

Abstract
Oxide-bridged phenylmorphans were conceptualized as topologically distinct, structurally rigid ligands with 3-dimensional shapes that could not be appreciably modified on interaction with opioid receptors. An enantiomer of the N-phenethyl-substituted ortho-f isomer was found to have high affinity for the μ-receptor (Ki = 7 nM) and was about four times more potent than naloxone as an antagonist. In order to examine the effect of introduction of a small amount of flexibility into these molecules, we have replaced the rigid 5-membered oxide ring with a more flexible 6-membered carbon ring. Synthesis of the new N-phenethyl-substituted tricyclic N-substituted-2,3,4,9,10,10a-hexahydro-1H-1,4a-(epiminoethano)phenanthren-6- and 8-ols resulted in a two carbon-bridged relative of the f-isomers, the dihydrofuran ring was replaced by a cyclohexene ring. The carbocyclic compounds had much higher affinity and greater selectivity for the μ-receptor than the f-oxide-bridged phenylmorphans. They were also much more potent μ-antagonists, with activities comparable to naltrexone in the [35S]GTPγS assay.
Keywords: Synthesis; opioid receptor binding and efficacy; N-substituted-2,3,4,9,10,10a-hexahydro-1H-1,4a-(epiminoethano)phenanthren-6- and 8-ols; f-oxide-bridged phenylmorphans; opioid antagonist
1. Introduction
We have reported the synthesis of all of the possible a- through f-oxide-bridged phenylmorphans (1, Figure 1)[2-16]. The f- and the e-oxide bridged phenylmorphans were of interest because an enantiomer of the N-phenethyl-substituted ortho-f isomer had high affinity to the μ-receptor (Ki = 7 nM) and was about 4 times more potent than naloxone as an antagonist (2, Fig. 1). In contrast, an enantiomer of the para-e isomer had morphine-like agonist activity. The N-phenethyl ortho-b isomer was a moderately selective kappa antagonist, and the N-phenethyl substituted ortho-c isomer had very high affinity for μ-receptors (Ki = 1 nM), and was a very potent μ- and κ-antagonist (Ke = 0.7 and 3 nM, respectively). Prior to the determination of the structures of the opioid receptors, we believed that a structurally rigid ligand that was selective and had high affinity to an opioid receptor would give us insight into the spatial requirements necessary for interaction with the amino acids in the receptor binding pocket. Since the oxide-bridged phenylmorphans were rigid ligands, an element of ambiguity introduced by the flexibility of most ligands would be removed. The recent determination of the crystal structures of all of the opioid receptors[17-20] will allow us to examine the ligand-receptor interaction differently in the future.
Fig. 1.

Structures of oxide-bridged phenylmorphans and cannabinoid receptor ligands
Having examined the affinity and activity of the rigid oxide-bridged phenylmorphans, we considered the possibility of allowing a small amount of determinable flexibility into some of these compounds to see whether that would alter their affinity or activity. It has been previously shown that the affinity and selectivity of cannabinoids for the CB1 and CB2 receptors could be very much altered, by enlarging a 5-membered ring to a 6-membered ring (3, Fig. 1)[21]. We thought it would be of interest to see whether this modification could also be applied to the f-isomers of the oxide-bridged phenylmorphans, where the 5-membered oxide ring would be changed to a 6-membered carbocyclic structure. The f-oxide-bridged phenylmorphans were initially chosen because they were pharmacologically interesting and synthetically more accessible than many of the other oxide-bridged phenylmorphans. Also, a path to the N-methyl substituted 2,3,4,9,10,10a-hexahydro-1H-1,4a-(epiminoethano)phenanthren-6-ols had been previously reported[22, 23].
2. Chemistry
The synthesis of the target compounds was straightforward and started from the known 9-oxomorphan 4 (5-(3-methoxyphenyl)-2-methyl-9-oxo-morphan), which was prepared in six steps using modified literature procedures (Scheme 1)[12, 24-27]. The condensation of compound 4 and malononitrile proceeded normally to give the unsaturated dinitrile 5 in good yield[22]. Catalyzed hydrogenation of 5 with platinum oxide was not chemically selective and gave two diastereomers in a 7:1 ratio. Fortunately, the desired isomer was separable by flash chromatography and the stereochemically pure, saturated dinitrile 6 was obtained in good yield. Palladium on charcoal was unsuccessfully used to attempt to improve the ratio of the two isomers, at both atmosphere pressure and higher pressure (50 psi). Platinum oxide worked under both conditions, although the reaction was slower (40 h or longer) at atmospheric pressure. Under higher pressure (50 psi), and lengthy reaction times (> 4 h), a very polar uncharacterized by-product was obtained. Saturated dinitrile 6 was smoothly cyclized in refluxing hydrochloric acid (10 M) to ketophenanthrene-type structures, in good yield, aided by the electron-donating methoxy group. 3-O-Demethylation was achieved with refluxing hydrobromic acid, to afford cyclized regioisomers 7 and 8, easily isolated by chromatography. The ortho-hydroxyphenyl isomer 7 was much less polar than the para-isomer 8, possibly due to hydrogen bonding between the phenolic hydroxyl and the C9-ketone. N-Demethylation of 7 and 8 with ethyl chloroformate and subsequent realkylation with phenethyl bromide afforded the corresponding N-phenenthyl analogues 9 (Scheme 2) and 18 (Scheme 3). Because the reduction of these ketones with NaBH4 was very sluggish, a number of reducing agents and conditions were explored (Table 1).
Scheme 1.
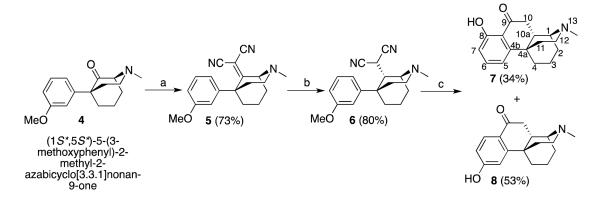
Synthesis of ortho- and para-hydroxyphenylketones 7 and 8. Reagents and conditions: a) malononitrile, NH4OAc, AcOH, toluene, reflux, 2 h; b) H2 (60 psi) PtO2, EtOH, 4 h; c) (1) 10 M HCl,reflux, overnight; (2) 48% HBr, reflux, 48 h.
Scheme 2.

Synthesis of ortho-hydroxyphenyl 9α- and 9β-hydroxy compounds and the saturated and unsaturated carbocyclics. Reagents and conditions: a) (1) ClCO2Et, K2CO3, ClCH2CH2Cl, reflux, overnight; (2) 48% HBr, reflux, overnight; (3) phenethyl bromide, Et3N, EtOH, reflux, 48 h; b) superhydride, THF, −78 °C to rt, overnight; c) LiAlH , THF, 0 °C, 1 h; d) 2 M HCl, MeOH, rt; e) H2 (60psi), 5% Pd/C, EtOH, overnight; f) H2 (60 psi), 5% Pd/C, 2 M HCl, EtOH.
Scheme 3.
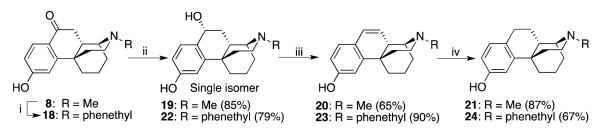
Preparation of the C9α-OH compound, and the unsaturated and saturated carbocyclic compounds in the para-hydroxyphenyl series. Reagents and conditions: i) (1) ClCO2Et, K2CO3, ClCH2CH2Cl, reflux, overnight; (2) 48% HBr, reflux, overnight; (3) phenethyl bromide, Et3N, EtOH, reflux, 48 h; ii) LiAlH , THF, 0 °C, 1 h; iii) 2 M HCl, MeOH, rt; iv) H2 (60 psi), 5% Pd/C, EtOH,overnight.
Table 1.
Attempts to introduce a C9β-OH in the para-hydroxyphenyl series
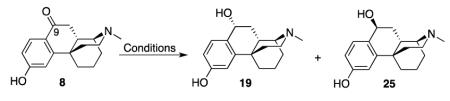
| Conditions | Results |
|---|---|
| 1) NaBH4, MeOH, rt | 19 (25%, ~57% SM recovered) |
| 2) Superhydride, −78 °C to rt, overnight | 19 (43%) |
| 3) LiAlH4, THF, O °C, 2h | 19 (93%) |
| 4) Formamidinesulfinic acid, aq NaOH | SM, no reaction |
| 5) K-selectride, KH, THF, rt, overnight | Decomposed |
| 6) PtO2, H2 | 2h: 19 + SM; 48h: 20 + SM |
| 7) Al(Oi-Pr)3, i-PrOH, toluene, 50 °C | 20 (67%) |
| 8) Li, NH3 (liquid), t-BuOH, THF, −78 °C | 19 (58%) + 25 (17%) |
We found that superhydride and LiAlH4 (conditions 2 and 3 in Table 1) at a low temperature rapidly reduced 7 and 8 in the N-methyl series and 9 and 18 in the N-phenethyl series. Interestingly, the reduction of ortho-hydroxyphenyl 7 gave two stereoisomers, 10 with an 9α-OH group, and 11 with the 9β-OH (Scheme 2), while reduction of the para--hydroxyphenyl compound 8 under the same conditions only gave the single isomer 19 with a 9α-OH (Scheme 3). The low yields of 10 and 11 may be due to the repeated purification that was needed.
The difference in reactivity of the ortho- and para-hydroxyphenyl compounds might be attributed to intramolecular hydrogen bonding between the phenolic hydroxyl and the carbonyl group in 7, the ortho-hydroxyphenyl compound, that is not possible in 8. Compounds 12, 13, and 22 were obtained similarly. The relative configurations of compounds 12, 19 and 22 were confirmed by X-ray crystallographic analyses (Figure 1), and the relative configurations of compounds 11 and 13 were confirmed by comparing the polarity and the chemical shift of the benzyl proton with those of compound 12. Compounds 10 and 12 were less polar (by tlc) than corresponding isomers 11 and 13. On the other hand, the chemical shifts of the benzyl proton in the proton NMR spectra of 10 and 12 with 9α-OH (δ 4.92 ppm and 4.99 ppm, respectively) were at higher field than those of compounds 11 and 13 with a 9β-OH (δ 5.02 ppm and 5.23 ppm, respectively), possibly because of the deshielding effect of the ortho-hydroxyphenyl group. These benzylic alcohols were very susceptible to dehydration under acidic conditions to give the corresponding phenanthrene-like compounds 14, 16, 20 and 23 in good to excellent yield. The double bond was saturated by catalytic hydrogenation to give the more flexible corresponding compounds 15, 17, 21 and 24, in good yield (Schemes 2 and 3).
3. Results and discussion
The μ, δ, and κ-opioid receptor binding affinities of 7-25 were determined by an initial screen (see experimental, section 5.3.2). Ten of these compounds (10, 12-14, 16-17, 20, 22-24) were estimated to have a Ki < 60 nM. These were examined in the full binding assay (Table 2). Only a few of the remaining compounds, 18 and 20, had an estimated Ki (μ) of < 100 nM (80 and 67 nM, respectively), and 9, 11, and 25 had an estimated Ki (μ) of > 200 nM (estimated Ki (μ) = 297, 258, and 227, respectively); these were not examined further, nor were the remaining compounds that were found to have an estimated Ki > 1 μM.
Table 2.
Opioid binding affinity (Ki, nM) and efficacy ([35S]GTPγS, Ke, nM) of selected antagonists
| # | Ki μa | Ki δb | μ | Ki κc | κ/μ | Ke μ | |
|---|---|---|---|---|---|---|---|
| 10 |
|
30 ± 6 | 3220 ± 332 | 107 | 2020 ± 169 | 24 | 706 ± 135 |
| 12 |
|
14 ± 2 | >10,000 | 714 | 525 ± 39 | 38 | 6 ± 1 |
| 13 |
|
179 ± 14 | >10,000 | 56 | 923 ± 32 | 5 | ND |
| 14 |
|
67 ± 7 | >10,000 | 149 | 3890 ± 441 | 58 | ND |
| 16 |
|
19 ± 4 | 1840 ±212 | 97 | 369 ± 26 | 19 | 1.7 ± 0.26 |
| 17 |
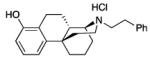
|
21 ± 5 | 2830 ± 223 | 135 | 873 ± 101 | 42 | 7 ± 1.8 |
| 20 |
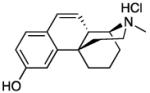
|
57 ± 6 | >10,000 | 175 | 4040 ± 363 | 71 | ND |
| 22 |
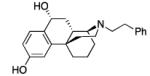
|
72 ± 10 | >10,000 | 139 | 1810 ± 95 | 25 | ND |
| 23 |
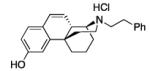
|
2.6 ± 0.35 | 127 ± 31 | 49 | 51 ± 4 | 20 | 0.34 ± 0.07 |
| 24 |
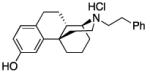
|
2.3 ± 0.2 | 186 ± 25 | 81 | 61 ± 6 | 27 | 0.71 ± 0.13 |
Opiate receptor binding assays were performed as indicated in section 5.3.2 using CHO hMOR, CHO hDOR and CHO hKOR cells. The data of three experiments were pooled (N=30 data points) and fit to the two-parameter logistic equation for the best-fit estimates (±SD) of the IC50, the value of which was then used to calculate the Ki value, as described in section 5.3.2; the Ke values from the [35S]GTPgS assay was carried out as indicated in section 5.3.3. ND = not done. In these assays, the μKi of morphine = 2.6 ± 0.01 nM, DAMGO μKi = 1.2 ± 0.1, and the μKe of naloxone = 2.3 ± 0.3 nM.
[3H]-DAMGO
[3H]-DADLE
[3H]-U69,593
In Table 2, only those compounds that were found to have Ki < 50 nM in the full binding assay were examined for their efficacy in the [35S]GTPγS assay. It is apparent in Table 2 that the N-methyl substituted compounds had less affinity than the comparable N-phenethyl substituted relatives. Compound 10, the best of the N-methyl compounds, had, for example, half the affinity of the comparable N-phenethyl-substituted relative, compound 12, and, unlike 12, had almost no efficacy in the [35S]GTPγS assay (μKe > 700 nM). Among the N-phenethyl substituted compounds, those with a C9- OH showed less μ-receptor affinity than the comparable C9-OH compounds. Compound 31, for example, had less than a tenth of the affinity of 12, a similar compound, but with a C9-OH moiety. All of the C9-OH substituted compounds in both the ortho-hydroxyphenyl series or in the para-hydroxyphenyl series, except for 12, were generally seen to have less affinity and efficacy than comparable compounds lacking that C9-OH. At that C9-position in this new tricyclic carbocyclic opioid series, both the carbonyl and, usually, the hydroxyl group appear to have a deleterious effect. The effect of oxygen at C9 seems to become more damaging when it is present in the para-hydroxyphenyl compounds (e.g., compound 22). This observation has precedent in that introduction of a keto group at the benzyl position has been found to generally decrease the affinity of morphinans at μ-, δ-, and κ-opioid receptors[28]. Removal of the C9-OH moiety in the ortho-hydroxyphenyl series (12), to form the unsaturated compound 16, or its saturated relative 17, does not appreciably improve μ-affinity. Both 16 and 17 have about the same affinity as the C9-OH compound 21. Indeed, both 12 and 17 also have about the same antagonist efficacy in the [35S]GTPγS assay (μKe = 6 or 7 nM). The unsaturated relative, the ortho-hydroxyphenyl compound 16, however, was found to have surprisingly good efficacy (μKe = 1.7 nM); efficacy was improved in that compound by removal of the C9-OH group.
A considerable improvement in affinity (μKi = ca. 2 nM), and a remarkable increase in efficacy (μKe = 0.3 to 0.7 nM), was found in the unsaturated and saturated carbocyclic compounds 23 and 24, in the para-hydroxyphenyl series. Thus, removal of the C9-OH group in the para-hydroxyphenyl series, where the presence of a C9-OH group is greatly disadvantageous, gave us μ-antagonists of great potency, comparable to naltrexone. Unfortunately, like all of the known μ-receptor antagonists, selectivity for the μ-opioid receptor was compromised in 23 and 24 in that they had some affinity (Ki = 50 to 60 nM) for the κ-opioid receptor, and the μ/κ ratio was 20 to 27 (Table 2). Thus, the carbocyclic compounds had reasonable, but not excellent selectivity for the μ-receptor.
4. Conclusion
A tricyclic ring system was conceptualized to have greater flexibility than the f-isomers of the oxidebridged phenymorphans, and a few of the synthesized compounds were found to have high μ-opioid receptor affinity and exceptionally potent antagonist activity. These carbocyclic compounds had greater affinity for μ-opioid receptors and were much more potent as μ-opioid antagonists than the original f-oxide-bridged phenymorphans with a dihydrofuran ring.
It was initially thought that the slight relaxation of the rigid framework of the oxide-bridged phenylmorphan that was allowed by substituting a 6-membered carbocyclic ring for the 5- membered dihydrofuran ring in the oxide-bridged phenylmorphans would be sufficient to permit more extensive interaction with the amino acids in the binding site of the μ-opioid receptor; however, the fact that both the unsaturated compound 23 and its less flexible saturated relative 24 had similar affinity and efficacy did not lend weight to that hypothesis. Further exploration will be undertaken in the future to determine if the slightly different spatial positions of the heteroatoms in the carbocyclic molecules caused by increasing the size of the ring from a 5- to a 6-membered ring caused the observed increase in affinity and potency. Alternatively, alteration of the polarity and H-bonding characteristics of the original dihydrofuro compound by the substitution of two carbon atoms for an oxygen could also affect affinity and potency.
5. Experimental section
5.1. Synthetic chemistry
5.1.1. General
Melting points were determined on a Thomas-Hoover melting point apparatus and are uncorrected. Proton nuclear magnetic resonance (1H NMR, 400 or 500 MHz) and carbon nuclear magnetic resonance (13C NMR, 100 or 125 MHz) spectra were recorded on a Bruker DMX500 wide-bore spectrometer in CDCl3 (unless otherwise noted) with the values given in ppm and J (Hz) assignments of 1H resonance coupling. For 1H NMR spectra (CDCl3), the residual solvent peak was used as the reference (7.26 ppm) while the central solvent peak was used as the 13C NMR reference (77.0 ppm in CDCl3). The high-resolution electrospray ionization (ESI) mass spectra were obtained on a Waters LCT Premier time-of-flight (TOF) mass spectrometer. Thin-layer chromatography (TLC) was performed on 0.25 mm Analtech GHLF silica gel and used to determine the completion of the reaction (solvent system: chloroform/methanol/ammonia (19:0.9:0.1 or 9:0.9:0.1) depending on the polarity of the compounds. Flash column chromatography was performed with Bodman silica gel LC 60 A. Elemental analyses were performed by Micro-Analysis, Inc, Wilmington, DE, and were within 0.4% for C, H, and N. IR spectra were recorded on a PerkinElmer Spectrum One FT-IR spectrometer.
5.1.2. 2-(5-(3-methoxyphenyl)-2-methyl-2-azabicyclo[3.3.1]nonan-9-ylidene)malononitrile (5)
A mixture of 5-(3-methoxyphenyl)-2-methyl-2-azabicyclo[3.3.1]nonan-9-one hydrochloride 4 (2.96g, 10 mmol), malononitrile (0.92 g, 14 mmol), ammonium acetate (0.23 g, 3 mmol) and acetic acid (0.5 mL) in toluene was heated azeotropically for 2 h. After cooling to ambient temperature, the mixture was extracted with 2 N HCl. The aqueous layer was cooled in ice-bath, basified with 28% NH4OH to pH 9 and extracted with CHCl3. The combined extracts were washed with brine and dried over anhydrous Na2SO4. The crude product was purified by flash chromatography (silica, gradient: CHCl3 to 3% MeOH/NH4OH/CHCl3) to afford the racemic 5-(3-methoxyphenyl)-2-methyl-2-azabicyclo[3.3.1]nonan-9-ylidenemalononitrile (5) in 73% yield (2.26 g) as a yellow oil. 1H NMR (CDCl3, 500 MHz) δ 7.31 (t, J = 7.5 Hz, 1H), 6.98-6.88 (m, 3H), 4.06 (s, 1 H), 3.81 (s, 3H), 3.23-3.19 (m, 1H), 2.73-2.70 (m, 1H), 2.63-2.57 (m, 1H), 2.54 (s, 3H), 2.44-2.41 (m, 2H), 2.31-2.15 (m, 3H), 1.79-1.76 (m, 1H), 1.72-1.66 (m, 1H); 13C NMR (CDCl3, 125 MHz) (mixture of two rotamers) δ 188.6, 188.5, 161.0, 160.0, 154.6, 154.5, 130.1, 129.8, 121.5, 119.1, 115.4, 114.2, 113.8, 113.4, 112.3, 109.4, 82.4, 64.5, 64.4, 55.44, 55.36, 50.1, 50.0, 47.9, 47.8, 43.9, 43.8, 41.6, 41.4, 40.4, 32.4, 32.1, 20.3, 20.2; IR (film) νmax = 3017, 2937, 2854, 2226, 1722, 1602, 1583, 1449, 1219, 1046, 1033, 772, 701 cm−1; ESI-MS 308.2 (M++1); HRMS (ES+) calcd for C19H22N3O (M + H)+ 308.1673; found 308.1671.
5.1.3.5-(3-Methoxyphenyl)-2-methyl-2-azabicyclo[3.3.1]nonan-9-yl)-malononitrile (6)
Compound 5 (6 g, 19.5 mmol) was dissolved in MeOH (100 mL) in a glass bomb and PtO2 (0.44 g,1.95 mmol) was added. The bomb was placed in a Parr apparatus, evacuated and backfilled with H2 three times, then filled with H2 to 50 psi and shaken for 4 h. The reaction mixture was filtered through a pad of celite and the filtrate was concentrated. The residue was purified by flash chromatography (silica, gradient: CHCl3 to 5% MeOH/0.5%NH4OH/CHCl3) to yield 5-(3-methoxyphenyl)-2-methyl-2-azabicyclo[3.3.1]nonan-9-ylmalononitrile (6) as a yellow foam (4.8 g, 80%). 1H NMR (CDCl3, 500 MHz) δ 7.31 (t, J = 8.0 Hz, 1H), 6.98 (d, J = 7.5 Hz, 1H), 6.94 (s, 1H), 6.83 (dd, J = 8.0, 2.0 Hz, 1H), 3.80 (s, 3 H), 3.66 (d, J = 6.5 Hz, 1H), 3.23 (s, 1H), 3.08 (td, J = 12.5, 5.0 Hz, 1H), 2.87-2.83 (m, 2H), 2.52 (s, 3H), 2.42-2.33 (m, 2H), 2.21 (dd, J = 16.5, 6.0 Hz, 1H), 2.06-1.92 (m, 3H), 1.79 (dd, J = 14.5, 4.5 Hz, 1H), 1.71-1.66 (m, 1H); 13C NMR (CDCl3, 125 MHz) δ 160.2, 147.2, 130.2, 118.1, 112.9, 112.7, 112.1, 112.0, 55.8, 55.4, 50.9, 48.1, 43.2, 41.6, 37.8, 28.0, 22.5, 21.0, 17.7; ESI-MS 310.2 (M++1); HRMS (ES+) calcd for C19H24N3O (M + H)+ 310.1919; found 310.1922. The free base was converted into its HCl salt for analysis, mp 253.0~256.8 °C. IR (film) νmax = 3408, 2937, 2836, 2485, 2409, 1608, 1583, 1493, 1454, 1434, 1219, 1049, 771 cm−1. Anal. calcd for (C19H23N3O·HCl·0.3H2O) C, 64.97; H, 7.08; N, 11.96; found C, 64.91; H, 7.38; N, 11.84.
5.1.4 8-Hydroxy-13-methyl-3,4,10,10a-tetrahydro-1H-1,4a-(epiminoethano)phenanthren-9(2H)-one (7) and 6-hydroxy-13-methyl-3,4,10,10a-tetrahydro-1H-1,4a-(epiminoethano)phenanthren-9(2H)-one (8)
The free base 6 (7.1 g, 22.9 mmol) was dissolved in 10 N HCl (80 mL) and the resulting solution was heated to reflux under argon for 16 h. The solvent was removed and the residue was redissolved in 48% HBr (40 mL) and heated to reflux under argon for 24 h. After the solvent was removed, the residue was treated with crushed ice and basified with 28% NH4OH to pH 9-9.5. The mixture was extracted with a mixed solvent of CHCl3 and MeOH (20:1, v/v) several times. The combined extracts were washed with brine and dried over Na2SO4. After filtration and concentration, the crude product was purified by flash chromatography (silica, gradient: 5-10% MeOH/1% NH4OH/CHCl3) to give 8-hydroxy-13-methyl-3,4,10,10a-tetrahydro-1H-1,4a-(epiminoethano)phenanthren-9(2H)-one (7) as a brown oil (2.1 g, 34%) and 6-hydroxy-13-methyl-3,4,10,10a-tetrahydro-1H-1,4a-(epiminoethano)-phenanthren-9(2H)-one (8) as a brown foam (3.29 g, 53%).
7
1H NMR (CDCl3, 500 MHz) δ 12.7 (s, 1H, -OH), 7.37 (t, J = 8.0 Hz, 1H), 6.79-6.74 (m, 2H), 3.09 (td, J = 12.5, 5.0 Hz, 1H), 2.96-2.88 (m, 2H), 2.67 (d, J = 1.5 Hz, 1H), 2.48-2.33 (m, 3H), 2.42 (s, 3H), 2.03 (dd, J = 14.0, 4.0 Hz, 1H), 1.96-1.89 (m, 1H), 1.88-1.78 (m, 2H), 1.63-1.60 (m, 2H), 1.45-1.38 (m, 1H); 13C NMR (CDCl3, 125 MHz) δ 204.4, 163.1, 154.4, 136.8, 115.6, 115.4, 115.3, 57.0, 51.6, 42.9, 41.5, 38.6, 36.8, 34.5, 33.3, 21.7, 17.8; ESI-MS 272.2 (M++1); HRMS (ES+) calcd for C17H22NO2 (M + H)+ 272.1651; found, 272.1639. The free base was converted into its HCl salt for analysis, mp 205.2~207.6 °C. IR (film) νmax = 3706, 3681, 3474, 2967, 2938, 2475, 1635, 1615, 1453, 1345, 1230, 1055, 1033, 1012, 801, 749 cm−1; Anal. calcd for (C17H22NO2·HCl·0.5H2O) C, 64.45; H, 7.32; N, 4.42; found C, 64.24; H, 7.44; N, 4.38.
8
1H NMR (CDCl3, 500 MHz) δ 11.0 (s, 1H, -OH), 7.82 (d, J = 8.0 Hz, 1H), 6.56 (s, 1H), 6.52 (d, J = 8.5 Hz, 1H), 3.10 (s, 1H), 2.94 (s, 1H), 2.74-2.70 (m, 2H), 2.42 (s, 4H), 2.32 (d, J = 14.5 Hz, 1H), 2.19 (d, J = 9.0 Hz, 1H), 1.98-1.97 (m, 1H), 1.85 (s, 1H), 1.75 (s, 1H), 1.57-1.51 (m, 3H); 13C NMR (CDCl3, 125 MHz) δ 195.9, 164.5, 155.4, 130.2, 122.2, 155.0, 111.8, 57.3, 51.4, 42.4, 41.1, 38.2, 35.9, 34.0, 32.7, 21.3, 17.5; ESI-MS 272.2 (M++1); HRMS (ES+) calcd for C17H22NO2 (M + H)+ 272.1651; found, 272.1645. The free base was converted into its HCl salt for analysis, mp 279.6~284.5 °C. IR (film) νmax = 3126, 2945, 2642, 1670, 1606, 1571, 1462, 1285, 1260, 1248, 1234, 1198, 1052, 855, 729 cm−1. Anal. calcd for (C17H22NO2·HCl·0.2H2O) C, 65.57; H, 7.25; N, 4.50; found C, 65.29; H, 7.38; N, 4.40.
5.1.5. General procedure for synthesis of 8-hydroxy-13-phenethyl-3,4,10,10a-tetrahydro-1H-1,4a-(epiminoethano)phenanthren-9(2H)-one (9) and 6-hydroxy-13-phenethyl-3,4,10,10a-tetrahydro-1H-1,4a-(epiminoethano)phenanthren-9(2H)-one (18): represented by synthesis of 18
Compound 8 (2.2 g, 8.1 mmol) was dissolved in 1,2-dichloroethane (50 mL). To the solution was added K2CO3 (7.85 g, 56.8 mmol) and ethyl chloroformate (5.28 g, 4.63 mL, 48.7 mmol). The reaction mixture was refluxed under argon for 24 h. Water (100 mL) was added to the reaction mixture after cooling to ambient temperature and the mixture was extracted with CHCl3. The combined extracts were washed with brine and dried over anhydrous Na2SO4. After filtration and concentration, the crude product was purified by flash chromatography (silica, gradient: CHCl3 to 3% MeOH/NH4OH/CHCl3) to afford the intermediate ethyl 6-((ethoxycarbonyl)oxy)-9-oxo-2,3,4,9,10,10a-hexahydro-1H-1,4a-(epiminoethano)phenanthrene-13-carboxylate in 94% yield as a brown oil (3.05 g). 1H NMR (CDCl3, 500 MHz) δ 8.03 (d, J = 8.5 Hz, 1H), 7.15 (d, J = 6.5 Hz, 1H), 7.10 (d, J = 9.0 Hz, 1H), 4.28 (q, J = 7.0 Hz, 2H), 4.16-4.05 (m, 3H), 3.87-3.81 (m, 1H), 3.76-3.71 (m, 1H), 2.82 (td, J = 16.5, 3.0 Hz, 1H), 2.54-2.48 (m, 1H), 2.42-2.35 (m, 1H), 1.96-1.76 (m, 4H), 1.70-1.65 (m, 3H), 1.36 (t, J = 7.0 Hz, 3H), 1.23 (t, J = 7.0 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 195.6, 155.9, 155.4, 154.8 152.8, 129.4, 128.5, 119.6, 118.0, 65.2, 61.3, 50.5, 41.3, 39.5, 38.4, 35.6, 34.9, 33.8, 23.7, 19.8, 14.8, 14.2; ESI-MS 402.2 (M++1); HRMS (ES+) calcd for C22H28NO6 (M + H)+ 402.1917; found, 402.1901.
The above intermediate carbamate (0.15 g, 0.37 mmol) was dissolved in 33% HBr-AcOH (3 mL) at room temperature. The resulting mixture was heated to reflux (110 °C) under argon for 18 h. The solvent was removed and the residue was redissolved in MeOH and basified with a small amount of NH4OH, and then evaporated to dryness. The product was purified by flash chromatography (silica, gradient: 10-40% MeOH/NH4OH/ CHCl3) to yield 6-hydroxy-3,4,10,10a-tetrahydro-1H-1,4a-(epiminoethano)phenanthren-9(2H)-one as a white solid (70 mg, 74%). 1H NMR (CDCl3+CD3OD, 500 MHz) δ 7.59 (d, J = 8.5 Hz, 1H), 6.48-6.46 (m, 2H), 3.49 (td, J = 13.5, 5.5 Hz, 1H), 3.31 (s, 1H), 3.18-3.14 (m, 1H), 2.54-2.48 (m, 1H), 2.39-2.32 (m, 2H), 2.18 (dd, J = 17, 3.5 Hz, 1H), 1.87-1.54 (m, 7H); ESI-MS 258.1 (M++1); HRMS (ES+) calcd for C16H20NO2 (M + H)+ 258.1494; found, 258.1494.
A mixture of the secondary amine prepared above (70 mg, 0.27 mmol), 2-phenylethyl bromide (61 mg, 46 μL, 0.33 mmol) and triethylamine (33 mg, 46 μL, 0.33 mmol) in EtOH (5 mL) was refluxed under argon for 48 h. The solvent was removed and the residue was purified by flash chromatography (silica, gradient: CHCl3 to 15% MeOH/1.5%NH4OH/CHCl3) to afford 6-hydroxy-13-phenethyl-3,4,10,10a-tetrahydro-1H-1,4a-(epiminoethano)phenanthren-9(2H)-one (18) as a white solid (50 mg, 52%). 1H NMR (CDCl3, 500 MHz) δ 7.94 (d, J = 7.5 Hz, 1H), 7.28-726 (m, 2H), 7.21-7.16 (m, 3H), 6.71-6.69 (m, 2H), 3.16-3.15 (m, 2H), 2.98 (s, 1H), 2.91-2.79 (m, 6H), 2.55 (d, J = 13.5 Hz, 1H), 2.45 (d, J = 14 Hz, 1H), 2.28 (d, J = 12.5 Hz, 1H), 2.03-1.94 (m, 2H), 1.84 (s, 2H), 1.66-1.58 (m, 2H); 13C NMR (CDCl3+CD3OD, 100 MHz) δ 197.2, 162.7, 156.1, 140.0, 130.2, 128.6 (2), 128.4 (2), 126.1, 123.3, 114.2, 111.3, 57.5, 55.3, 50.2, 41.5, 38.6, 36.4, 34.8, 33.9, 33.1, 21.7, 18.2; ESI-MS 362.2 (M++1); HRMS (ES+) calcd for C24H28NO2 (M + H)+ 362.2120; found, 362.2127; The free base was converted into its HCl salt for analysis, mp >300 °C (dec). IR (film) νmax = 3707, 3681, 3665, 2973, 2922, 2868, 1673, 1593, 1454, 1282, 1055, 1032, 1012, 870, 758, 703 cm−1. Anal. calcd for (C24H27NO2·HCl·0.3H2O) C, 71.47; H, 7.15; N, 3.47; found C, 71.48; H, 7.23; N, 3.38.
9: white solid (90%)
1H NMR (CDCl3, 400 MHz) δ 12.80 (s, 1H, -OH), 7.47 (t, J = 8.0 Hz, 1H), 7.39-7.28 (m, 5H), 6.82 (t, J = 8.0 Hz, 2H), 3.21-3.15 (m, 2H), 3.03-2.85 (m, 6H), 2.58-2.50 (m, 2H), 2.45-2.40 (m, 1H), 2.12-2.01 (m, 2H), 1.97-1.88 (m, 2H), 1.78-1.72 (m, 2H), 1.56-1.49 (m, 1H); 13C NMR (CDCl3, 100 MHz) δ 204.4, 163.2, 154.4, 140.4, 136.8, 128.7 (2), 128.4 (2), 126.0, 115.6, 115.5, 115.4, 57.6, 55.6, 50.0, 41.1, 38.7, 36.8, 35.0, 34.3, 33.4, 21.7, 18.6; ESI-MS 308.2 (M++1); HRMS (ES+) calcd for C19H22N3O, 308.1673 (M + H)+ found 308.1671. The free base was converted into its HCl salt for analysis, mp 298~302 °C (dec). IR (film) νmax = 3706, 3681, 3664, 2981, 2973, 2938, 2923,2866, 1631, 1576, 1454, 1055, 1032, 1012, 796, 754 cm−1. Anal. calcd for (C24H27NO2·HCl·0.5H2O) C, 70.83; H, 7.18; N, 3.44; found C, 70.88; H, 7.37; N, 3.43.
5.2.1. General procedure for synthesis of 13-methyl-2,3,4,9,10,10a-hexahydro-1H-1,4a-(epiminoethano)-phenanthrene-8,9α-diol (10), 13-methyl-2,3,4,9,10,10a-hexahydro-1H-1,4a-(epiminoethano)-phenanthrene-8,9β-diol (11), 13-phenethyl-2,3,4,9,10,10a-hexahydro-1H-1,4a-(epiminoethano)-phenanthrene-8,9α-diol (12) and 13-phenethyl-2,3,4,9,10,10a-hexahydro-1H-1,4a-(epiminoethano)phenanthrene-8,9β-diol (13), 13-methyl-2,3,4,9,10,10a-hexahydro-1H-1,4a-(epiminoethano)phenanthrene-6,9-diol (19), and 13-phenethyl-2,3,4,9,10,10a-hexahydro-1H-1,4a-(epiminoethano)-phenanthrene-6,9α-diol (22): represented by synthesis of 19
Compound 8 (0.49 g, 1.8 mmol) was dissolved in THF (10 mL) under argon. The solution was cooled to −78 °C in a dry ice-acetone bath. A solution of Superhydride (1 M, 2.72 mL, 2.72 mmol) was added dropwise and the reaction solution was stirred at −78 °C for 1 h and gradually warmed to room temperature overnight. The reaction was quenched with H2O and the pH was adjusted to 9 with 28% NH4OH. The aqueous layer was extracted several times with a mixed solvent of CHCl3 and MeOH (20:1, v/v). The combined extracts were washed with brine and dried over anhydrous Na2SO4. After filtration and concentration, the crude product was purified by flash chromatography (silica, gradient: 10-20% MeOH/NH4OH/CHCl3) to afford a single product, 13-methyl-2,3,4,9,10,10a-hexahydro-1H-1,4a-(epiminoethano)phenanthrene-6,9-diol (19) as a white solid (0.21 g, 43%). 1H NMR (CDCl3+CD3OD, 500 MHz) δ 7.39 (d, J = 7.5 Hz, 1H), 6.68 (d, J = 8.0 Hz, 1H), 6.62 (s, 1H), 4.80 (t, J= 7.5 Hz, 1H), 3.23-3.21 (m, 1H), 3.06 (s, 1H), 2.88 (s, 1H), 2.54 (s, 3H), 2.31 (d, J = 11.5 Hz, 1H), 2.11-1.80 (m, 7H), 1.72-1.61 (m, 3H); 13C NMR (CDCl3, 125 MHz) δ 155.8, 146.8, 129.2, 128.2, 113.6, 111.1, 68.8, 59.0, 51.7, 41.8, 40.2, 36.6, 34.2, 33.8, 33.2, 21.3, 18.4; ESI-MS 274.2 (M++1); HRMS (ES+) calcd for C17H24NO2 (M + H)+ 274.1807; found, 274.1809. IR (film) νmax = 3185, 2942, 2910, 2873, 1618, 1579, 1219, 771 cm−1. Anal. calcd for (C17H23NO2·0.5H2O) C, 72.31; H, 8.57; N, 4.96; found C, 72.42; H, 8.59; N, 4.78.
10: white solid (24%)
1H NMR (CDCl3, 400 MHz) δ 6.93 (t, J = 7.9 Hz, 1H), 6.58 (d, J = 7.6 Hz, 1H), 6.50 (d, J = 7.9 Hz, 1H), 4.92 (m, 1H), 3.00 (td, J = 12.6, 5.0 Hz, 1H), 2.78 (dd, J = 12.0, 7.2 Hz, 1H), 2.59 (m, 1H), 2.30 (s, 3H), 2.17 (dd, J = 13.4, 4.2 Hz, 1H), 1.99-1.63 (m, 7H), 1.57-1.44 (m, 2H), 1.43-1.38 (m, 1H); 13C NMR (CDCl3, 100 MHz) δ 156.8, 148.1, 128.9, 123.4, 116.9, 113.9, 69.4, 58.7, 52.1, 42.6, 40.7, 37.9, 34.7, 34.3, 32.6, 22.2, 18.9; ESI-MS 274.2 (M++1); HRMS (ES+) calcd for C17H24NO2 (M + H)+ 274.1807; found, 274.1805. IR (film) νmax = 3055, 2933, 2866, 2628, 1612, 1578, 1452, 1267, 1229, 1146, 1054, 1007, 797, 763 cm−1. Anal. calcd for (C17H23NO2) C, 74.69; H, 8.48; N, 5.12; found C, 74.56; H, 8.61; N, 5.22.
11: pale yellow solid (20%)
1H NMR (CDCl3+CD3OD, 500 MHz) δ 7.10 (t, J = 8.0 Hz, 1H), 6.73-6.70 (m, 2H), 5.02 (s, 1H), 3.18-3.14 (m, 1H), 2.91-2.88 (m, 1H), 2.82 (s, 1H), 2.50 (s, 3H), 2.34 (d, J = 13.5 Hz, 1H), 2.26 (t, J = 7.0 Hz, 1H), 2.14 (t, J = 12.5 Hz, 1H), 2.02 (d, J = 11.0 Hz, 1H), 1.80-1.62 (m, 6H), 1.54-1.51 (m, 1H); 13C NMR (CDCl3, 125 MHz) δ 155.6, 147.3, 128.4, 122.5, 115.9, 112.2, 61.9, 58.2, 51.4, 41.7, 36.6, 35.6, 33.6, 32.6, 30.8, 21.3, 18.0; ESI-MS 274.2 (M++1); HRMS (ES+) calcd for C17H24NO2 (M + H)+ 274.1807; found, 274.1804. IR (film) νmax = 3192, 2965, 2919, 2877, 2855, 1587, 1471, 1292, 1281, 1037, 931, 787, 747 cm−1. Anal. calcd for (C17H23NO2·i-PrOH) C,72.04; H, 9.37; N, 4.20; found C,72.10; H, 9.41; N, 4.26.
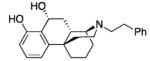
12: white solid (58%, crystallized from i-PrOH and Et2O)
1H NMR (CDCl3, 400 MHz) δ 7.30 (t, J = 8.0 Hz, 2H), 7.23-7.21 (m, 3H), 7.05 (t, J = 8.0 Hz, 1H), 6.67 (d, J = 8.0 Hz, 2H), 4.99 (m, 1H), 3.25-3.11 (m, 2H), 3.02 (s, 1H), 2.95-2.89 (m, 4H), 2.31(dd, J = 13.6, 4.0 Hz, 1H), 2.19 (s, 1H), 2.08-2.03 (m, 2H), 1.99-1.83 (m, 4H), 1.81-1.72 (m, 1H), 1.70-1.55 (m, 3H); 13C NMR (CDCl3, 100 MHz) δ 156.7, 147.3, 139.2, 128.8, 128.6, 128.5, 126.5, 123.2, 116.7, 114.5, 69.4, 57.1, 56.9, 50.5, 39.6, 37.2, 34.6, 34.0, 33.1, 32.4, 21.7, 18.9; ESI-MS 364.2 (M++1); HRMS (ES+) calcd for C24H30NO2 (M + H)+ 364.2277; found, 364.2271. IR (film) νmax = 3062, 3027, 3006, 2939, 2904, 2872, 2846, 2819, 1579, 1454, 1441, 1269, 724, 697 cm−1. Anal. calcd. for (C24H29NO2) C, 79.30; H, 8.04; N, 3.85; found C, 79.36; H, 8.12; N, 3.73.
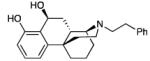
13: white solid (20%, crystallized from i-PrOH and Et2O)
1H NMR (CDCl3, 400 MHz) δ 7.36 (t, J = 7.6 Hz, 2H), 7.29-7.25 (m, 3H), 7.10 (t, J = 8.0 Hz, 1H), 6.75 (d, J = 6.0 Hz, 1H), 6.57 (t, J = 7.2 Hz, 1H), 5.23 (m, 1H), 3.29-3.25 (m, 2H), 3.09 (s, 1H), 2.97-2.92 (m, 4H), 2.71 (t, J = 11.2 Hz, 1H), 2.37 (d, J = 10.0 Hz, 1H), 2.16 (t, J = 11.2 Hz, 1H), 2.07 (d, J = 12.0 Hz, 1H), 1.90-1.83 (m, 3H), 1.75-1.59 (m, 4H); 13C NMR (CDCl3, 100 MHz) δ 156.0, 147.9, 140.0, 128.8 (2), 128.7, 128.5 (2), 126.3, 124.2, 116.0, 112.3, 62.3, 57.6, 56.8, 50.8, 37.3, 35.5, 34.6, 33.6, 33.2, 30.9, 22.2, 19.1; ESI-MS 364.1 (M++1); HRMS (ES+) calcd for C24H30NO2 (M + H)+ 364.2277; found, 364.2262. IR (film) νmax = 2981, 2940, 2923, 1583, 1470, 1455, 1286, 1220, 1055, 1033, 772, 700 cm−1. Anal. calcd for (C24H29NO2·i-PrOH·0.9H2O) C, 73.74; H, 8.89; N, 3.18; found C, 73.60; H, 8.57; N, 3.11.
22: white foam; yield 85.0%
1H NMR (CDCl3+CD3OD, 400 MHz) δ 7.31 (d, J = 8.4 Hz, 1H), 7.22 (t, J = 7.6 Hz, 2H), 7.16-7.12 (m, 3H), 6.60 (d, J = 8.4 Hz, 1H), 6.55 (s, 1H), 4.74 (t, J = 9.6 Hz, 1H), 3.33-2.74 (m, 1H), 3.09 (d, J = 8.4 Hz, 1H), 2.92 (s, 1H), 2.80-2.76 (m, 4H), 2.22 (d, J = 13.6 Hz, 1H), 2.04 (d, J = 12.6 Hz, 1H), 1.96-1.72 (m, 5H), 1.68-1.45 (m, 3H); 13C NMR (CDCl3+CD3OD, 100 MHz) δ 159.8, 151.4, 143.4, 133.3, 132.6, 132.4, 132.1, 130.2, 117.5, 115.2, 73.1, 61.1, 60.6, 54.5, 44.4, 41.1, 38.8, 38.1, 37.6, 37.3, 25.8, 23.0; ESI-MS 364.2 (M++1); HRMS (ES+) calcd for C24H30NO2 (M + H)+ 364.2277; found, 364.2266. IR (film) νmax = 3526, 3396, 3024, 2910, 2871, 2821, 1602, 1490, 1452, 1304, 1251, 1075, 834, 765, 709 cm−1. Anal. calcd for (C24H29NO2•H2O) C, 75.56; H, 8.19; N, 3.67; found C, 75.34; H, 8.29; N, 3.61.
5.2.2. General procedure for synthesis of 13-methyl-2,3,4,10a-tetrahydro-1H-1,4a-(epiminoethano)phenanthren-8-ol (14), 13-phenethyl-2,3,4,10a-tetrahydro-1H-1,4a-(epiminoethano)phenanthren-8-ol (16), 13-methyl-2,3,4,10a-tetrahydro-1H-1,4a-(epiminoethano)phenanthren-6-ol (20), and 13-phenethyl-2,3,4,10a-tetrahydro-1H-1,4a-(epiminoethano)phenanthren-6-ol (23): represented by synthesis of 16
To a solution of mixture of 12 and 13 (0.3 g, 0.82 mmol) in MeOH (10 mL) was added 2 M HCl (2 mL). The resulting solution was stirred at room temperature for 1 h and the solvent was removed. The residue was basified with 28% NH4OH and extracted with CHCl3 (3 × 20 mL). The combined extracts were washed with brine and dried over anhydrous Na2SO4. After filtration and evaporation, the crude product was purified by flash chromatography (silica, gradient: 5% MeOH/NH4OH/CHCl3) to afford 16 (234 mg, 82%) as a yellow foam. 1H NMR (CDCl3, 400 MHz) δ 7.32 (t, J = 7.6 Hz, 2H), 7.27-7.22 (m, 3H), 7.04 (t, J = 8.0 Hz, 1H), 6.88 (dd, J = 9.6, 3.2 Hz, 1H), 6.75 (d, J = 8.0 Hz, 1H), 6.67 (d, J = 8.0 Hz, 1H), 5.62 (dd, J = 9.6, 2.0 Hz, 1H), 3.27-3.14 (m, 3H), 2.96-2.86 (m, 4H), 2.65 (s, 1H), 2.35 (d, J = 11.2 Hz, 1H), 2.13-2.05 (m, 2H), 1.97-1.89 (m, 1H), 1.80-1.69 (m, 1H), 1.65-1.60 (m, 1H), 1.57-1.48 (m, 1H), 1.36-1.32 (m, 1H); 13C NMR (CDCl3, 100 MHz) δ 152.2, 146.2, 140.2, 128.9(2), 128.8(2), 128.4, 128.1, 126.1, 121.4, 121.2, 115.6, 114.9, 57.5, 55.4, 42.9, 34.4, 33.9, 31.4, 21.0, 20.3; ; ESI-MS 346.2 (M++1); HRMS (ES+) calcd for C24H28NO (M + H)+ 346.2171; found, 346.2172. IR (film) νmax = 3707, 3681, 3665, 3016, 2982, 2967, 2939, 2923, 2866, 2845, 2570, 1578, 1464, 1456, 1283, 1055, 1033, 1016, 763, 698 cm−1. Anal. calcd for (C24H27NO·HCl·0.8HCl) C, 72.73; H, 7.53; N, 3.53; found C, 72.66; H, 7.64; N, 3.41.
14: yellow powder (87)
1H NMR (CDCl3+CD3OD, 400 MHz) δ 6.78 (t, J = 8.0 Hz, 1H), 6.66 (dd, J = 9.6, 2.8 Hz, 1H), 6.48 (d, J = 7.6 Hz, 1H), 6.42 (d, J = 8.0 Hz, 1H), 5.40 (d, J = 9.6 Hz, 1H), 2.95 (td, J = 12.6, 5.2 Hz, 1H), 2.75 (dd, J = 12.0, 7.6 Hz, 1H), 2.69 (d, J = 2.0 Hz, 1H), 2.41 (m, 1H), 2.23 (s, 3H), 2.14 (dd, J = 13.6, 5.2 Hz, 1H), 1.86 (m, 2H), 1.68 (m, 1H), 1.51 (m, 1H), 1.38 (m, 1H),1.26 (m, 1H), 1.08 (dd, J = 13.6, 5.6 Hz, 1H); 13C NMR (CDCl3+CD3OD, 100 MHz) δ 156.6, 149.7, 132.1, 131.6, 125.3, 123.7, 118.4, 117.2, 61.5, 55.7, 46.8, 46.1, 38.7, 37.9, 35.2, 24.7, 23.5; ESI-MS, 256.2 (M++1); HRMS calcd for C17H22NO (M + H)+ 256.1701; found 256.1701. IR (film) νmax = 3135, 2956, 2928, 2890, 2681, 2606, 2549, 1577, 1465, 1288, 1272, 1013, 768, 701 cm−1. Anal. calcd for (C17H21NO·HCl· 0.2H2O) C 69.12, H 7.64, N 4.74; Found C 69.13, H 7.78, N 4.84.
20: yellow solid (65%)
1H NMR (CDCl3, 400 MHz) δ 6.87 (d, J = 8.0 Hz, 1H), 6.68 (s, 1H), 6.56 (d, J = 8.0 Hz, 1H), 6.40 (dd, J = 9.4, 2.0 Hz, 1H), 5.47 (d, J = 9.4 Hz, 1H), 3.20-3.12 (m, 1H), 3.07-3.04 (m, 2H), 2.79 (s, 1H), 2.51 (s, 3H), 2.24-2.04 (m, 1H), 1.88-1.83 (m, 1H), 1.75-1.60 (m, 2H), 1.53-1.50 (m, 1H), 1.28-1.20 (m, 1H); 13C NMR (CDCl3, 100 MHz) δ 156.9, 146.1, 128.4, 127.2, 126.4, 124.7, 113.2, 111.3, 57.6, 51.7, 42.8, 42.5, 34.5, 34.0, 31.6, 22.6, 20.9, 19.8; ESI-MS 256.2 (M++1); HRMS (ES+) calcd for C17H22NO (M + H)+ 256.1701; found, 256.1707. IR (film) νmax = 3134, 2948, 2930, 2855, 2493, 1602, 1441, 1247, 848 cm−1. Anal. calcd for (C17H21NO•HCl•0.9H2O) C, 66.29; H, 7.79; N, 4.55; found C, 66.12; H, 7.94; N, 4.39.
23: white foam (95%)
1H NMR (CDCl3, 400 MHz) δ 8.31 (br s, 1H), 7.32 (t, J = 7.6 Hz, 2H), 7.25-7.23 (m, 3H), 6.92 (dd, J = 9.6, 3.2 Hz, 1H), 6.72 (d, J = 4.8 Hz, 1H), 6.61 (d, J = 6.4 Hz, 1H), 6.44 (d, J = 9.6 Hz, 1H), 5.55 (d, J = 10.0 Hz, 1H), 3.22-3.13 (m, 3H), 2.92 (m, 4H), 2.80 (s, 1H), 2.28-2.15 (m, 2H), 2.07 (d, J = 11.2 Hz, 1H), 1.92-1.87 (m, 1H), 1.75-1.62 (m, 2H), 1.54-1.50 (m, 1H), 1.31-1.27 (m, 1H); 13C NMR (CDCl3, 100 MHz) δ 156.8, 146.3, 140.0, 128.8 (2), 128.5 (2), 128.4, 127.2, 126.7, 126.2, 124.9, 113.5, 111.6, 57.4, 55.4, 50.5, 42.7, 34.6, 34.5, 33.7, 31.9, 21.0, 20.3; ESI-MS 346.2 (M++1); HRMS (ES+) calcd for C24H28NO (M + H)+ 346.2171; found 346.2184. The free base was converted into its HCl salt for analysis. IR (film) νmax = 3128, 2948, 2929, 2915, 2851, 2838, 2538, 1603, 1569, 1492, 1442, 1292, 1249, 1232, 1178, 825, 752, 701 cm−1. Anal. calcd for (C24H27NO·HCl·0.3H2O) C, 74.42; H, 7.44; N, 3.62; found C, 74.26; H, 7.43; N, 3.48.
5.2.3. General procedure for synthesis of 13-methyl-2,3,4,9,10,10a-hexahydro-1H-1,4a-(epiminoethano)phenanthren-8-ol (15) and 13-phenethyl-2,3,4,9,10,10a-hexahydro-1H-1,4a-(epiminoethano)phenanthrene-8-ol (17), 13-methyl-2,3,4,9,10,10a-hexahydro-1H-1,4a-(epiminoethano)phenanthren-6-ol (21) and 13-phenethyl-2,3,4,9,10,10a-hexahydro-1H-1,4a-(epiminoethano)phenanthren-6-ol (24): represented by synthesis of 21
Compound 19 (0.16 g, 0.58 mmol) was dissolved into EtOH (15 mL) in a glass bomb and to the solution was added 1.5 mL of water, 0.2 mL of conc. HCl and catalyst 5% Pd/C. The bomb was placed in a Parr apparatus, evacuated and backfilled with H2 for three times, filled with H2 to 60 psi and shaken for 16 h. The reaction mixture was filtered on a pad of celite and washed with MeOH. The filtrate was concentrated and the residue was basified with NH4OH and extracted with CHCl3. The combined extracts were washed with brine and dried over anhydrous Na2SO4. After filtration and concentration, the residue was purified by flash chromatography (silica, gradient, CHCl3 to 10% MeOH/1%NH4OH/CHCl3) to give 21 as a white solid (0.13 g, 87%).
21
1H NMR (CDCl3+CD3OD, 500 MHz) δ 6.88 (d, J = 8.5 Hz, 1 H), 6.68 (s, 1 H), 6.59 (d, J = 8.0 Hz, 1 H), 3.18-3.16 (m, 1 H), 2.97-2.94 (m, 1 H), 2.83-2.80 (m, 3 H), 2.47 (s, 3 H), 2.33-2.30 (m, 1 H), 2.03-1.94 (m, 3 H), 1.89-1.78 (m, 3 H), 1.68-1.55 (m, 4H); 13C NMR (CDCl3+CD3OD, 125 MHz) δ 154.4, 147.1, 129.8, 112.8, 111.4, 58.6, 51.6, 42.8, 41.8, 37.2, 34.1, 34.0, 29.1, 23.3, 21.7, 18.5; ESI-MS 258.2 (M++1); HRMS (ES+) calcd for C17H24NO, 258.1858; found, 258.1857; The free base was converted into its HCl salt for analysis. HCl salt: mp 241.4~247.6 °C. IR (film) νmax = 3707, 3681, 3665, 2981, 2973, 2967, 2938, 2922, 2844, 1055, 1032, 1014 cm−1. Anal. calcd for (C17H24NO·HCl·0.7H2O) C, 66.63; H, 8.35; N, 4.57; found C, 66.49; H, 8.52; N, 4.50.
15: white solid (67%)
1H NMR (CDCl3+CD3OD, 500 MHz) δ 6.70 (t, J = 8.0 Hz, 1 H), 6.46 (d, J= 8.0 Hz, 1 H), 6.33 (d, J = 8.0 Hz, 1 H), 2.95 (td, J = 12.5, 5.0 Hz, 1 H), 2.73-2.66 (m, 2 H), 2.59 (s, 1 H), 2.31-2.20 (m, 4 H), 2.11 (dd, J = 13.5, 4.5 Hz, 1 H), 1.76-1.68 (m, 3 H), 1.61-1.53 (m, 3 H), 1.44 (dd, J = 12.5, 5.0 Hz, 1 H), 1.38-1.30 (m, 3 H); 13C NMR (CDCl3+CD3OD, 125 MHz) δ 154.1, 146.7, 125.8, 121.6, 116.0, 111.3, 59.2, 51.5, 41.7, 41.4, 36.8, 33.6, 23.3, 22.1, 21.1, 18.1; ESI-MS 258.2 (M++1); HRMS (ES+) calcd for C17H24NO, 258.1858; found, 258.1853; The free base was converted into its HCl salt for analysis, mp >300 °C (dec). IR (film) νmax = 3707, 3681, 3218, 2981, 2938, 2923, 2675, 1582, 1464, 1338, 1054, 1012, 1000, 789, 726 cm−1. Anal. calcd for (C17H24NO·HCl·0.25H2O) C, 68.44; H, 8.28; N, 4.69; found C, 68.43; H, 8.48; N, 4.58.
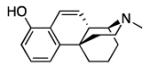
17: white foam (76%)
1H NMR (CDCl3+CD3OD, 400 MHz) δ 7.21-7.11 (m, 5H), 6.86 (t, J = 8.0 Hz, 1H), 6.53 (t, J = 6.0 Hz, 2H), 3.51 (s, 1H), 3.33 (s, 1H), 3.21-3.05 (m, 5H), 2.83 (dd, J = 17.6, 5.2 Hz, 1H), 2.53-2.44 (m, 1H), 2.38-2.34 (m, 2H), 2.04-1.99 (m, 1H), 1.88-1.65 (m, 8H); 13C NMR (CDCl3+CD3OD, 100 MHz) δ 155.2, 145.4, 136.7, 129.5 (2), 129.2 (2), 127.8, 127.1, 122.2, 116.6, 112.8, 59.2, 56.2, 51.6, 40.2, 36.0, 34.1, 33.6, 31.0, 23.7, 22.3, 20.9, 18.1; ESI-MS 348.3 (M++1); HRMS (ES+) calcd for C24H30NO (M + H)+ 348.2327; found, 348.2318. IR (film) νmax = 3237, 2927, 2871, 1581, 1466, 1268, 764, 762 cm−1. Anal. calcd for (C24H29NO•HCl•0.7H2O) C, 72.69; H, 7.98; N, 3.53; found C, 72.74; H, 7.96; N, 3.46.
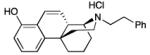
24: white solid; yield 43%
1H NMR (CDCl3+CD3OD, 400 MHz) δ 7.18 (t, J = 6.4 Hz, 2H), 7.13-7.08 (m, 3H), 6.78 (d, J = 8.0 Hz, 1H), 6.59 (s, 1H), 6.48 (d, J = 8.0 Hz, 1H), 3.03-3.00 (m, 2H), 2.85 (s, 1H), 2.73-2.66 (m, 6H), 2.22 (d, J = 13.0 Hz, 1H), 1.93-1.68 (m, 6H), 1.57-1.42 (m, 4H); 13C NMR (CDCl3+CD3OD, 100 MHz) δ 155.2, 148.2, 140.7, 130.7, 129.2 (2), 129.0 (2), 126.7, 126.5, 113.7, 112.4, 57.9, 57.3, 51.3, 43.3, 38.1, 35.3, 35.1, 34.3, 30.1, 24.2, 22.7, 19.8; ESI-MS 348.2 (M++1); HRMS (ES+) calcd for C24H30NO (M + H)+ 348.2327; found, 348.2332. IR (film) νmax = 3130, 2918, 2878, 2838, 2539, 1491, 1454, 1437, 1294, 1230, 754, 699 cm−1. Anal. calcd for (C24H29NO•HCl•0.4H2O) C, 73.69; H, 7.94; N, 3.58; found C, 73.61; H, 7.95; N, 3.52.
5.2.4. 13-Methyl-2,3,4,9,10,10a-hexahydro-1H-1,4a-(epiminoethano)phenanthrene-6,9β-diol (25)
A round flask charged with compound 8 (0.6 g) was evacuated and backfilled with argon. Potassium t-butoxide (1.2 M in THF, 2.65 mL), t-butanol (0.21 mL) and THF (10 mL) were added successively and the solution was cooled to −78 °C (dry ice-acetone bath) and NH3 (g) was bubbled into the mixture. Approximately 20 mL of NH3 (l) was condensed. Lithium metal was added in small pieces until a blue color appeared which persisted for 2 h whereupon the reaction was quenched with solid NH4Cl. The reaction was warmed to ambient temperature and H2O was added. The solution was extracted with CHCl3/MeOH (20/1) and the combined extracts were washed with brine and dried with Na2SO4. The crude product was purified by flash chromatography (silica, gradient: 10% to 15% MeOH/NH4OH/CHCl3) to give compound 10 (0.35 g, 58%) as a white solid and 25 (0.1 g, 17%) as yellow crystals (that were recrystallized in MeOH).
25: white solid
1H NMR (CD3OD, 400 MHz) δ 7.16 (d, J = 8.4 Hz, 1H), 6.70 (d, J = 2.0 Hz, 1H), 6.65 (dd, J = 8.4, 2.0 Hz, 1H), 4.71 (m, 1H), 3.26 (td, J = 12.8, 5.2 Hz, 1H), 2.98 (dd, J = 12.0, 7.2Hz, 1H), 2.82 (m, 1H), 2.51 (s, 3H), 2.43 (d, J = 14.0 Hz, 1H), 2.38 (dd, J = 13.6, 4.8 Hz, 1H), 2.22 (td, J = 14.0, 4.0 Hz, 1H), 2.07 (m, 1H), 1.90 (m, 2H), 1.64 (m, 5H); 13C NMR (CD3OD, 100 MHz) δ157.0, 147.8, 132.0, 126.8, 113.3, 110.9, 66.6, 58.9, 51.6, 41.3, 36.7, 35.8, 34.0, 33.2, 31.9, 21.4, 18.3; ESI-MS, 274.2 (M++1); HRMS calcd for (C17H24NO2) 274.1807 (M + H)+; found 274.1804. IR (film) νmax = 3202, 2942, 2868, 2685, 1606, 1436, 1293, 1242, 1196, 1030, 945, 866, 704 cm−1. Anal.calcd for (C17H23NO2·CH3OH·0.6H2O) C 68.37, H 8.99, N 4.43; found C 68.40, H 8.99, N 4.47.
5.3. Opioid binding assays
As previously described[29], the recombinant CHO cells (hMOR-CHO, hDOR-CHO and hKOR-CHO) were produced by stable transfection with the respective human opioid receptor cDNA, and provided by Dr. Larry Toll (SRI International, CA). The cells were grown in plastic flasks in DMEM (90%) (hDOR-CHO and hKOR-CHO) or DMEM/ F-12 (45%/ 45%) medium (hMOR-CHO) containing 5% FBS (Invitrogen), 5% FetalClone II (HyClone), and Geneticin (G-418: 0.10-0.2 mg/ml) (Invitrogen) under 95% air/5% CO2 at 37° C. Cell monolayers were harvested and frozen in −80 °C. The hKOR-CHO, hMOR-CHO and hDOR-CHO cells are used for opioid binding experiments. For the [35S]-GTP-γ-S binding experiments, we use hKOR-CHO and hMOR-CHO cells for assaying KOR and MOR receptor function. Currently, we use the NG108-15 neuroblastoma×glioma cell for the DOR [35S]-GTP-γ-S binding assay, and obtain an excellent signal-to-noise ratio. In summary, we use the hDOR-CHO cells for DOR binding assays, and the NG108-15 cells for the DOR [35S]-GTP-γ-S binding assay.
As noted formerly[30], [3H][D-Ala2-MePhe4,Gly-ol5]enkephalin ([3H]DAMGO, SA=44-48 Ci/mmol) was used to label MOR, [3H][D-Ala2,D-Leu5]enkephalin ([3H]DADLE, SA=40-50 Ci/mmol) to label DOR and [3H](-)-U69,593 (SA=50 Ci/mmol) to label KOR binding sites. On the day of the assay, cell pellets were thawed on ice for 15 min then homogenized with a polytron in 10 mL/pellet of ice-cold 10 mM Tris-HCl, pH 7.4. Membranes were then centrifuged at 30,000 × g for 10 min, resuspended in 10 ml/pellet ice-cold 10mM Tris-HCl, pH 7.4 and again centrifuged 30,000 × g for 10 min. Membranes were then resuspended in 25 °C 50 mM Tris-HCl, pH 7.4 (~100 mL/pellet hMOR-CHO, 50 mL/pellet hDOR-CHO and 120 mL/pellet hKOR-CHO). All assays took place in 50 mM Tris-HCl, pH 7.4, with a protease inhibitor cocktail [bacitracin (100 μg/mL), bestatin (10 μg/mL), leupeptin (4 μg/mL) and chymostatin (2 μg/mL)], in a final assay volume of 1.0 mL. All drug dilution curves were made up with buffer containing 1 mg/mL BSA. Nonspecific binding was determined using 20 μM levallorphan ([3H]DAMGO and [3H]DADLE) and 1 μM (-)-U69,593 (for [3H]U69,593 binding). [3H]Radioligands were used at ~ 2 nM concentrations. Triplicate samples were filtered with Brandel Cell Harvesters (Biomedical Research & Development Inc., Gaithersburg, MD), over Whatman GF/B filters, after a 2 h incubation at 25 °C. The filters were punched into 24-well plates to which was added 0.6 mL of LSC-cocktail (Cytoscint). Samples were counted, after an overnight extraction, in a Trilux liquid scintillation counter at 44% efficiency. Opioid binding assays had ~30 μg protein per assay tube. Inhibition curves were generated by displacing a single concentration of radioligand by 10 concentrations of drug. The pooled data of three experiments (typically 30 data points) were fit to the two-parameter logistic equation for the best-fit estimates of the IC50 and N values: Y=100/(1+([INHIBITOR]/IC50)N), where “Y” is the percent of control value. Ki values for test drugs are calculated according to the standard equation: Ki = IC50/(1+[radioligand]/Kd]). For the [3H]radioligands, the following Kd values (nM ± SD, n = 3) were used in the Ki calculation: [3H]DAMGO (0.93 ± 0.04), [3H]DADLE (1.9 ± 0.3) and [3H](-)-U69,593 (11 ± 0.6).
5.3.1. [35S]GTP-γ-S binding assays
As noted formerly[30], the assays were conducted with minor modifications of published methods[31]. In this description, buffer “A” is 50 mM Tris-HCl, pH 7.4, containing 100 mM NaCl, 10 mM MgCl2, 1 mM EDTA and buffer “B” is buffer A plus 1.67 mM DTT and 0.15% BSA. On the day of the assay, cells were thawed on ice for 15 min and homogenized using a polytron in 50 mM Tris-HCl, pH 7.4, containing 4 μg/mL leupeptin, 2 μg/mL chymostatin, 10 μg/mL bestatin and 100 μg/mL bacitracin. The homogenate was centrifuged at 30,000 × g for 10 min at 4 °C, and the supernatant discarded. The membrane pellets were resuspended in buffer B and used for [35S]GTP-γ-S binding assays. Test tubes received the following additions: 50 μL buffer A plus 0.1% BSA, 50 μL GDP in buffer A/0.1% BSA (final concentration = 40 μM), 50 μL drug in buffer A/0.1% BSA, 50 μL [35S]-GTP-γ-S in buffer A/0.1% BSA (final concentration = 50 pM), and 300 μL of cell membranes (50 μg of protein) in buffer B. The final concentrations of reagents in the [35S]GTP-γ-S binding assays were: 50 mM Tris-HCl, pH 7.4, containing 100 mM NaCl, 10 mM MgCl2, 1 mM EDTA, 1 mM DTT, 40 μM GDP and 0.1% BSA. Incubations proceeded for 3 h at 25 °C. Nonspecific binding was determined using GTP-γ-S (40 μM). Bound and free [35S]GTP-γ-S were separated by vacuum filtration (Brandel) through GF/B filters. The filters were punched into 24-well plates to which was added 0.6 mL LSC-cocktail (Cytoscint). Samples were counted, after an overnight extraction, in a Trilux liquid scintillation counter at 27% efficiency.
For the [35S]GTP-γ-S binding experiments, Ke values were determined using the “shift” experimental design, agonist (DAMGO) dose-response curves were generated, using hMOR-CHO cells, in the absence and presence (ten points/curve) of a test compound. The data of several experiments, 3 or more, were pooled, and the Ke values were calculated according to the equation: [Test Drug]/(EC50-2/EC50-1 – 1), where EC50-2 is the EC50 value in the presence of the test drug and EC50-1 is the value in the absence of the test drug.
5.3.2 Data analysis of estimated Ki in opioid binding experiments
Formerly, initial dose-ranging experiments (n=1) were conducted using 1 nM, 10 nM, 100 nM, 1000 nM and 10000 nM test drug to facilitate the selection of an appropriate concentration range to be used in the final inhibition curves. We have now adopted a modified approach that we term the “sextuplet” method. The binding inhibition produced by 1 μM of a test compound was determined using six test tubes, rather than three, in order to increase the precision of the result. The “total” and nonspecific binding conditions were also determined as sextuplets. Given the known concentration of radioligand and its Kd value, an estimated Ki value was calculated according to the following equation, which was derived from standard binding equations:
Where “Kd” = the Kd value of the radioligand at the receptor of interest; “[I]” = the concentration of the inhibitor “I” used in the binding assay; “[L]” = the concentration of the radioligand used in the assay; “total binding” = the specific binding of the radioligand in the absence of the inhibitor”; “inhibited binding” = the specific binding of the radioligand in the presence of the inhibitor, I; “Fc” = the fraction of control = “inhibited binding”/“total binding”; “A” = ([L] × (1-Fc))/(Fc × Kd); “B” = A + (1/Fc) – 1. The Ki value is the calculated as follows: Ki = [I]/B.
In general, full dose-response curves were generated only with compounds that had estimated Ki values indicative of high to moderate potency. In this case, the estimated Ki was then used to select the concentration range to be used in the final inhibition curves. In the event, full inhibition curves were not run, the estimated Ki values might be reported. The reported estimated SD was calculated as follows: estimated KI × CV, where CV is the coefficient of variation of the % of control observed with the 1 μM test compound. When full inhibition curves were generated, the pooled data of three experiments (typically 30 data points) were fit to the two-parameter logistic equation for the best-fit estimates of the IC50 and N values: Y=100/(1+([INHIBITOR]/IC50)N), where “Y” is the percent of control value. Ki values for test drugs are calculated according to the standard equation: Ki = IC50/(1+[radioligand]/Kd])[32, 33]. For the [3H]radioligands, the following Kd values (nM ± SD, n = 3) were used in the Ki calculation: [3H]DAMGO (0.93±0.04), [3H]DADLE (1.9±0.3) and [3H](−)-U69,593 (11 ± 0.6). The corresponding Bmax values were (fmol/mg protein ± SD, n = 3): [3H]DAMGO (1912 ± 68), [3H]DADLE (3655 ± 391) and [3H](−)-U69,593 (3320 ± 364)[32, 33].
5.4. X-ray crystal data on compounds 12, 19, and 22
Single-crystal X-ray diffraction data on compounds 12 and 22 were collected using MoKα radiation and a Bruker APEX-2 CCD area detector. Single-crystal X-ray diffraction data on compound 19 were collected using CuKα radiation and a Bruker Platinum 135 CCD area detector. Crystals were prepared for data collection by coating with high viscosity microscope oil. The oil-coated crystal was mounted on a micro-mesh mount (Mitergen, Inc.) and transferred to the diffractometer. The structures were solved by direct methods and refined by full-matrix least squares on F2 values using the programs found in the SHELXTL suite (Bruker, SHELXTL v6.10, 2000, Bruker AXS Inc., Madison, WI). Corrections were applied for Lorentz, polarization, and absorption effects. Parameters refined included atomic coordinates and anisotropic thermal parameters for all non-hydrogen atoms. Hydrogen atoms on carbons were included using a riding model [coordinate shifts of C applied to H atoms] with C-H distance set at 0.96 Å. Complete information on data collection and refinement is available in the supplemental material.
For compound 12 a 0.612 × 0.141 × 0.071 mm3 crystal was prepared for data collection and a data set collected at 100°K. The crystal was triclinic in space group P -1, with unit cell dimensions a = 10.3229(5), b = 10.6517(5), c = 10.8196(5) Å, α = 76.573(2), β = 83.215(2), and γ = 77.843(2). Data was 98.5% complete to 68.26° θ (~ 0.73 Å) with an average redundancy of 2.13. The final anisotropic full matrix least-squares refinement on F2 with 288 variables converged at R1 = 4.46%, for the observed data and wR2 = 12.00% for all data.
For compound 19 a 0.583 × 0.188 × 0.145 mm3 crystal was prepared for data collection and a data set collected at room temperature. The crystal was orthorhombic in space group P 212121, with unit cell dimensions a = 7.4350(2), b = 12.9968(5), and c = 14.6395(6) Å. Data was 95.5% complete to 69.49° θ (~ 0.83 Å) with an average redundancy of 5.45. The final anisotropic full matrix least-squares refinement on F2 with 187 variables converged at R1 = 2.75%, for the observed data and wR2 = 7.49% for all data.
For compound 22 a 0.453 × 0.101 × 0.054 mm3 crystal was prepared for data collection and a data set collected at 100 °K. The crystal was triclinic in space group P -1, with unit cell dimensions a = 10.4069(6), b = 10.5010(6), c = 10.5882(7) Å, α = 81.084(2), β = 68.581(2), and γ = 72.607(2). Data was 97.4% complete to 29.16° θ (~ 0.73 Å) with an average redundancy of 2.16. The final anisotropic full matrix least-squares refinement on F2 with 265 variables converged at R1 = 4.34%, for the observed data and wR2 = 10.19% for all data.
Atomic coordinates for 12, 19, and 22 have been deposited with the Cambridge Crystallographic Data Centre (deposition numbers CCDC 897094, CCDC 897095, and CCDC 897096, respectively). Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK [fax: +44(0)-1223-336033 or deposit@ccdc.cam.ac.uk.
Supplementary Material
Highlights.
 Synthesis of an N-phenethyl substituted carbocyclic tricyclic system with high affinity for μ-opioid receptors.
Synthesis of an N-phenethyl substituted carbocyclic tricyclic system with high affinity for μ-opioid receptors. Potent (Ke < 1 nM) and reasonably μ-selective (μ/κ ratio 20 to 27) opioid antagonists.
Potent (Ke < 1 nM) and reasonably μ-selective (μ/κ ratio 20 to 27) opioid antagonists. Oxygen atoms (carbonyl and OH) on the carbon bridge of the inner ring do not improve affinity.
Oxygen atoms (carbonyl and OH) on the carbon bridge of the inner ring do not improve affinity.
Figure 2.

X-ray crystallographic structure of (1S*,4aR*,9R*,10aS*)-13-phenethyl-2,3,4,9,10,10a-hexahydro-1H-1,4a-(epiminoethano)phenanthrene-8,9-diol (12), (1R,4aS,9S,10aR)-13-methyl-2,3,4,9,10,10a-hexahydro-1H-1,4a-(epiminoethano)phenanthrene-6,9-diol (19) and (1S*,4aR*,9R*,10aS*)-13-phenethyl-2,3,4,9,10,10a-hexahydro-1H-1,4a-(epiminoethano)phenanthrene 6,9-diol (22). Displacement ellipsoids are shown at the 30% level for compounds 19 and 22 and at the 50% level for compound 12.
Acknowledgement
This research was supported by the NIH Intramural Research Programs of the National Institute of Diabetes and Digestive and Kidney Diseases, the National Institute on Drug Abuse, and the National Institute of Alcohol Abuse and Alcoholism, NIH, DHHS. The X-ray crystallographic work was supported by NIDA through an Interagency Agreement #Y1-DA1101 with the Naval Research Laboratory (NRL). We thank Noel Whittaker and Dr. John Lloyd (Mass Spectrometry Facility, NIDDK) for the mass spectral data, and Drs. Klaus Gawrisch and Walter Teague (Laboratory of Membrane Biochemistry and Biophysics, NIAAA) for NMR spectroscopic data.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Probes 45 – see reference[1].
References
- [1].Lim HJ, Deschamps JR, Jacobson AE, Rice KC. Diastereoselective one-pot synthesis of 7- and 8-substituted 5-phenylmorphans. Org. Lett. 2011;13:5322–5325. doi: 10.1021/ol2021862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Burke TR, Jr., Jacobson AE, Rice KC, Weissman BA, Silverton JV. Probes for narcotic receptor mediated phenomena 3. Oxide bridged 5-phenylmorphans. In: Harris LS, editor. Problems of Drug Dependence 1983. Vol. 49. National Institute on Drug Abuse Research Monograph; Washington DC: 1984. pp. 109–113. DHHS ((ADM) 84-1316) [PubMed] [Google Scholar]
- [3].Burke TR, Jr., Jacobson AE, Rice KC, Silverton JV. Probes for narcotic receptor mediated phenomena. 4. Synthesis of (±)-2,3,4,5,6,6a-hexahydro-3-methyl-8-hydroxy-1H-4,11b-methanobenzofuro[2,3-d]azocine, an oxide-bridged 5-(m-hydroxyphenyl)morphan. J. Org. Chem. 1984;49:1051–1056. [Google Scholar]
- [4].Burke TR, Jr., Jacobson AE, Rice KC, Silverton JV. Probes for narcotic receptor mediated phenomena. 6. Synthesis of (±)-(1α,4aα,9aβ)-1,3,4,9a-tetrahydro-2-methyl-2H-1,4-propanobenzofuro[2,3-c]pyridin-8-ol, an oxide-bridged 5-(3-hydroxyphenyl)morphan. J. Org. Chem. 1984;49:2508–2510. [Google Scholar]
- [5].Burke TR, Jr., Jacobson AE, Rice KC, Weissman BA, Huang HC, Silverton JV. Probes for narcotic receptor mediated phenomena. 9. Synthesis of (±)-(3α,6aα,11aβ)-1,3,4,5,6,11a-hexahydro-2-methyl-2H-3,6a-methanobenzofuro[2,3-c]azocin-10-ol, an oxide-bridged 5-(m-hydroxyphenyl)morphan. J. Med. Chem. 1986;29:748–751. doi: 10.1021/jm00155a026. [DOI] [PubMed] [Google Scholar]
- [6].Ohshima E, Kodato S, Yamada K, Rothman RB, Xu H, Jacobson AE, Rice KC. New Synthetic approaches to oxide-bridged 5-phenylmorphans as probes for opioid receptors. In: Harris LS, editor. Problems of Drug Dependence 1994. Vol. 153. National Institute on Drug Abuse Research Monograph; Washington, DC: 1995. p. 126. NIH Publication No 95-3883. 1995. [Google Scholar]
- [7].Yamada K, Flippen-Anderson JL, Jacobson AE, Rice KC. Probes for Narcotic Receptor Mediated Phenomena; 29: Synthesis of rac-(4R,6aR,11bR)-3-methyl 2,3,4,5,6,6a hexahydro-1H 4,11b-methanobenzofuro[3,2-d]azocin-10-ol, the para-a oxide-bridged phenylmorphan isomer, and a new route to rac-(4R,6aR,11bR)-3-methyl-2,3,4,5,6,6a-hexahydro-1H-4,11b-methanobenzofuro[3,2-d]azocin-8-ol, the ortho-a oxide-bridged phenylmorphan isomer. Synthesis-Stuttgart. 2002:2359–2364. [Google Scholar]
- [8].Tadic D, Linders JTM, Flippen-Anderson JL, Jacobson AE, Rice KC. Probes for narcotic receptor mediated phenomena. Part 31: Synthesis of rac-(3R,6aS,11aS)-2-methyl-1,3,4,5,6,11a-hexahydro-2H-3,6a-methanobenzofuro[2,3-c]azocine-10-ol, and azocine-8-ol, the ortho-c and the para-c oxide-bridged phenylmorphan isomers. Tetrahedron. 2003;59:4603–4614. [Google Scholar]
- [9].Linders JTM, Mirsadeghi S, Flippen-Anderson JL, George C, Jacobson AE, Rice KC. Probes for narcotic receptor mediated phenomena. Part 30. Synthesis of rac-(3R,6aS,11aR)-2-methyl-1,3,4,5,6,11a-hexahydro-2H-3,6a-methanobenzofuro[2,3-c]azocin-8-ol, an epoxy isomer of 5-phenylmorphan. Helv. Chim. Acta. 2003;86:484–493. [Google Scholar]
- [10].Kodato S, Linders JTM, Gu X-H, Yamada K, Flippen-Anderson JL, Deschamps JR, Jacobson AE, Rice KC. Synthesis of rac-(1R,4aR,9aR)-2-methyl-1,3,4,9a-tetrahydro-2H-1,4a-propanobenzofuro[2.3-c]pyridin-6-ol. An unusual double rearrangement leading to the ortho- and para–f oxide-bridged phenylmorphan isomers. Org. & Biomolec. Chem. 2004;2:330–336. doi: 10.1039/b312633c. [DOI] [PubMed] [Google Scholar]
- [11].Hashimoto A, Przybyl AK, Linders JTM, Kodato S, Tian XR, Deschamps JR, George C, Flippen-Anderson JL, Jacobson AE, Rice KC. Probes for narcotic receptor-mediated phenomena. 33. Construction of a strained trans-5,6-ring system by displacement of a nitro-activated aromatic fluorine. Synthesis of the penultimate oxide-bridged phenylmorphans. J. Org. Chem. 2004;69:5322–5327. doi: 10.1021/jo040159k. [DOI] [PubMed] [Google Scholar]
- [12].Zezula J, Jacobson AE, Rice KC. A novel divergent synthesis of ortho-hydroxy-e and -f oxidebridged 5-phenylmorphans. Heterocycles. 2007;71:881–889. [Google Scholar]
- [13].Zezula J, Singer LB, Przybyl AK, Hashimoto A, Dersch CM, Rothman RB, Deschamps J, Lee YS, Jacobson AE, Rice KC. Synthesis and pharmacological effects of the enantiomers of the N-phenethyl analogues of the ortho and para e- and f-oxide-bridged phenylmorphans. Org. & Biomol. Chem. 2008;6:2868–2883. doi: 10.1039/b803433h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kurimura M, Liu H, Sulima A, Przybyl AK, Ohshima E, Kodato S, Deschamps JR, Dersch C, Rothman RB, Lee YS, Jacobson AE, Rice KC. Probes for narcotic receptor mediated phenomena. 37. Synthesis and opioid binding affinity of the final pair of oxide-bridged phenylmorphans, the ortho- and para-b isomers and their N-phenethyl analogues, and the synthesis of the N-phenethyl analogues of the ortho- and para-d isomers. J. Med. Chem. 2008;51:7866–7881. doi: 10.1021/jm800913d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kim J-H, Deschamps JR, Rothman RB, Dersch CM, Folk JE, Cheng K, Jacobson AE, Rice KC. Probes for narcotic receptor mediated phenomena. 42. Synthesis and in vitro pharmacological characterization of the N-methyl and N-phenethyl analogues of the racemic ortho-c and para-c oxidebridged phenylmorphans. Bioorg. Med. Chem. 2011;19:3434–3443. doi: 10.1016/j.bmc.2011.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Li F, Folk J, Cheng K, Kurimura M, Deck J. Probes for narcotic receptor mediated phenomena. 43. Synthesis of the ortho-a and para-a, and improved synthesis and optical resolution of the ortho-b and para-b oxide-bridged phenylmorphans: Compounds with moderate to low opioid-receptor affinity. Bioorg. Med. Chem. 2011;19:4330–4337. doi: 10.1016/j.bmc.2011.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang X-P, Carroll FI, Mascarella SW, Westkaemper RB, Mosier PD, Roth BL, Cherezov V, Stevens RC. Structure of the human κ-opioid receptor in complex with JDTic. Nature. 2012;485:327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, Kobilka BK. Structure of the δ-opioid receptor bound to naltrindole. Nature. 2012;485:400–404. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Thompson AA, Liu W, Chun E, Katritch V, Wu H, Vardy E, Huang X-P, Trapella C, Guerrini R, Calo G, Roth BL, Cherezov V, Stevens RC. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature. 2012;485:395–399. doi: 10.1038/nature11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Murineddu G, Ruiu S, Loriga G, Manca I, Lazzari P, Reali R, Pani L, Toma L, Pinna GA. Tricyclic pyrazoles. 3. Synthesis, biological evaluation, and molecular modeling of analogues of the cannabinoid antagonist 8-chloro-1-(2′,4′,-dichlorophenyl)-N-piperidin-1-yl-1,4,5,6- tetrahydrobenzo[6,7]cyclohepta[1,2-c]pyrazole-3-carboxamide. J. Med. Chem. 2005;48:7351–7362. doi: 10.1021/jm050317f. [DOI] [PubMed] [Google Scholar]
- [22].May EL. Structures related to morphine. X. A position isomer of (±)-3-hydroxy-N-methylmorphinan (racemorphan) J. Org. Chem. 1958;23:947–949. [Google Scholar]
- [23].May EL, Murphy JG. Structures related to morphine. II. An isomer of N-methylmorphinan. J. Org. Chem. 1954;19:618–622. [Google Scholar]
- [24].May EL, Murphy JG. Structures related to morphine. IV. m-Substituted phenylcyclohexane derivatives. J. Org. Chem. 1955;20:1197–1201. [Google Scholar]
- [25].Awaya H, May EL, Aceto MD, Merz H, Rogers ME, Harris LS. Racemic and optically active 2,9-dimethyl-5-(m-hydroxyphenyl)morphans and pharmacological comparison with the 9-demethyl homologues. J. Med. Chem. 1984;27:536–539. doi: 10.1021/jm00370a019. [DOI] [PubMed] [Google Scholar]
- [26].Awaya H, May EL, Jacobson AE, Aceto MD. 9α- and 9β-Hydroxyphenylmorphans. J. Pharm. Sci. 1984;73:1867–1868. doi: 10.1002/jps.2600731261. [DOI] [PubMed] [Google Scholar]
- [27].Hiebel AC, Lee YS, Bilsky EJ, Giuvelis D, Deschamps JR, Parrish DA, Aceto MD, May EL, Harris EM, Coop A, Dersch CM, Partilla JS, Rothman RB, Jacobson AE, Rice KC. Probes for narcotic receptor mediated phenomena. 34. synthesis and structure-activity relationships of a potent -agonist δ-antagonist and an exceedingly potent antinociceptive in the enantiomeric C9- substituted 5-(3-hydroxyphenyl)-N-phenylethylmorphan Series. J. Med. Chem. 2007;50:3765–3776. doi: 10.1021/jm061325e. [DOI] [PubMed] [Google Scholar]
- [28].Zhang A, Xiong W, Bidlack JM, Hilbert JE, Knapp BI, Wentland MP, Neumeyer JL. 10-Ketomorphinan and 3-substituted-3-desoxymorphinan analogues as mixed κ and μ opioid ligands:Synthesis and biological evaluation of their binding affinity at opioid receptors. J. Med. Chem. 2003;47:165–174. doi: 10.1021/jm0304156. [DOI] [PubMed] [Google Scholar]
- [29].Fontana G, Savona G, Rodriguez B, Dersch CM, Rothman RB, Prisinzano TE. Synthetic studies of neoclerodane diterpenoids from Salvia splendens and evaluation of opioid receptor affinity. Tetrahedron. 2008;64:10041–10048. doi: 10.1016/j.tet.2008.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Iyer MR, Lee YS, Deschamps JR, Dersch CM, Rothman RB, Jacobson AE, Rice KC. Probes for narcotic receptor mediated phenomena. 44. Synthesis of an N-substituted 4-hydroxy-5-(3-hydroxyphenyl)morphan with high affinity and selective μ-antagonist activity. Eur. J. Med. Chem. 2012;50:44–54. doi: 10.1016/j.ejmech.2012.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Xu H, Hashimoto A, Rice KC, Jacobson AE, Thomas JB, Carroll FI, Lai J, Rothman RB. Opioid peptide receptor studies. 14. Stereochemistry determines agonist efficacy and intrinsic efficacy in the [35S]GTP-γ-S functional binding assay. Synapse. 2001;39:64–69. doi: 10.1002/1098-2396(20010101)39:1<64::AID-SYN9>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- [32].Rothman RB, Murphy DL, Xu H, Godin JA, Dersch CM, Partilla JS, Tidgewell K, Schmidt M, Prisinzano TE. Salvinorin A: Allosteric interactions at the μ-opioid receptor. J. Pharmacol. Exp. Ther. 2007;320:801–810. doi: 10.1124/jpet.106.113167. [DOI] [PubMed] [Google Scholar]
- [33].Xu H, Partilla JS, Wang X, Rutherford JM, Tidgewell K, Prisinzano TE, Bohn LM, Rothman RB. A comparison of noninternalizing (herkinorin) and internalizing (DAMGO) μ-opioid agonists on cellular markers related to opioid tolerance and dependence. Synapse. 2007;61:166–175. doi: 10.1002/syn.20356. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.