang JG, Lin TH, et al. Epigenetic changes of tumor suppressor genes, P15, P16, VHL and P53 in oral cancer. Oncol Rep. 2003;10(3):659–663. [PubMed] [Google Scholar]
- 31.Nakahara Y, Shintani S, Mihara M, Ueyama Y, Matsumura T. High frequency of homozygous deletion and methylation of p16(INK4A) gene in oral squamous cell carcinomas. Cancer Lett. 2001;163(2):221–228. doi: 10.1016/s0304-3835(00)00699-6. [DOI] [PubMed] [Google Scholar]
- 32.Cao J, Zhou J, Gao Y, et al. Methylation of p16 CpG island associated with malignant progression of oral epithelial dysplasia: a prospective cohort study. Clin Cancer Res. 2009;15(16):5178–5183. doi: 10.1158/1078-0432.CCR-09-0580. [DOI] [PubMed] [Google Scholar]
- 33.Kresty LA, Mallery SR, Knobloch TJ, et al. Alterations of p16(INK4a) and p14(ARF) in patients with severe oral epithelial dysplasia. Cancer Res. 2002;62(18):5295–5300. [PubMed] [Google Scholar]
- 34.Ruesga MT, Acha-Sagredo A, Rodriguez MJ, et al. p16(INK4a) promoter hypermethylation in oral scrapings of oral squamous cell carcinoma risk patients. Cancer Lett. 2007;250(1):140–145. doi: 10.1016/j.canlet.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 35.Ohta S, Uemura H, Matsui Y, et al. Alterations of p16 and p14ARF genes and their 9p21 locus in oral squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009;107(1):81–91. doi: 10.1016/j.tripleo.2008.08.027. [DOI] [PubMed] [Google Scholar]
- 36.Kaur J, Demokan S, Tripathi SC, et al. Promoter hypermethylation in Indian primary oral squamous cell carcinoma. Int J Cancer. 2010;127(10):2367–2373. doi: 10.1002/ijc.25377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dong Y, Wang J, Dong F, Wang X, Zhang Y. The correlations between alteration of p16 gene and clinicopathological factors and prognosis in squamous cell carcinomas of the buccal mucosa. J Oral Pathol Med. 2012;41(6):463–469. doi: 10.1111/j.1600-0714.2012.01132.x. [DOI] [PubMed] [Google Scholar]
- 38.McGregor F, Muntoni A, Fleming J, et al. Molecular changes associated with oral dysplasia progression and acquisition of immortality: potential for its reversal by 5-azacytidine. Cancer Res. 2002;62(16):4757–4766. [PubMed] [Google Scholar]
- 39.Timmermann S, Hinds PW, Munger K. Re-expression of endogenous p16ink4a in oral squamous cell carcinoma lines by 5-aza-2′-deoxycytidine treatment induces a senescence-like state. Oncogene. 1998;17(26):3445–3453. doi: 10.1038/sj.onc.1202244. [DOI] [PubMed] [Google Scholar]
- 40.Li J, Warner B, Casto BC, Knobloch TJ, Weghorst CM. Tumor suppressor p16(INK4A)/Cdkn2a alterations in 7,12-dimethylbenz(a)anthracene (DMBA)-induced hamster cheek pouch tumors. Mol Carcinog. 2008;47(10):733–738. doi: 10.1002/mc.20424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakahara Y, Shintani S, Mihara M, Matsumura T, Hamakawa H. High frequency methylation of p16INK4A gene during 4-nitroquinoline 1-oxide-induced rat tongue carcinogenesis. Oncol Rep. 2004;12(1):101–106. [PubMed] [Google Scholar]
- 42.Youssef EM, Lotan D, Issa JP, et al. Hypermethylation of the retinoic acid receptor-β(2) gene in head and neck carcinogenesis. Clin Cancer Res. 2004;10(5):1733–1742. doi: 10.1158/1078-0432.ccr-0989-3. [DOI] [PubMed] [Google Scholar]
- 43.Imai T, Toyota M, Suzuki H, et al. Epigenetic inactivation of RASSF2 in oral squamous cell carcinoma. Cancer Sci. 2008;99(5):958–966. doi: 10.1111/j.1349-7006.2008.00769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang KH, Huang SF, Chen IH, Liao CT, Wang HM, Hsieh LL. Methylation of RASSF1A, RASSF2A, and HIN-1 is associated with poor outcome after radiotherapy, but not surgery, in oral squamous cell carcinoma. Clin Cancer Res. 2009;15(12):4174–4180. doi: 10.1158/1078-0432.CCR-08-2929. [DOI] [PubMed] [Google Scholar]
- 45.Tran TN, Liu Y, Takagi M, Yamaguchi A, Fujii H. Frequent promoter hypermethylation of RASSF1A and p16INK4a and infrequent allelic loss other than 9p21 in betel-associated oral carcinoma in a Vietnamese non-smoking/non-drinking female population. J Oral Pathol Med. 2005;34(3):150–156. doi: 10.1111/j.1600-0714.2004.00292.x. [DOI] [PubMed] [Google Scholar]
- 46▪ ▪.Viet CT, Schmidt BL. Methylation array analysis of preoperative and postoperative saliva DNA in oral cancer patients. Cancer Epidemiol Biomarkers Prev. 2008;17(12):3603–3611. doi: 10.1158/1055-9965.EPI-08-0507. This methylation array analysis looked at 807 cancer-associated genes using saliva of healthy subjects, and preoperative saliva, postoperative saliva and oral squamous cell carcinoma (OSCC) tissue of cancer patients. The analysis identified 41 gene loci from 34 genes that were highly methylated in tumor tissue and preoperative saliva samples but unmethylated in postoperative saliva and healthy subjects. These are promising biomarkers for the early detection of OSCC in the future. [DOI] [PubMed] [Google Scholar]
- 47.Guerrero-Preston R, Soudry E, Acero J, et al. NID2 and HOXA9 promoter hypermethylation as biomarkers for prevention and early detection in oral cavity squamous cell carcinoma tissues and saliva. Cancer Prev Res (Phila) 2011;4(7):1061–1072. doi: 10.1158/1940-6207.CAPR-11-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nagata S, Hamada T, Yamada N, et al. Aberrant DNA methylation of tumor-related genes in oral rinse: a noninvasive method for detection of oral squamous cell carcinoma. Cancer. 2012;118(17):4298–4308. doi: 10.1002/cncr.27417. [DOI] [PubMed] [Google Scholar]
- 49.Subbalekha K, Pimkhaokham A, Pavasant P, et al. Detection of LINE-1s hypomethylation in oral rinses of oral squamous cell carcinoma patients. Oral Oncol. 2009;45(2):184–191. doi: 10.1016/j.oraloncology.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 50.von Zeidler SV, Miracca EC, Nagai MA, Birman EG. Hypermethylation of the p16 gene in normal oral mucosa of smokers. Int J Mol Med. 2004;14(5):807–811. doi: 10.3892/ijmm.14.5.807. [DOI] [PubMed] [Google Scholar]
- 51.Lopez M, Aguirre JM, Cuevas N, et al. Gene promoter hypermethylation in oral rinses of leukoplakia patients – a diagnostic and/or prognostic tool? Eur J Cancer. 2003;39(16):2306–2309. doi: 10.1016/s0959-8049(03)00550-1. [DOI] [PubMed] [Google Scholar]
- 52.Hall GL, Shaw RJ, Field EA, et al. p16 Promoter methylation is a potential predictor of malignant transformation in oral epithelial dysplasia. Cancer Epidemiol Biomarkers Prev. 2008;17(8):2174–2179. doi: 10.1158/1055-9965.EPI-07-2867. [DOI] [PubMed] [Google Scholar]
- 53.Shaw RJ, Hall GL, Woolgar JA, et al. Quantitative methylation analysis of resection margins and lymph nodes in oral squamous cell carcinoma. Br J Oral Maxillofac Surg. 2007;45(8):617–622. doi: 10.1016/j.bjoms.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 54.Sinha P, Bahadur S, Thakar A, et al. Significance of promoter hypermethylation of p16 gene for margin assessment in carcinoma tongue. Head Neck. 2009;31(11):1423–1430. doi: 10.1002/hed.21122. [DOI] [PubMed] [Google Scholar]
- 55.Nakahara Y, Shintani S, Mihara M, Hino S, Hamakawa H. Detection of p16 promoter methylation in the serum of oral cancer patients. Int J Oral Maxillofac Surg. 2006;35(4):362–365. doi: 10.1016/j.ijom.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 56.Suzuki M, Shinohara F, Nishimura K, Echigo S, Rikiishi H. Epigenetic regulation of chemosensitivity to 5-fluorouracil and cisplatin by zebularine in oral squamous cell carcinoma. Int J Oncol. 2007;31(6):1449–1456. [PubMed] [Google Scholar]
- 57.Suzuki M, Shinohara F, Endo M, Sugazaki M, Echigo S, Rikiishi H. Zebularine suppresses the apoptotic potential of 5-fluorouracil via cAMP/PKA/CREB pathway against human oral squamous cell carcinoma cells. Cancer Chemother Pharmacol. 2009;64(2):223–232. doi: 10.1007/s00280-008-0833-4. [DOI] [PubMed] [Google Scholar]
- 58.Murakami J, Lee YJ, Kokeguchi S, et al. Depletion of O6-methylguanine-DNA methyltransferase by O6-benzylguanine enhances 5-FU cytotoxicity in colon and oral cancer cell lines. Oncol Rep. 2007;17(6):1461–1467. [PubMed] [Google Scholar]
- 59.Tsao AS, Liu D, Martin J, et al. Phase II randomized, placebo-controlled trial of green tea extract in patients with high-risk oral premalignant lesions. Cancer Prev Res (Phila) 2009;2(11):931–941. doi: 10.1158/1940-6207.CAPR-09-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kato K, Long NK, Makita H, et al. Effects of green tea polyphenol on methylation status of RECK gene and cancer cell invasion in oral squamous cell carcinoma cells. Br J Cancer. 2008;99(4):647–654. doi: 10.1038/sj.bjc.6604521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Volkel P, Angrand PO. The control of histone lysine methylation in epigenetic regulation. Biochimie. 2007;89(1):1–20. doi: 10.1016/j.biochi.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 62.Yamamoto D, Shima K, Matsuo K, et al. Ornithine decarboxylase antizyme induces hypomethylation of genome DNA and histone H3 lysine 9 dimethylation (H3K9me2) in human oral cancer cell line. PLoS ONE. 2010;5(9):E12554. doi: 10.1371/journal.pone.0012554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lachner M, Jenuwein T. The many faces of histone lysine methylation. Curr Opin Cell Biol. 2002;14(3):286–298. doi: 10.1016/s0955-0674(02)00335-6. [DOI] [PubMed] [Google Scholar]
- 64.Piyathilake CJ, Bell WC, Jones J, et al. Patterns of global DNA and histone methylation appear to be similar in normal, dysplastic and neoplastic oral epithelium of humans. Dis Markers. 2005;21(3):147–151. doi: 10.1155/2005/285134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mancuso M, Matassa DS, Conte M, et al. H3K4 histone methylation in oral squamous cell carcinoma. Acta Biochim Pol. 2009;56(3):405–410. [PubMed] [Google Scholar]
- 66.Chang CC, Lin BR, Chen ST, Hsieh TH, Li YJ, Kuo MY. HDAC2 promotes cell migration/invasion abilities through HIF-1α stabilization in human oral squamous cell carcinoma. J Oral Pathol Med. 2011;40(7):567–575. doi: 10.1111/j.1600-0714.2011.01009.x. [DOI] [PubMed] [Google Scholar]
- 67.Sakuma T, Uzawa K, Onda T, et al. Aberrant expression of histone deacetylase 6 in oral squamous cell carcinoma. Int J Oncol. 2006;29(1):117–124. [PubMed] [Google Scholar]
- 68.Hubbert C, Guardiola A, Shao R, et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417(6887):455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 69.Arif M, Vedamurthy BM, Choudhari R, et al. Nitric oxide-mediated histone hyperacetylation in oral cancer: target for a water-soluble HAT inhibitor, CTK7A. Chem Biol. 2010;17(8):903–913. doi: 10.1016/j.chembiol.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 70.Das BR. Increased ADP-ribosylation of histones in oral cancer. Cancer Lett. 1993;73(1):29–34. doi: 10.1016/0304-3835(93)90184-b. [DOI] [PubMed] [Google Scholar]
- 71.Mascolo M, Ilardi G, Romano MF, et al. Overexpression of chromatin assembly factor-1 p60, poly(ADP-ribose) polymerase 1 and nestin predicts metastasizing behaviour of oral cancer. Histopathology. 2012 doi: 10.1111/j.1365-2559.2012.04313.x. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Staibano S, Mignogna C, Lo ML, et al. Chromatin assembly factor-1 (CAF-1)-mediated regulation of cell proliferation and DNA repair: a link with the biological behaviour of squamous cell carcinoma of the tongue? Histopathology. 2007;50(7):911–919. doi: 10.1111/j.1365-2559.2007.02698.x. [DOI] [PubMed] [Google Scholar]
- 73.Chang HH, Chiang CP, Hung HC, Lin CY, Deng YT, Kuo MY. Histone deacetylase 2 expression predicts poorer prognosis in oral cancer patients. Oral Oncol. 2009;45(7):610–614. doi: 10.1016/j.oraloncology.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 74.Kusumoto T, Hamada T, Yamada N, et al. Comprehensive epigenetic analysis using oral rinse samples: a pilot study. J Oral Maxillofac Surg. 2012;70(6):1486–1494. doi: 10.1016/j.joms.2011.04.021. [DOI] [PubMed] [Google Scholar]
- 75.Sato T, Suzuki M, Sato Y, Echigo S, Rikiishi H. Sequence-dependent interaction between cisplatin and histone deacetylase inhibitors in human oral squamous cell carcinoma cells. Int J Oncol. 2006;28(5):1233–1241. [PubMed] [Google Scholar]
- 76.Zhang L, Fang H, Xu W. Strategies in developing promising histone deacetylase inhibitors. Med Res Rev. 2010;30(4):585–602. doi: 10.1002/med.20169. [DOI] [PubMed] [Google Scholar]
- 77.Jung M. Inhibitors of histone deacetylase as new anticancer agents. Curr Med Chem. 2001;8(12):1505–1511. doi: 10.2174/0929867013372058. [DOI] [PubMed] [Google Scholar]
- 78.Anh TD, Ahn MY, Kim SA, Yoon JH, Ahn SG. The histone deacetylase inhibitor, Trichostatin A, induces G2/M phase arrest and apoptosis in YD-10B oral squamous carcinoma cells. Oncol Rep. 2012;27(2):455–460. doi: 10.3892/or.2011.1496. [DOI] [PubMed] [Google Scholar]
- 79.Suzuki T, Yokozaki H, Kuniyasu H, et al. Effect of trichostatin A on cell growth and expression of cell cycle- and apoptosis-related molecules in human gastric and oral carcinoma cell lines. Int J Cancer. 2000;88(6):992–997. doi: 10.1002/1097-0215(20001215)88:6<992::aid-ijc24>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 80.Yao J, Duan L, Fan M, Wu X. NF-κB signaling pathway is involved in growth inhibition, G2/M arrest and apoptosis induced by Trichostatin A in human tongue carcinoma cells. Pharmacol Res. 2006;54(6):406–413. doi: 10.1016/j.phrs.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 81.Chung YL, Lee MY, Pui NN. Epigenetic therapy using the histone deacetylase inhibitor for increasing therapeutic gain in oral cancer: prevention of radiation-induced oral mucositis and inhibition of chemical-induced oral carcinogenesis. Carcinogenesis. 2009;30(8):1387–1397. doi: 10.1093/carcin/bgp079. [DOI] [PubMed] [Google Scholar]
- 82.Miki Y, Mukae S, Murakami M, et al. Butyrate inhibits oral cancer cell proliferation and regulates expression of secretory phospholipase A2-X and COX-2. Anticancer Res. 2007;27(3B):1493–1502. [PubMed] [Google Scholar]
- 83.Wang A, Zeng R, Huang H. Retinoic acid and sodium butyrate as cell cycle regulators in the treatment of oral squamous carcinoma cells. Oncol Res. 2008;17(4):175–182. doi: 10.3727/096504008785114129. [DOI] [PubMed] [Google Scholar]
- 84.Bai LY, Omar HA, Chiu CF, Chi ZP, Hu JL, Weng JR. Antitumor effects of (S)-HDAC42, a phenylbutyrate-derived histone deacetylase inhibitor, in multiple myeloma cells. Cancer Chemother Pharmacol. 2011;68(2):489–496. doi: 10.1007/s00280-010-1501-z. [DOI] [PubMed] [Google Scholar]
- 85.Bertino EM, Otterson GA. Romidepsin: a novel histone deacetylase inhibitor for cancer. Expert Opin Investig Drugs. 2011;20(8):1151–1158. doi: 10.1517/13543784.2011.594437. [DOI] [PubMed] [Google Scholar]
- 86.Murakami J, Asaumi J, Maki Y, et al. Effects of demethylating agent 5-aza-2(′)-deoxycytidine and histone deacetylase inhibitor FR901228 on maspin gene expression in oral cancer cell lines. Oral Oncol. 2004;40(6):597–603. doi: 10.1016/j.oraloncology.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 87.Murakami J, Asaumi J, Kawai N, et al. Effects of histone deacetylase inhibitor FR901228 on the expression level of telomerase reverse transcriptase in oral cancer. Cancer Chemother Pharmacol. 2005;56(1):22–28. doi: 10.1007/s00280-004-0976-x. [DOI] [PubMed] [Google Scholar]
- 88.Perego P, Zuco V, Gatti L, Zunino F. Sensitization of tumor cells by targeting histone deacetylases. Biochem Pharmacol. 2012;83(8):987–994. doi: 10.1016/j.bcp.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 89.Rikiishi H, Shinohara F, Sato T, Sato Y, Suzuki M, Echigo S. Chemosensitization of oral squamous cell carcinoma cells to cisplatin by histone deacetylase inhibitor, suberoylanilide hydroxamic acid. Int J Oncol. 2007;30(5):1181–1188. [PubMed] [Google Scholar]
- 90.Shen J, Huang C, Jiang L, et al. Enhancement of cisplatin induced apoptosis by suberoylanilide hydroxamic acid in human oral squamous cell carcinoma cell lines. Biochem Pharmacol. 2007;73(12):1901–1909. doi: 10.1016/j.bcp.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 91▪ ▪.Wong TS, Liu XB, Wong BY, Ng RW, Yuen AP, Wei WI. Mature miR-184 as potential oncogenic microRNA of squamous cell carcinoma of tongue. Clin Cancer Res. 2008;14(9):2588–2592. doi: 10.1158/1078-0432.CCR-07-0666. miRNA profiling study on tongue squamous cell carcinoma and normal tissues examining the expression level of 156 miRNAs. [DOI] [PubMed] [Google Scholar]
- 92.Li J, Huang H, Sun L, et al. MiR-21 indicates poor prognosis in tongue squamous cell carcinomas as an apoptosis inhibitor. Clin Cancer Res. 2009;15(12):3998–4008. doi: 10.1158/1078-0432.CCR-08-3053. [DOI] [PubMed] [Google Scholar]
- 93.Liu X, Wang A, Heidbreder CE, et al. MicroRNA-24 targeting RNA-binding protein DND1 in tongue squamous cell carcinoma. FEBS Lett. 2010;584(18):4115–4120. doi: 10.1016/j.febslet.2010.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wong TS, Ho WK, Chan JY, Ng RW, Wei WI. Mature miR-184 and squamous cell carcinoma of the tongue. Sci World J. 2009;9:130–132. doi: 10.1100/tsw.2009.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jiang L, Liu X, Kolokythas A, et al. Downregulation of the Rho GTPase signaling pathway is involved in the microRNA-138-mediated inhibition of cell migration and invasion in tongue squamous cell carcinoma. Int J Cancer. 2010;127(3):505–512. doi: 10.1002/ijc.25320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96▪ ▪.Cervigne NK, Reis PP, Machado J, et al. Identification of a microRNA signature associated with progression of leukoplakia to oral carcinoma. Hum Mol Genet. 2009;18(24):4818–4829. doi: 10.1093/hmg/ddp446. miRNA profiling study on oral premalignant lesions. This study classified 109 miRNAs that are highly expressed in progressive leukoplakia and invasive OSCC only. Therefore, it helped to discover markers that identify lesions at high risk for malignant transformation. [DOI] [PubMed] [Google Scholar]
- 97.Kozaki K, Imoto I, Mogi S, Omura K, Inazawa J. Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res. 2008;68(7):2094–2105. doi: 10.1158/0008-5472.CAN-07-5194. [DOI] [PubMed] [Google Scholar]
- 98.Yu T, Wang XY, Gong RG, et al. The expression profile of microRNAs in a model of 7,12-dimethyl-benz[a]anthrance-induced oral carcinogenesis in Syrian hamster. J Exp Clin Cancer Res. 2009;28:64. doi: 10.1186/1756-9966-28-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Reis PP, Tomenson M, Cervigne NK, et al. Programmed cell death 4 loss increases tumor cell invasion and is regulated by miR-21 in oral squamous cell carcinoma. Mol Cancer. 2010;9:238. doi: 10.1186/1476-4598-9-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chang KW, Liu CJ, Chu TH, et al. Association between high miR-211 microRNA expression and the poor prognosis of oral carcinoma. J Dent Res. 2008;87(11):1063–1068. doi: 10.1177/154405910808701116. [DOI] [PubMed] [Google Scholar]
- 101.Wong TS, Liu XB, Chung-Wai HA, Po-Wing YA, Wai-Man NR, Ignace WW. Identification of pyruvate kinase type M2 as potential oncoprotein in squamous cell carcinoma of tongue through microRNA profiling. Int J Cancer. 2008;123(2):251–257. doi: 10.1002/ijc.23583. [DOI] [PubMed] [Google Scholar]
- 102.Jakymiw A, Patel RS, Deming N, et al. Overexpression of dicer as a result of reduced let-7 microRNA levels contributes to increased cell proliferation of oral cancer cells. Genes Chromosomes Cancer. 2010;49(6):549–559. doi: 10.1002/gcc.20765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liu CJ, Kao SY, Tu HF, Tsai MM, Chang KW, Lin SC. Increase of microRNA miR-31 level in plasma could be a potential marker of oral cancer. Oral Dis. 2010;16(4):360–364. doi: 10.1111/j.1601-0825.2009.01646.x. [DOI] [PubMed] [Google Scholar]
- 104.Lu YC, Chen YJ, Wang HM, et al. Oncogenic function and early detection potential of miRNA-10b in oral cancer as identified by microRNA profiling. Cancer Prev Res (Phila) 2012;5(4):665–674. doi: 10.1158/1940-6207.CAPR-11-0358. [DOI] [PubMed] [Google Scholar]
- 105.Park NJ, Zhou H, Elashoff D, et al. Salivary microRNA: discovery, characterization, and clinical utility for oral cancer detection. Clin Cancer Res. 2009;15(17):5473–5477. doi: 10.1158/1078-0432.CCR-09-0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wiklund ED, Gao S, Hulf T, et al. MicroRNA alterations and associated aberrant DNA methylation patterns across multiple sample types in oral squamous cell carcinoma. PLoS ONE. 2011;6(11):E27840. doi: 10.1371/journal.pone.0027840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ogi K, Toyota M, Ohe-Toyota M, et al. Aberrant methylation of multiple genes and clinicopathological features in oral squamous cell carcinoma. Clin Cancer Res. 2002;8(10):3164–3171. [PubMed] [Google Scholar]
- 108.Baba S, Hara A, Kato K, et al. Aberrant promoter hypermethylation of the CHFR gene in oral squamous cell carcinomas. Oncol Rep. 2009;22(5):1173–1179. doi: 10.3892/or_00000552. [DOI] [PubMed] [Google Scholar]
- 109.Kulkarni V, Saranath D. Concurrent hypermethylation of multiple regulatory genes in chewing tobacco associated oral squamous cell carcinomas and adjacent normal tissues. Oral Oncol. 2004;40(2):145–153. doi: 10.1016/s1368-8375(03)00143-x. [DOI] [PubMed] [Google Scholar]
- 110.Shiah SG, Chang LC, Tai KY, Lee GH, Wu CW, Shieh YS. The involvement of promoter methylation and DNA methyltransferase-1 in the regulation of EpCAM expression in oral squamous cell carcinoma. Oral Oncol. 2009;45(1):E1–E8. doi: 10.1016/j.oraloncology.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 111.Chang KW, Kao SY, Tzeng RJ, et al. Multiple molecular alterations of FHIT in betel-associated oral carcinoma. J Pathol. 2002;196(3):300–306. doi: 10.1002/path.1047. [DOI] [PubMed] [Google Scholar]
- 112.Gao S, Krogdahl A, Eiberg H, Liu CJ, Sorensen JA. LOH at chromosome 9q34.3 and the Notch1 gene methylation are less involved in oral squamous cell carcinomas. J Oral Pathol Med. 2007;36(3):173–176. doi: 10.1111/j.1600-0714.2007.00520.x. [DOI] [PubMed] [Google Scholar]
- 113.Long NK, Kato K, Yamashita T, et al. Hypermethylation of the RECK gene predicts poor prognosis in oral squamous cell carcinomas. Oral Oncol. 2008;44(11):1052–1058. doi: 10.1016/j.oraloncology.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 114.Gao F, Huang C, Lin M, et al. Frequent inactivation of RUNX3 by promoter hypermethylation and protein mislocalization in oral squamous cell carcinomas. J Cancer Res Clin Oncol. 2009;135(5):739–747. doi: 10.1007/s00432-008-0508-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sogabe Y, Suzuki H, Toyota M, et al. Epigenetic inactivation of SFRP genes in oral squamous cell carcinoma. Int J Oncol. 2008;32(6):1253–1261. doi: 10.3892/ijo_32_6_1253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.