

Abstract
Phosphorodiamidate morpholino oligomers (PMO) are neutrally charged, sequence-specific antisense agents that interfere with targeted gene expression. PMO have been shown to be highly specific and potent therapies after cellular uptake, yet methods to detect PMO in tissue and inside the cell are limited. We offer in this report novel methods for the detection of cellular resident PMO using flow cytometry-fluorescence in situ hybridization (flow FISH) and a sandwich hybridization technique to quickly and sensitively quantify tissue resident PMO. These methods rely on oligonucleotide probes complementary to a PMO to specifically detect and quantify cell-associated and tissue resident PMO after in vitro or in vivo administration. Using the sandwich hybridization technique, we show that intranasally delivered PMO demonstrates zero-order clearance kinetics from the lung. Furthermore, PMO was detected in nonhematopoietic and hematopoietic cells of the lung regardless of influenza virus infection, although an increase in PMO uptake in infected hematopoietic cells was observed. Coincident measurement of target knock-down to cell-associated influenza A PMO concentration allowed for the calculation of an EC50.
Key words: drug discovery, gene regulation, nucleic acids, virology
Introduction
Antisense oligonucleotides offer the promise of specific and effective therapy to a wide range of disease states. Phosphorodiamidate morpholino oligomers (PMO) were designed to improve water solubility, increase nuclease resistance and stability, and strengthen target affinity of antisense agents. PMO are designed with the same four bases as DNA but contain nonionic morpholine rings and phosphorodiamidate intersubunit linkages.1 PMO can function by steric blockade of ribosomal or spliceosomal assembly, thereby leading to translational arrest or alteration of splicing.2,3 PMO therapies have been shown to be effective against many RNA viruses including lethal filovirus challenge4 and in inducing novel dystrophin protein expression through exon skipping in Duchenne muscular dystrophy.5 In addition, PMO show an excellent safety profile with only mild renal tubular basophillia/vacuolation in mice and no test-article related effects in nonhuman primates and humans.6–8 Despite the promise of PMO technology as effective therapies, there remains a fundamental challenge of PMO delivery into certain target cells such as immune cells.9 While pharmacokinetic analysis of PMO shows accumulation in kidney and liver, and to a lesser extent spleen, lung, and heart,10 these data do not specify what cell types internalize these molecules. The analytical method that has provided the most accurate information about PMO distribution is the high performance liquid chromatography–duplex method, which employs the use of a 5′-fluorescein-labeled DNA probe that is complementary to the sequence of the analyte PMO. The main limitation to this method is the requirement for whole tissue homogenization, which prevents determination of PMO concentration on a per cell basis. Many diseases affect distinct cell populations within an organ, and a whole tissue analysis does not provide the resolution to determine if the desired cells are permissive to PMO uptake. Previous data has shown that factors such as the activation state of cells can determine PMO uptake9 demonstrating the importance of more specifically identifying specific cell populations for PMO penetration. Hence, new methods to detect PMO are required. Therefore, we have developed two proof-of-concept methods for the detection of PMO. First, we have adapted the “sandwich hybridization” method from Efler et al.11 for analysis by flow cytometry. The advantages of our method are a quick processing through analysis time (4–8 h) and the fluorescent readout by the flow cytometer, which is more sensitive and has a broader detection range than colorimetric and chemiluminescent detection systems. Secondly, we combined the analytical methods of fluorescent in situ hybridization (FISH) and flow cytometric analysis (flow FISH) for detection of cell-associated PMO or various chemical modifications to PMO, which have improved efficacy including the addition of arginine rich cell penetrating peptides (PPMO) or piperazine linkages (PMOplus).4,12,13 Finally, combining this method with detection of protein product from PMO gene target provides critical information linking PMO concentration to efficacy, thereby establishing a meaningful EC50 value.
Material and Methods
PMO design and synthesis
PMO were synthesized at Sarepta Therapeutics (Corvallis, OR) by methods previously described.1 Influenza PMOplus sequence targets the segment 7 of influenza A (CGG T+TAG AAG AC+TCA TC+TTT, where + signifies piperazine linkage). Arenavirus PMOplus sequence is GCC TAG GAT CC+ACG G+TGC GC. Dengue PMOplus sequence is CGG TCC+ACG+TAG AC+TAA CAA CT. For peptide PMO, peptide (RXR)4XB (X stands for 6-aminohexanoic acid and B stands for β-alanine) was covalently conjugated to the 5′ end of each PMO.
Virus
Influenza A/PR/8/34 (H1N1) were obtained from Manoj Pastey (College of Veterinary Medicine, Oregon State University, Corvallis, OR) and grown on MDCK cells. Virus was harvested and stored at −80°C for future use. Virus was titered by standard TCID50 assay.
Mouse infections and PMOplus treatments
All animal experiments were performed with female Balb/C mice (Jackson Laboratories, Bar Harbor, ME). Mice were infected with 8.4 TCID50 in 50 μL of phosphate-buffered saline (PBS) through the intranasal route. Mice were then treated with the indicated amount of PMOplus in 50 μL through the intranasal route. All animal experiments were carried out under biosafety level 2 conditions with protocols approved by the Institutional Animal Care and Use Committee of Oregon State University.
TCID50
Weighed tissues were homogenized with a steel bead (Qiagen, Valencia, CA) in a tissue lyser (Qiagen) for 2 min. Samples were centrifuged at 14,000 g for 3 min and supernatant was taken for TCID50 and sandwich hybridization assay. Six dilution series were made from lung homogenates in tissue culture media. One hundred microliters of each dilution was placed in a 96-well plate and 5×104 AMJ macrophage cells (ATCC CRL-2456) were added in 100 μL/well. Five replicates of each dilution were made. Cells and lung homogenate dilutions were incubated overnight at 37°C. The next day, medium was removed and replaced with 200 μL of fresh medium. Cells were incubated an additional 72 h at 37°C. One hundred microliters of medium was removed and replaced with 100 μL of a 1% solution of chicken red blood cells (RBCs) in PBS. RBCs were incubated for 1 h at 4°C and hemagglutination pattern was read. TCID50 values were calculated using the Reed and Muench method14 and then normalized to input tissue weight.
Sandwich hybridization assay
Lung homogenates were prepared as described for TCID50. A biotinylated capture LNA (5Biosg/+A+A+A+ GA+T+G+A+G+T+C; Exiqon, Woburn, MA) that spanned 11 base pairs of the 3′ end of the PMO was bound in 200 μL at 100 nM to magnetic SA-Dyna Beads (Invitrogen, Grand Island, NY) per manufacturer's protocol. After washing in B&W buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA, 2 M NaCl, 0.1% Tween-20), 50-μL samples or standards of a known concentration of influenza PMOplus were added in B&W buffer in a 96-well plate. Lung homogenate samples were diluted 1:100 in B&W buffer. Samples were incubated at 37°C for 30 min. The beads were washed one time with B&W buffer and then again with B&W buffer without Tween using an Ambion 96-magnet (Invitrogen) to capture the beads each time. A TYE665 fluorescently tagged detector LNA (+C+T+A+A+C+C+G/3TYE665) that spanned the seven base pairs of the 5′ end of the PMO was then added in 50 μL at 100 μM and incubated for 30 min at 37°C. Beads were washed two times with B&W buffer and resuspended for flow cytometry (FC500, Beckman Coulter, Brea, CA). Analysis was conducted using FlowJo 9.0 (Treestar, Ashland, OR).
Macrophage cell infection
Macrophage cells (AMJ2-C11) were plated at 5×105 per well and infected in 25 μL of influenza at 0.1 multiplicity of infection and incubated for 1 h at 4°C. Cells were washed twice with Dulbecco's modified Eagle's medium+0.1% bovine serum albumin and 200 μL of tissue culture media with PMOplus and PPMO were added for overnight incubation.
Flow FISH
Primary lung cells from BALB/c were stained with CD45-PE (BD Biosciences, San Jose, CA) prior to fixation. AMJ macrophages, lung cells, and splenocytes were washed in PBS and fixed with cytofix/cytoperm (BD Biosciences) for 15 min at 4°C and then washed two times with perm/wash solution (BD Biosciences). M2 was detected using 14C2 antibody (Santa Cruz Biotechnology, Santa Cruz, CA) at 10 μg/mL in perm/wash solution. Antibody incubation was conducted at 37°C for 30 min. Secondary antibody (rabbit anti-mouse IgG-Alexa647, Invitrogen) was added at a final concentration of 1:500 in perm/wash and incubated for 15 min at room temp. Cells were washed again with perm/wash followed by an additional fixation for 15 min at 4°C. After washing, cells were treated with RNase (RNase ONE, Promega, Madison, WI) and DNase (RQ1 RNase Free DNase, Promega) in PBS+50 mM MgCl2+0.5% Tween for 30 min at 37°C. To stop the reaction, 4 μL of 50 mM EDTA solution was added for 30 min at 4°C. Cells were washed in hybridization buffer (60% deionized formamide [Sigma, St. Louis, MO], 20×SSC [Sigma], 1×Denhardt Solution [Sigma], 5% dextran sulfate [Sigma], 250 μg/mL sheared salmon sperm DNA [Ambion], 5 mM Na3PO4 [Sigma], 0.1% sodium dodecyl sulfate solution [Sigma]). The cells were then resuspended in 200 μL of hybridization buffer with added probe at 5 μM. The LNA probe was conjugated with biotin tags on the 3′ and 5′ ends from Exiqon (5Biosg/+AAA+GAT+GAG+TCT+TCT+AAC+CG/3Bio). After 2 h of incubation at 42°C, 100 μL of 0.01×SSC was added to the probe mixture for wash. Two hundred microliters of 2×SSC/50% formamide was added and incubated for 30 min at 42°C. Cells were washed in 2×SSC and for biotinylated LNA probes a streptavidin-Alexa 647 (Invitrogen) incubation was conducted at 1:500 in perm/wash for 20 min at 37°C. Cells were washed again in perm/wash prior to flow acquisition. The number of PMO molecules was determined by running standardized beads with known amounts of conjugated fluorochrome as per manufacturer's instructions (Bangs Laboratories, Fishers, IN). Analysis was conducted using FlowJo 9.0.
Results
The sandwich hybridization assay consists of two 8-10mer LNA oligonucleotide probes that are complementary to the PMO sequence (Fig. 1A). The capture probe is biotinylated and the detector probe is linked to a fluorophore. The capture probe was bound to streptavidin-coated magnetic beads for ease of washing and detection by the flow cytometer. After incubation with PMOplus tissue samples, the LNA-fluorophore detector probe was added for quantification of tissue resident PMOplus. To test the robustness of the assay, we intranasally delivered influenza PMOplus in half-log increments from 10 to 1000 μg to influenza-infected mice the day of infection and 24 h post-infection. Lungs were harvested 72 h post-infection and assayed for PMOplus concentrations. To determine the PMOplus concentrations in samples, a standard curve was run on known amounts of PMOplus (Fig. 2A, B). The detection range of the assay is 0.4–400 nM. The molecular weight of influenza PMOplus is 7076, so the upper end of the detection limit is equivalent to 142 ng of PMOplus per 50 μL. Lungs were homogenized in 500 μL, which allows a detection limit up to 1.42 μg per lung. Therefore, lung samples were diluted 1:100 in assay buffer to accommodate the upper end of the dose response. As can be seen in Figure 2C, the flow sandwich hybridization assay showed a linear detection of PMOplus from doses of 10 to 1000 μg (slope=0.7723±0.05537; R2=0.9197). Animals dosed with 10 μg of compound demonstrated 0.011±0.002 μM of PMOplus in lungs, while animals dosed at 1000 μg had 0.378±0.033 μM PMOplus in lungs (Fig. 2C). In animals dosed with an arenavirus PMOplus, which was not complementary to the probes, as well as in untreated animals, the PMO tissue concentration was below the detection threshold (Fig. 2C). Viral lung burden was also quantified in these animals and plotted against tissue resident PMOplus from Figure 2B. A threshold appeared to be reached at 0.3 μM tissue resident PMOplus wherein 1 log reduction in viral load was observed (Fig. 2D). Lastly, kinetics of lung resident PMOplus was assessed using the sandwich hybridization assay. PMOplus was delivered intranasally to naïve mice, and lungs were harvested at the indicated times for PMOplus quantification (Fig. 2E). The lung clearance was zero order (concentration at any time is not a proportion of the lung concentration or first order elimination) based on the finding that the slope of lung concentration versus time relationship was linear and best modeled by the following equation:
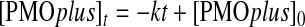 |
FIG. 1.

Schematic of phosphorodiamidate morpholino oligomers (PMO) detection methods. (A) A biotinylated capture LNA was designed to the 3′ end of PMO which is bound to a streptavidin-labeled magnetic bead. After PMO is captured, a fluorescently labeled detector LNA is utilized. (B) A fully complementary fluorescently tagged LNA is used to detect cellular PMO by flow cytometry-fluorescence in situ hybridization (flow FISH).
FIG. 2.

Tissue resident PMO detection using a sandwich hybridization assay by flow cytometry. (A) Flow cytometry histograms of standard curve for PMOplus. Shaded gray histogram represents 4 μM PMOplus input, orange line represents 0.4 μM, yellow line represents 0.04 μM, green line represents 0.004 μM, blue line represents 0.0004 μM, purple line represents 0.00004 μM, and black line signifies no PMO. (B) Standard curve of known amounts of PMO, PMOplus, and PPMO. Data are presented as the mean±SD of two to four replicates of each concentration. (C, D) Influenza-infected mice were dosed with PMOplus the day of infection and 24 h post-infection. Lungs were harvested 72 h post-infection for quantification of PMOplus by flow sandwich hybridization assay and influenza viral RNA. Each data point represents a separate mouse and results are representative of three independent experiments. (C) PMO tissue concentration is plotted with the dose of PMO administered. Slope was determined by linear regression. PBS-treated and arenavirus PMOplus–treated mice had background levels of influenza PMOplus. (D) Influenza PMOplus tissue concentration from (C) was replotted with lung influenza viral burden. (E) PMOplus was delivered intranasally at 0.5 mg/mouse at time=0 and lungs were harvested at the indicated times for PMOplus quantification. Each data point represents a single mouse.
Hence, the rate of PMOplus elimination from the lung is independent of the PMOplus concentration in the lung and the zero order rate constant k=3.5±0.9 nmol/L/h (p<0.0024).
While the sandwich hybridization assay was useful for examining tissue distribution of PMO, it did not yield quality information about cellular distribution. We, therefore, employed the use of a flow FISH assay for detection of cell-associated PMO (Fig. 1B). Fluorescently tagged LNA probe was designed that was complementary to PMO and used to determine the cell-associated concentrations of influenza PMOplus after intranasal delivery. Mice were infected with influenza and treated with influenza-specific PMOplus or dengue PMOplus 4 h prior to infection and 24 h post-infection. Lungs were harvested 48 h post-infection for detection of PMOplus using flow FISH and detection of influenza matrix protein using flow cytometry. To quantify PMOplus levels, standardized beads with known amounts of fluorophore were run (Fig. 3A). Background fluorescent signal from PBS-treated mice was 0.083±0.039 fg/cell and was subtracted from samples. PMOplus was detected in both hematopoietic and nonhematopoietic cells, although higher amounts of PMOplus were found in hematopoietic cells (0.434±0.129 fg and 0.053±0.03 fg for hematopoietic and nonhematopoietic, respectively; Fig. 3D, E). Interestingly, influenza PMOplus was more effective at reducing influenza matrix protein in nonhematopoietic cells than hematopoietic cells (Fig. 3B, C). Influenza PMOplus reduced viral matrix protein in nonhematopoietic cells to background levels (Fig. 3C) with an EC50 of 0.031 fg. Influenza PMOplus reduced matrix protein from a median fluorescence intensity (MFI) of 6.1±0.2 to 5.3±0.07 (background MFI 2.9±0.3) in hematopoietic cells. The higher influenza matrix protein MFI in hematopoietic cells suggests more viral replication in these cells, which could set the bar higher for antiviral activity for the influenza PMOplus.
FIG. 3.

In vivo detection of PMOplus using flow FISH. (A–D) Mice were infected with influenza A virus and treated with influenza-specific PMOplus or a dengue PMOplus 4 h prior to infection and 24 h post-infection. Lungs were harvested 48 h post-infection and hematopoietic (A, B, D) and nonhematopoietic cells (C, E) were probed for influenza A matrix protein with anti-matrix antibody and cell-associated PMO using a complementary LNA sequence as a probe. (A) Flow cytometry histograms of influenza probe fluorescence in hematopoietic cells. Black line represents influenza PMOplus, red line represents dengue PMOplus, and blue line represents PBS. (B–E) Probe fluorescence was transformed into femtograms per cell using standardized microbeads with known amounts of fluorophore. Background signal of PBS-treated mice was subtracted from all samples. Data represent the mean±standard deviation of two animals per group run in duplicate.
Discussion
Currently, there are four different drugs in clinical trials using the phosphorodiamidate morpholino oligmer chemistry ranging from the treatment of Duchenne muscular dystrophy to infectious diseases (www.clinicaltrials.gov). The PMO antisense technology is uniquely versatile in that PMO can utilize both splice switching and translational blockade to up-regulate or down-regulate targeted genes. Furthermore, the neutral structure of the PMO allows vast opportunities to enhance potency, bioavailability, and tissue or cell selectivity. However, more efficient and robust bioanalytical techniques are required to verify potential beneficial characteristics of novel chemical modifications to PMO.
We have developed two novel methods for the detection of cell-associated and tissue resident PMO using LNA probes. The currently used method for PMO detection consists of detecting PMO with a fluorescein-labeled complementary DNA probe after liquid chromatography. This method is time consuming and requires assay development for each new PMO sequence. The sandwich hybridization assay is very quick (2 h) and processing of tissue is simple and straightforward. We have tested two PMO sequences and multiple PMO chemical variations with this assay and found no issues (Fig. 2, data not shown). The drawback to the sandwich hybridization assay is that it only yields tissue distribution data. The main hurdle for PMO as a therapeutic platform is cellular delivery and a technique that can provide specific cellular uptake data will streamline the development of new, more potent delivery modalities. Hence, we developed a novel method for detecting cellular PMO using FISH which provides information of PMO delivery on a single-cell basis.
Acknowledgments
This research was funded in part through contract awards received from the Defense Threat Reduction Agency for H1N1 Countermeasures Development (HDTRA-1-09-0046 and HDTRA-10-C- 0079).
Author Disclosure Statement
All authors receive compensation in the form of salary and benefits from Sarepta Therapeutics, Inc., which provided funding for the studies reported in this manuscript.
References
- 1.Summerton J. Weller D. Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997;7:187–195. doi: 10.1089/oli.1.1997.7.187. [DOI] [PubMed] [Google Scholar]
- 2.Ghosh C. Stein D. Weller D. Iversen P. Evaluation of antisense mechanisms of action. Methods Enzymol. 2000;313:135–143. doi: 10.1016/s0076-6879(00)13008-3. [DOI] [PubMed] [Google Scholar]
- 3.Sazani P. Kole R. Therapeutic potential of antisense oligonucleotides as modulators of alternative splicing. J Clin Invest. 2003;112:481–486. doi: 10.1172/JCI19547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Warren TK. Warfield KL. Wells J, et al. Advanced antisense therapies for postexposure protection against lethal filovirus infections. Nat Med. 2010;16:991–994. doi: 10.1038/nm.2202. [DOI] [PubMed] [Google Scholar]
- 5.Kinali M. Arechavala-Gomeza V. Feng L, et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009;8:918–928. doi: 10.1016/S1474-4422(09)70211-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iversen PL. Arora V. Acker AJ, et al. Efficacy of antisense morpholino oligomer targeted to c-myc in prostate cancer xenograft murine model and a Phase I safety study in humans. Clin Cancer Res. 2003;9:2510–2519. [PubMed] [Google Scholar]
- 7.Sazani P. Ness KP. Weller DL, et al. Chemical and mechanistic toxicology evaluation of exon skipping phosphorodiamidate morpholino oligomers in mdx mice. Int J Toxicol. 2011;30:322–333. doi: 10.1177/1091581811403504. [DOI] [PubMed] [Google Scholar]
- 8.Sazani P. Weller DL. Shrewsbury SB. Safety pharmacology and genotoxicity evaluation of AVI-4658. Int J Toxicol. 2010;29:143–156. doi: 10.1177/1091581809359206. [DOI] [PubMed] [Google Scholar]
- 9.Marshall NB. Oda SK. London CA, et al. Arginine-rich cell-penetrating peptides facilitate delivery of antisense oligomers into murine leukocytes and alter pre-mRNA splicing. J Immunol Methods. 2007;325:114–126. doi: 10.1016/j.jim.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 10.Arora V. Devi GR. Iversen PL. Neutrally charged phosphorodiamidate morpholino antisense oligomers: uptake, efficacy and pharmacokinetics. Curr Pharm Biotechnol. 2004;5:431–439. doi: 10.2174/1389201043376706. [DOI] [PubMed] [Google Scholar]
- 11.Efler SM. Zhang L. Noll BO, et al. Quantification of oligodeoxynucleotides in human plasma with a novel hybridization assay offers greatly enhanced sensitivity over capillary gel electrophoresis. Oligonucleotides. 2005;15:119–131. doi: 10.1089/oli.2005.15.119. [DOI] [PubMed] [Google Scholar]
- 12.Moulton HM. Wu B. Jearawiriyapaisarn N, et al. Peptide-morpholino conjugate: a promising therapeutic for Duchenne muscular dystrophy. Ann NY Acad Sci. 2009;1175:55–60. doi: 10.1111/j.1749-6632.2009.04976.x. [DOI] [PubMed] [Google Scholar]
- 13.Swenson DL. Warfield KL. Warren TK, et al. Chemical modifications of antisense morpholino oligomers enhance their efficacy against Ebola virus infection. Antimicrob Agents Chemother. 2009;53:2089–2099. doi: 10.1128/AAC.00936-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reed L. Meunch H. A simple method of estimating fifty percent endpoints. Am J Hygiene. 1838;27:493–497. [Google Scholar]