

Abstract
Mutations of the intrinsic lysosomal membrane protein SCARB2 cause action myoclonus-renal failure syndrome (AMRF syndrome), a rare disease characterized by renal and neurological manifestations. In this study, examination of Cos7 cells transfected with SCARB2 cDNA derived from two patients with AMRF syndrome showed that the resultant protein was truncated and was not incorporated into vesicular structures, as occurred with full-length SCARB2 cDNA. Mutant SCARB2 protein failed to colocalize with lysosomes and was found in the endoplasmic reticulum or the cytosol indicating a loss of function. Cultured skin fibroblast and Epstein–Barr virus–transformed lymphoblastoid B cell lines (LCLs) were created from these two patients. Despite the loss of SCARB2 function, studies with lysosomal-associated membrane protein (LAMP) 1 and LAMP2 demonstrated normal lysosomal numbers in fibroblasts and LCLs. Immunofluorescence microscopy using anti-LAMP1 and anti-LAMP2 antibodies also showed normal lysosomal structures in fibroblasts. There was no change in the morphology of fibroblasts examined by electron microscopy compared with cells from unaffected individuals. By contrast, LCLs from individuals bearing SCARB2 mutations had large intracellular vesicles that resembled autophagosomes and contained heterogeneous cellular debris. Some of the autophagosomes were seen to be extruding cellular contents into the media. Furthermore, LCLs had elevated levels of microtubule-associated protein light chain 3-II, consistent with increased autophagy. These data demonstrate that SCARB2 mutations are associated with an inability to process autophagosomes in B lymphocytes, suggesting a novel function for SCARB2 in immune function.
Key words: biochemistry, cellular biology, genetics, physiology
Introduction
The intrinsic lysosomal protein SCARB2 (lysosomal integral membrane protein [Limp]-2 in mice) is an intrinsic lysosomal protein with cytosolic N- and C-termini and a heavily glycosylated loop within the lysosome.1 SCARB2 is present within late endosomes and lysosomes2 and may be expressed on the cell surface of some cells. It is a member of the class B scavenger receptor family, which includes the polygamous cell surface receptor CD36. SCARB2 itself is involved in binding and intracellular trafficking of the lysosomal hydrolase β-glucocerebrosidase,3,4 the abnormal protein in Gaucher disease,5 and is also the receptor for enterovirus 71 and Coxsackievirus A16, viruses responsible for hand, foot, and mouth disease.6
Recently, mutations in the SCARB2 gene were identified as the cause of action myoclonus-renal failure syndrome (AMRF syndrome), an autosomal recessive disorder characterized by severe neurological manifestations and renal failure.7,8 Subsequent studies have identified a group of patients with neurological manifestations but apparently normal kidney function.9
During the course of these studies, fibroblast and lymphoblastoid cell lines were obtained from two of the patients. In this study, we have used these cell lines to examine the effect of SCARB2 mutations on human cell morphology.
Materials and Methods
Patients and plasmids
Two patients from the original description7 were studied and agreed to provide blood samples and skin biopsies. Case A is homozygous for the splice site mutation c.1239+1G>T predicted to truncate the protein at amino acid 433. The usual length of SCARB2 is 478 amino acids. Case B is a compound heterozygote, with a frameshift mutation (c.296delA N99IfsX33) inherited from his unaffected mother that predicts truncation of the protein to 131 amino acids. A second mutation was identified in intron 5 of the SCARB2 gene, c.704+5G>A, predicted to affect mRNA splicing. This mutation was not further examined. The Austin Health Human Research Ethics Committee approved all procedures.
RNA isolation, cDNA synthesis and polymerase chain reaction
Total RNA was isolated from lymphoblastoid B cell lines (LCL) cells using the RNeasy kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. cDNA was prepared from the RNA using the Superscript (Invitrogen, Carlsbad, CA). Ten microliters of RNA, 1 μL of 10 mM dNTP (New England Biolabs, Ipswich, MA), and 1 μL of oligo dT primer (Promega, Madison, WI) were incubated at 65°C for 5 min and immediately chilled on ice, after which 4 μL of reaction buffer, 2 μL of 0.1 mM DTT, and 1 μL of RNAsin (Promega) were added, and the mixture heated to 42°C for 2 min. One microliter of superscript was then added and the reaction incubated at 42°C for 50 min. The enzymes were inactivated at 70°C for 15 min and any single-stranded RNA remaining was digested using RNAseH (Promega) for 20 min at 37°C. Two microliters of the cDNA reaction was used in a 50-μL polymerase chain reaction (PCR) volume. PCR was carried out using forward and reverse primers to the human SCARB2 sequence: F5′-TTG CTA AAG CTT CAT GGG CCG ATG CTG CTT CTA-3′ and R5′-CGA ACT GAA TTC TTA GGT TCG AAT GAG GGG TGC T-3′. PCR was performed using Accusure Polymerase (Bioline, London, United Kingdom) and with an initial denaturation at 95°C for 10 min and 30 cycles of denaturation at 95°C/20 sec annealing/extension at 70°C/4 min. PCR products were resolved on 1% agarose gels and bands corresponding to the LIMP2 sequence were excised, the DNA extracted using QIAquick gel extraction kit (Qiagen) and the PCR product was digested by HindIII and EcoRI then cloned in frame into the pEGFP-C2 vector (Clontech, Mountain View, CA). Ligations were transformed into competent Escherichia coli and colonies were grown, then plasmid DNA was extracted using the QIAprep spin mini prep kit (Qiagen). Positive colonies were then sequenced to confirm the identity of the PCR product. For case A and case B, a discrete band was not seen after the first round of PCR; therefore, the bands were excised, the DNA extracted, and this material was used as a template for a second round of PCR. In both cases, a band at around 1.4 kb, corresponding to the size of SCARB2 cDNA coding region, was seen; this was prepared, cloned, and sequenced as described above.
Cell lines and maintenance
Cos7 cells lines were maintained in RPMI-1640 with 20% heat-inactivated fetal calf serum (FCS) and 2 mM glutamine. LCLs were generated from peripheral blood lymphocytes, as previously described.10 They were maintained in RPMI with 20% FCS, 2 mM glutamine, and 50 U/mL benzylpenicillin and 50 μg/mL streptomycin. For culture of human fibroblasts, a 3-mm skin biopsy excised from the upper arm was cut finely and transferred to a tissue culture flask, as previously described.11 Control skin fibroblasts and LCLs were purchased from the Murdoch Childrens Research Institute, Melbourne. Fibroblasts were maintained in RPMI, 20% FCS and 2 mM glutamine, 50 U/mL benzylpenicillin, and 50 μg/mL streptomycin.
Transfection of Cos7 cells
Transfection of cell lines was carried out using Effectene transfection reagent (Qiagen). Normal and mutant SCARB2 was amplified by PCR, using RNA isolated from LCLs, and cloned into pGEM-T with a N-terminal GFP tag. Inserts were checked by cDNA sequencing.
Immunofluorescence microscopy
Cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) at 4°C for 30 min, washed three times in PBS, incubated with 150 mM glycine for 30 min, washed three times in PBS, then incubated with 5% bovine serum albumin (BSA)/0.5% TritonX100/0.025% CHAPS. Cells were incubated in primary antibody in 10% BSA for 16 h at 4°C, washed three times in PBS, then incubated in secondary antibodies conjugated with Alexa 594 (red) or Alexa 488 (green) in 10% BSA for 30 min. Following three further washes in PBS, sections were mounted with UltraCruz Mounting Media (Santa Cruz Biotechnology, Santa Cruz, CA) or fluorescent mounting medium (Dako, Glostrup, Denmark). Images were generated and collected on a confocal laser microscope (Leica Microsystems, Heidelberg, Germany).
Antibodies for Western blotting and immunofluorescence microscopy
Antibodies used were rabbit anti-GRP78BiP (Abcam, Cambridge, United Kingdom), rabbit anti-Limp2 (SCARB2; Novus Biologicals, Littleton, CO, USA), mouse anti–lysosomal-associated membrane protein (Lamp)1 and mouse anti-Lamp2 (Developmental Studies Hybridoma Bank, Iowa City, Iowa), rabbit anti–early endosome antigen 1 (EEA1; Abcam), mouse anti-β-tubulin (Sigma-Aldrich, St. Louis, MO), goat anti-GFP (Rockland, Gilbertsville, PA) and rabbit anti-microtubule-associated protein light chain 3 (LC3; Abcam). For Western blotting, secondary antibodies conjugated with horseradish peroxidase against appropriate species were used (Dako). For immunofluorescence, secondary antibodies conjugated with Alexa 594 or Alexa 488 against appropriate species were used (Invitrogen).
Western blot analysis
Samples were separated by SDS-PAGE and electrically transferred to a polyvinylidene difluoride membrane. The membrane was blocked in 5% casein in Tris-buffered saline (TBS) for 1 hour and then incubated in primary antibody overnight. After washing in TBS 0.05% Tween20 (TBS-T) the membrane was then incubated for 30 minutes in secondary antibody at 1/2500 dilution. After further washing, immunoreactive proteins were detected with West Pico Chemiluminescent Supersignal (Pierce, Rockford, IL). Western blots were quantified by densitometry (Scion Image for Windows, Scion Corporation, Frederick, MD).
Quantification of colocalization
Colocalization of fluorescence of GFP and GRP78BiP in Cos7 cells was quantified using the software NIH ImageJ 1.34 (http://rsbweb.nih.gov/ij). The software was used to measure the area of pixels of SCARB2 (GFP in the green channel) and GRP78BiP (red channel) after adjustment of the threshold to minimize background noise while retaining the signal. This process was performed in the same manner for all analysis of transfected Cos7 samples by the same operator.12 Using JACoP (http://rsbweb.nih.gov/ij/plugins/track/jacop.html), a plugin for the software ImageJ, the Manders' coefficient, as a measure of the ratio of the “area of pixels of GFP colocalizing with GRP78BiP” to that of the total GFP, for each cell was generated. All images were acquired under the same settings, in particular the same pinhole for an identical section thickness of 0.8 μm, by the Zeiss LSM 510 Meta confocal microscope (Göttingen, Germany). Images were analyzed by z-series to ensure that the areas of colocalization existed in all three dimensions and not due to pixels from two different channels at various levels of the sections.
Electron microscopy
Cells were suspended in 2.5% glutaraldehyde in PBS for 2 h at 4°C. Electron microscopy was performed by the Department of Anatomical Pathology, Austin Health (Melbourne, Australia).
Statistical analysis
GraphPad InStat v.3.06 (GraphPad Software, La Jolla, CA) was used for data analysis. Statistical differences between individual experimental groups were compared by Mann-Whitney U-test. Results were expressed as mean±SD. A p value of<0.05 was considered significant.
Results
SCARB2 mutations in patients with AMRF syndrome
GFP-tagged SCARB2 cDNA from a normal individual (wild type [WT]) and two patients with AMRF syndrome (cases A and B) was transfected into Cos7 cells. Western blots of cell lysates probed with anti-GFP (Fig. 1A) showed expression of a GFP-tagged SCARB2 protein that was truncated in case B compared to the WT species. The shorter case B mutant did not bind anti-SCARB2 antibody and there was reduced expression of endogenous SCARB2 (Fig. 1B). In case A, the transfected protein was of similar size to the endogenous SCARB2 due to the presence of the GFP. Therefore it was not possible to determine the effect on endogenous SCARB2 expression.
FIG. 1.

Expression of SCARB2 from two cases of action myoclonus-renal failure syndrome (AMRF syndrome). cDNA encoding GFP-tagged SCARB2 from a normal individual (wild-type [WT]), cases A or B, or GFP alone was transiently transfected into Cos7 cells and the cell lysates separated by SDS-PAGE and analyzed by Western blot. Blots were probed with antibodies to GFP (A), SCARB2 (B), β-tubulin, or lysosomal-associated membrane protein (LAMP)2 lysates from nontransfected cells (NTF) were also used.
Failure of localization of mutant SCARB2 protein to lysosomes
To determine the intracellular distribution of the mutant proteins, transfected cells were examined by immunofluorescence microscopy. It has previously been shown that transfection of cDNA encoding the murine homologue of SCARB2, called Limp2, results in the formation of vesicular structures that contain other lysosomal proteins.2 In Cos7 cells transfected with WT SCARB2 cDNA tagged with GFP, SCARB2 colocalized with vesicular structures, consistent with lysosomal expression (Fig. 2). In contrast, this was not observed in cells transfected with cDNA from case A or B. In both instances, the cells transfected with cDNA from case A or B, GFP fluorescence was less intense and more diffusely expressed throughout the cell. To determine whether the mutant proteins were colocalized to the endoplasmic reticulum (ER), co-staining was performed with Texas Red fluorescent ER marker GRP78. In case A analysis of the immunofluorescence using Mander's coefficient was consistent with colocalization of SCARB2 and the ER (Fig. 2D), and had a significantly greater amount colocalization than WT protein. In cells transfected with cDNA encoding case B, GFP immunofluorescence was detected in only a few samples (n=4), making quantification of these samples technically difficult to perform and interpret. However, in cells with detectable GFP, the mutant protein was diffusely expressed and did not colocalize with the ER, consistent with cytosolic expression.
FIG. 2.

Confocal microscopy of Cos7 cells transfected with GFP-tagged SCARB2 cDNA (green) co-stained with anti-GRP78BiP antibody (red) and DAPI (blue). Vesicular structures are seen in cells transfected with normal SCARB2 cDNA (C), but cDNA from case A (A) or case B (B) was associated with diffuse expression. (D) Quantification of colocalization showed significant colocalization in case A (n=7, *p<0.005) when compared with the control (n=8) but not in case B (n=4). Scale bar=10 μm.
Analysis of lysosomal morphology in SCARB2-deficient cells
Western blots demonstrated that LCLs isolated from cases A and B did not express SCARB2 but had normal levels of LAMP2 and EEA (Fig. 3A). Fibroblast cell lines from affected individuals did not express SCARB2, as has previously been demonstrated,7 but had normal levels of LAMP1 and LAMP2 (Fig. 3B). The inability to detect SCARB2 in either of the cell lines was contrary to the transfected GFP-tagged protein where the SCARB2 antibody demonstrated immunoreactivity with SCARB2 from case A (Fig. 1). Since the transfected protein was tagged on the N-terminal with GFP, we would expect that the effect would stabilize the protein and prevent rapid degradation. With regard to case B the truncation was more severe and the antibody recognition site was missing.
FIG. 3.
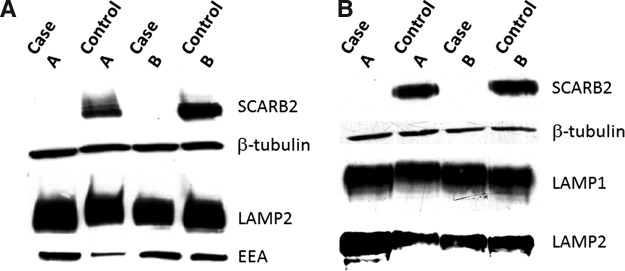
Western blots of cell lysates from patients with AMRF syndrome. (A) Epstein–Barr virus (EBV)-transformed lymphoblastoid cell lines from cases A and B, and control; (B) dermal fibroblasts from cases A and B, and normal individuals. Blots were probed with antibodies to SCARB2, β-tubulin, LAMP2, and LAMP1 or early endosome antigen (EEA).
Immunofluorescence microscopy of fibroblasts from SCARB2-deficient individuals using anti-LAMP1 and anti-LAMP2 antibody showed normal lysosomal structures and no difference when compared with control cells from normal individuals (Fig. 4).
FIG. 4.

Identification of lysosomes in SCARB2 null fibroblast. Immunofluorescence microscopy of fibroblasts from cases (A, B) and controls (C, D) using anti-LAMP1 and LAMP2 Ab. Scale=20 μm. Quantification of LAMP1 (E) and LAMP2 (F) immunofluorescence showed no significant difference between controls (n=19 and 15, respectively) when compared with the cases (n=25 and 21, respectively).
Electron microscopy of SCARB2-deficient cells
To determine whether there was any ultrastructural abnormality in SCARB2-deficient cells, they were grown in normal media and then fixed for electron microscopy. Although there was no difference in the appearance of fibroblasts (data not shown), approximately 30% of LCLs from the two SCARB2-deficient individuals showed large intracellular vesicles (Fig. 5A, B-i–ii) compared with the control (Fig. 5C). In some instances these were in the process of exocytosis (Fig. 5B-i–ii) and contained debris from cellular organelles, consistent with the appearance of autophagosomes (Fig. 5B-iii–iv).
FIG. 5.

Transmission electron microscopy of lymphoblastoid cell lines. Cells from cases (A, B) or control (C) were examined. For case B, an autophagosome-like structure is labeled with an arrow. (B-iii–iv) Magnified images of the autophagosome-like structures of case B. Large autophagosome-like structures are seen in the cell lines from the affected individuals but not the control. Scale bar=15 μm.
Autophagy in SCARB2-deficient cells
In view of the presence of large autophagosomes in LCLs bearing SCARB2 mutations, autophagy was quantified by Western blot for LC3-I and LC3-II. Basal LC3-I and LC3-II levels were increased in both patients when compared to controls (Fig. 6A). Quantified LC3-II/I ratio showed a significantly higher fold increase in patients than controls (Fig. 6B). An increase in the LC3-II/1 ratio is consistent with increased autophagy.
FIG. 6.
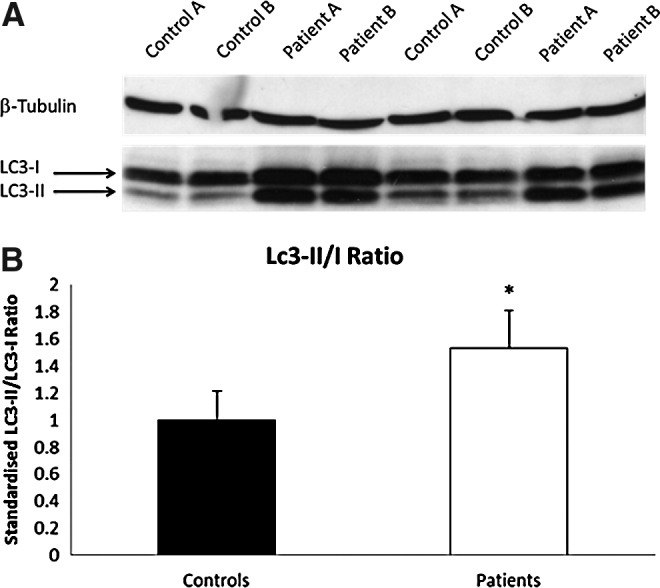
Autophagy is up-regulated in SCARB2-deficient LCLs. (A) Western blots of EBV-transformed lymphoblastoid cell lines from cases A and B and two control samples showing an increase in the amount of LC3-I and LC3-II. (B) Autophagy measured as a significant increase in LC3-II/LC3-I ratio in cases (n=7, *p<0.005) and is represented as a fold increased from control cells (n=8). LC3, microtubule-associated protein light chain 3.
Discussion
Numerous mutations in SCARB2 have been associated with AMRF syndrome in humans.7,8 These comprise splice site, missense, and nonsense mutations. In addition, a further seven mutations were identified in patients who had progressive myoclonus epilepsy with no renal involvement.9,13 This study demonstrates that two of the mutations associated with AMRF syndrome lead to a loss of SCARB2 function and a failure of mutant SCARB2 protein to localize to lysosomes. Cells lacking SCARB2 were, however, able to form lysosomes, as determined by the expression of LAMP1 and LAMP2 in intracellular vesicles. Blanz et al.3 have recently shown that four other SCARB2 mutations fail to exit the ER and reduce expression of endogenous SCARB2 when overexpressed in cells.
In view of the different effects of absence of SCARB2 on various cell types, with most showing no detectable abnormality, there must be different function of SCARB2 in various cell types that cannot be replaced by other molecules. While the morphology of fibroblast cell lines was similar in patients and controls, there were large vacuolar structures in approximately 30% of the LCLs from affected individuals that were not seen in controls. These had the morphological appearance of autophagosomes, since they contained partly degraded intracellular organelles as has been described for early autophagosomes.14,15 Some of these structures appeared to be in the process of exocytosis into the media of the cell cultures. To confirm an up-regulation of autophagy, we examined LC3-I and LC3-II, widely accepted to correlate with increased levels of autophagy.16 This analysis confirmed increased autophagy in SCARB2-deficient LCLs. This is consistent with an important role for SCARB2 in facilitating the fusion of lysosomes with autophagosomes, such that the absence of SCARB2 results in a compensatory up-regulation of autophagy.
These data suggest that fusion of lysosomes with autophagosomes requires SCARB2 in LCLs but not fibroblasts. Whether this requirement relates to the role of B lymphocytes in antigen presentation during the immune response is unknown. Patients with AMRF syndrome have thus far not been reported to suffer any form of immune deficiency or autoimmune disease. Recent data from Carrasco-Marin et al.,17 however, has demonstrated increased susceptibility to infection with Listeria monocytogenes in mice lacking Limp-2, the murine homologue of SCARB2. This was associated with impaired fusion of late endosomes–lysosomes with phagosomes in macrophages, as well as greatly reduced interaction with MHC class II molecules. The origin of the structures identified in the present study is not likely to be phagosomes, because B lymphocytes are not generally capable of phagocytosis.18 In addition, the cells were not challenged with an external pathogen, and phagocytic activity was not observed by electron microscopy.
Conclusions
In summary, this study has demonstrated that cDNA encoding SCARB2 mutations do not affect lysosome formation but give rise to a protein that does not incorporate itself into endosome- and lysosome-like structures, as occurs with the overexpressed wild type protein. In Epstein-Barr virus–transformed B cells, but not fibroblasts from individuals bearing SCARB2 mutations, there was formation of large intracellular vacuoles with the morphological appearances of autophagosomes. These observations indicate an unsuspected role for SCARB2 in B-cell function that could be relevant to the emerging role of autophagy in immunity.19
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Fujita H. Takata Y. Kono A, et al. Isolation and sequencing of a cDNA clone encoding the 85 kDa human lysosomal sialoglycoprotein (hLGP85) in human metastatic pancreas islet tumor cells. Biochem Biophys Res Commun. 1992;184:604–611. doi: 10.1016/0006-291x(92)90632-u. [DOI] [PubMed] [Google Scholar]
- 2.Kuronita T. Eskelinen EL. Fujita H, et al. A role for the lysosomal membrane protein LGP85 in the biogenesis and maintenance of endosomal and lysosomal morphology. J Cell Sci. 2002;115:4117–4131. doi: 10.1242/jcs.00075. [DOI] [PubMed] [Google Scholar]
- 3.Blanz J. Groth J. Zachos C, et al. Disease-causing mutations within the lysosomal integral membrane protein type 2 (LIMP-2) reveal the nature of binding to its ligand beta-glucocerebrosidase. Hum Mol Genet. 2010;19:563–572. doi: 10.1093/hmg/ddp523. [DOI] [PubMed] [Google Scholar]
- 4.Reczek D. Schwake M. Schroder J, et al. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell. 2007;131:770–783. doi: 10.1016/j.cell.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 5.Velayati A. DePaolo J. Gupta N, et al. A Mutation in SCARB2 is a modifier in Gaucher disease. Hum Mutat. 2011;32:1232–1238. doi: 10.1002/humu.21566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamayoshi S. Iizuka S. Yamashita T, et al. Human SCARB2-dependent infection by coxsackievirus A7, A14, and A16 and enterovirus 71. J Virol. 2012;86:5686–5696. doi: 10.1128/JVI.00020-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berkovic SF. Dibbens LM. Oshlack A, et al. Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis. Am J Hum Genet. 2008;82:673–684. doi: 10.1016/j.ajhg.2007.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balreira A. Gaspar P. Caiola D, et al. A nonsense mutation in the LIMP-2 gene associated with progressive myoclonic epilepsy and nephrotic syndrome. Hum Mol Genet. 2008;17:2238–2243. doi: 10.1093/hmg/ddn124. [DOI] [PubMed] [Google Scholar]
- 9.Dibbens LM. Michelucci R. Gambardella A, et al. SCARB2 mutations in progressive myoclonus epilepsy (PME) without renal failure. Ann Neurol. 2009;66:532–536. doi: 10.1002/ana.21765. [DOI] [PubMed] [Google Scholar]
- 10.Neitzel H. A routine method for the establishment of permanent growing lymphoblastoid cell lines. Hum Genet. 1986;73:320–326. doi: 10.1007/BF00279094. [DOI] [PubMed] [Google Scholar]
- 11.Dibbens LM. Tarpey PS. Hynes K, et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet. 2008;40:776–781. doi: 10.1038/ng.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bolte S. Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 13.Dibbens LM. Karakis I. Bayly MA, et al. Mutation of SCARB2 in a patient with progressive myoclonus epilepsy and demyelinating peripheral neuropathy. Arch Neurol. 2011;68:812–813. doi: 10.1001/archneurol.2011.120. [DOI] [PubMed] [Google Scholar]
- 14.Burman C. Ktistakis NT. Autophagosome formation in mammalian cells. Semin Immunopathol. 2010;32:397–413. doi: 10.1007/s00281-010-0222-z. [DOI] [PubMed] [Google Scholar]
- 15.Mehrpour M. Esclatine A. Beau I. Codogno P. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010;20:748–762. doi: 10.1038/cr.2010.82. [DOI] [PubMed] [Google Scholar]
- 16.Klionsky DJ. Cuervo AM. Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy. 2007;3:181–206. doi: 10.4161/auto.3678. [DOI] [PubMed] [Google Scholar]
- 17.Carrasco-Marin E. Fernandez-Prieto L. Rodriguez-Del Rio E, et al. Limp-2 links late phagosomal trafficking with the onset of the innate immune response to Listeria monocytogenes: a role in macrophage activation. J Biol Chem. 2011;286:3332–3341. doi: 10.1074/jbc.M110.146761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chiang SF. Lin TY. Chow KC. Chiou SH. SARS spike protein induces phenotypic conversion of human B cells to macrophage-like cells. Mol Immunol. 2010;47:2575–2586. doi: 10.1016/j.molimm.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saitoh T. Akira S. Regulation of innate immune responses by autophagy-related proteins. J Cell Biol. 2010;189:925–935. doi: 10.1083/jcb.201002021. [DOI] [PMC free article] [PubMed] [Google Scholar]