

Abstract
γ-Tocotrienol is a natural vitamin E that displays potent anticancer activity, and previous studies suggest that these effects involve alterations in PPARγ activity. Treatment with 0.5–6 μM γ-tocotrienol, 0.4–50 μM PPARγ agonists (rosiglitazone or troglitazone), or 0.4–25 μM PPARγ antagonists (GW9662 or T0070907) alone resulted in a dose-responsive inhibition of MCF-7 and MDA-MB-231 breast cancer proliferation. However, combined treatment of 1–4 μM γ-tocotrienol with PPARγ agonists reversed the growth inhibitory effects of γ-tocotrienol, whereas combined treatment of 1–4 μM γ-tocotrienol with PPARγ antagonists synergistically inhibited MCF-7 and MDA-MB-231 cell growth. Combined treatment of γ-tocotrienol and PPARγ agonists caused an increase in transcription activity of PPARγ along with increased expression of PPARγ and RXR, and decrease in PPARγ coactivators, CBP p/300, CBP C-20, and SRC-1, in both breast cancer cell lines. In contrast, combined treatment of γ-tocotrienol with PPARγ antagonists resulted in a decrease in transcription activity of PPARγ, along with decreased expression of PPARγ and RXR, increase in PPARγ coactivators, and corresponding decrease in PI3K/Akt mitogenic signaling in these cells. These findings suggest that elevations in PPARγ are correlated with increased breast cancer growth and survival, and treatment that decreases PPARγ expression may provide benefit in the treatment of breast cancer.
1. Introduction
Peroxisome proliferator activated receptor γ (PPARγ) belongs to the nuclear receptor superfamily and functions as a ligand-activated transcription factor that forms a heterodimer complex with retinoid X receptor (RXR). This complex then binds to a specific DNA sequence called the peroxisome proliferator response element and initiates the recruitment of coactivator proteins such as CBP p/300, SRC-1, and CBP C-20, which further modulate gene transcription [1–3]. Studies have shown that PPARγ is overexpressed in many types of breast cancer cells [4–7]. Experimental evidence in rodents has shown that overexpression of PPARγ is associated with an increased incidence and growth in mammary tumors, whereas knockdown of PPARγ expression was found to significantly inhibit spontaneous mammary tumor development [8, 9]. Taken together these results suggest that inhibition of PPARγ expression and/or activity may be beneficial in the treatment of breast cancer. However, other studies have shown that treatment with the PPARγ agonist rosiglitazone and troglitazone, or conversely with PPARγ antagonists GW9662 and T0070907, were both found to significantly inhibit the growth of a wide variety of cancer cell lines [10, 11]. An explanation for these conflicting findings is not clearly evident, especially since some of the anticancer effects of these agents may be mediated through PPARγ-independent mechanisms. Interpretation of these findings is further complicated by the fact that PPARγ transcriptional activity can be modulated when phosphorylation by Akt and other kinases, which can occur from crosstalk with other mitogenic signaling pathways [12].
γ-Tocotrienol is a member of the vitamin E family of compounds that displays potent anticancer activity [13, 14]. The mechanism(s) involved in mediating the anticancer activity of γ-tocotrienol appear to involve the suppression of growth-factor-dependent mitogenic signaling, particularly the PI3K/Akt signaling pathway [15–18]. PI3K is a lipid signaling kinase that activates PDK-1, which subsequently phosphorylates and activates Akt. Activated Akt phosphorylates various proteins associated with cell proliferation and survival [19]. PDK-1 and Akt activity is terminated by phosphatases such as PTEN [20].
Recent studies have shown that tocotrienols activate specific PPARs in reporter-based assays [21], whereas other studies have shown that γ-tocotrienol increases intracellular levels of 15-lipoxygenase-2, the enzyme responsible for the conversion of arachidonic acid to the PPARγ activating ligand, 15-S-hydroxyeicosatrienooic acid, in prostate cancer cells [22]. Therefore, it was hypothesized that the anticancer effects of γ-tocotrienol may be mediated, at least in part, through a PPARγ-dependent mechanism. Studies were conducted to characterize the effects of γ-tocotrienol treatment alone and in combination with specific PPARγ agonists and antagonists on the growth and survival of MCF-7 and MDA-MB-231 human breast cancer cells. Additional studies evaluated treatment effects on the expression of PPARγ and PPARγ coactivators, and PI3K/Akt mitogenic signaling in these breast cancer cell lines. Results from these studies further characterize the anticancer mechanism of action of γ-tocotrienol, as well as PPARγ agonist and antagonists, and provides insights as to potential benefits of these therapies in the treatment of breast cancer.
2. Materials and Methods
2.1. Reagents and Antibodies
All reagents were purchased from Sigma Chemical Company (St. Louis, MO) unless otherwise stated. Purified γ-tocotrienol (>98% purity) was generously provided as a gift by First Tech International Ltd (Hong Kong). The PPARγ agonists, rosiglitazone and troglitazone, and the PPARγ antagonists, GW9662 and T0070907, were purchased from Cayman Chemicals (San Diego, CA). Fetal bovine serum was purchased from American Type Culture Collection (Manassas, VA). Antibodies for β-actin, PPARγ, Akt, phospho-Akt, PTEN, phospho-PTEN, PDK-1, PI3K, cleaved caspase-3, and cleaved PARP were purchased from Cell Signaling Technology (Beverly, MA). Antibodies for RXR, CBP C-20, SRC-1, and CBP p/300 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Goat anti-rabbit and anti-mouse secondary antibodies were purchased from PerkinElmer Biosciences (Boston, MA).
2.2. Cell Lines and Culture Conditions
The estrogen-receptor negative MDA-MB-231, and the estrogen-receptor positive MCF-7 breast carcinoma cell lines were purchased from American Type Culture Collection (Manassas, VA). MDA-MB-231 and MCF-7 breast cancer cells were cultured in modified Dulbecco's modified Eagle Medium (DMEM)/F12 supplemented with 10% fetal bovine serum, 10 μg/mL insulin, 100 U/mL penicillin, 0.1 mg/mL streptomycin at 37°C in an environment of 95% air and 5% CO2 in a humidified incubator. For subculturing, cells were rinsed twice with sterile Ca2+- and Mg2+-free phosphate-buffered saline (PBS) and incubated in 0.05% trypsin containing 0.025% EDTA in PBS for 5 min at 37°C. The released cells were centrifuged, resuspended in serum containing media, and counted using a hemocytometer.
2.3. Experimental Treatments
The highly lipophilic γ-tocotrienol was suspended in a solution of sterile 10% BSA as described previously [13, 14]. Briefly, an appropriate amount of γ-tocotrienol was first dissolved in 100 μL of 100% ethanol, then added to a small volume of sterile 10% BSA in water and incubated overnight at 37°C with continuous shaking. This stock solution was then used to prepare various concentrations of treatment media. Stock solutions of rosiglitazone, troglitazone, GW9662 and T0070907 were prepared in DMSO. Ethanol and/or DMSO was added to all treatment media such that the final concentration was the same in all treatment groups within any given experiment and was always less than 0.1%.
2.4. Growth Studies
MCF-7 and MDA-MB-231 cells were plated at a density of 5 × 104 cells/well (6 replicates/group) in 24 well culture plates and 1 × 104 cells/well in 96 well culture plate, respectively and allowed to adhere overnight. The next day, cells were divided into different treatment groups, culture media was removed, washed with sterile PBS, then fed fresh media containing their respective treatments, and then returned to the incubator. Cells were treated with media containing 0–50 μM rosiglitazone, troglitazone, GW9662, T0070907 or 0–8 μM γ-tocotrienol alone or a combination for a 4-day culture period. Cells in each treatment group were fed fresh media every other day throughout the experimental period. For apoptosis experiments, MCF-7 and MDA-MB-231 cells were plated as described above. Cells were allowed to grow in control media for 3 days, after which they were exposed to the various treatments for a 24 h period. Treatment with 20 μM γ-tocotrienol has previous been shown to induce apoptosis in breast cancer cells [13, 14] and was used as a positive control in this study.
2.5. Measurement of Viable Cell Number
MCF-7 and MDA-MB-231 viable cell number was determined using the 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyl tetrazolium bromide (MTT) colorimetric assay as described previously [13, 14]. At the end of the treatment period, treatment media was removed and all cells were exposed for 3 h (96 well plates) or 4 h (24 well/plates) to fresh control media containing 0.41 mg/mL MTT at 37°C. Afterwards, media was removed and MTT crystals were dissolved in 1 mL of isopropanol for 24 culture plate or 100 μL of DMSO for 96 culture plate assays. The optical density of each sample was measured at 570 nm at a microplate reader (Spectracount; Packard Bioscience Company, Meriden, CT) zeroed against a blank prepared from cell-free medium. The number of cells per well was calculated against a standard curve prepared by plating known cell densities, as determined by hemocytometer, in triplicate at the start of each experiment.
2.6. Western Blot Analysis
MCF-7 and MDA-MB-231 cells were plated at a density of 1 × 106 cells/100 mm culture dish and exposed to control or treatment media for a 4-day culture period. Afterwards, cells were washed with PBS, isolated with trypsin, and whole cell lysates were prepared in Laemmli buffer [23] as described previously [24]. The protein concentration in each sample was determined using Bio-Rad protein assay kit (Bio-Rad, Hercules, CA). Equal amounts of protein from each sample in a given experiment was loaded onto SDS-polyacrylamide minigels and electrophoresed through 5%–15% resolving gel. Proteins separated on each gel were transblotted at 30 V for 12–16 h at 4°C onto a polyvinylidene fluoride (PVDF) membrane (PerkinElmer Lifesciences, Wellesley, MA) in a Trans-Blot Cell (Bio-Rad, Hercules, CA) according to the method of Towbin et al. [25]. The membranes were then blocked with 2% BSA in 10 mM Tris HCl containing 50 mM NaCl and 0.1% Tween 20 pH 7.4 (TBST) and then incubated with specific primary antibodies against PPARγ, Akt, phospho-Akt, PTEN, phospho-PTEN, PDK-1, PI3K, RXR, CBP C-20, SRC-1, CBP p/300, cleaved capase-3, cleaved PARP or β-actin, diluted 1 : 500 to 1 : 5000 in TBST/2% BSA for 2 h. Membranes are washed 5 times with TBST followed by incubation with the respective horseradish peroxide-conjugated secondary antibodies diluted 1 : 3000 to 1 : 5000 in TBST/2% BSA for 1 h followed by rinsing with TBST. Protein bands bound to the antibody were visualized by chemiluminescence (Pierce, Rockford, IL) according to the manufacturer's instructions and images were obtained using a Kodak Gel Logic 1500 Imaging System (Carestream Health Inc, Rochester, NY). The visualization of β-actin was performed to confirm equal sample loading in each lane. Images of protein bands on the film were acquired and scanning densitometric analysis was performed with Kodak molecular imaging software version 4.5 (Carestream Health Inc, Rochester, NY). All experiments were repeated at least three times and a representative western blot image from each experiment is shown in the figures.
2.7. Transient Transfection and Luciferase Reporter Assay
MCF-7 and MDA-MB-231 cells were plated at a density of 2 × 104 per well in 96-well plates and allowed to adhere overnight. After this cells were transfected with 32 ng of PPRE X3-TK-luc (Addgene plasmid no. 1015) [26] and 3.2 ng of renilla luciferase plasmid per well (Promega, Madison, WI) using 0.8 μL of lipofectamine 2000 transfection reagent for each well (Invitrogen, Grand Island, NY). After 6 h transfection, the media was removed; the cells were washed once and exposed to 100 μL of control or treatment media for a 4-day culture period. Afterwards, cells were lysed with 75 μL of passive lysis buffer and treated according to manufacturer's instructions using dual-glo luciferase assay system (Promega, Madison, WI). Luciferase activity of each sample was normalized by the level of renilla activity. Data is represented as mean fold changes in treated cells as compared to control cells.
2.8. Statistical Analysis
The level of interaction between PPARγ ligands and γ-tocotrienol was evaluated by isobologram method [27]. A straight line was formed by plotting IC50 doses of γ-tocotrienol and individual PPARγ ligands on the x-axes and y-axes, respectively as determined by non-linear regression curve fit analysis using GraphPad Prism 4 (GraphPad Software inc. La Jolla, CA). The data point in the isobologram corresponds to the actual IC50 dose of combined γ-tocotrienol and PPARγ ligands treatment. If a data point is on or near the line, this represents an additive treatment effect, whereas a data point that lies below or above the line indicates synergism or antagonism, respectively. Differences among the various treatment groups in growth studies and western blot studies were determined by analysis of variance followed by Dunnett's multiple range test. Differences were considered statistically significant at a value of P < 0.05.
3. Results
3.1. Antiproliferative Effects of γ-Tocotrienol, PPARγ Agonists (Rosiglitazone and Troglitazone), and PPARγ Antagonists (GW9662 and T0070907)
Treatment with 3–6 μM γ-tocotrienol, 1.6–12 μM rosiglitazone, 6.4–25 μM troglitazone, 1.6–6.4 μM GW9662, or 6.4–25 μM T0070907 was found to significantly inhibit growth of MCF-7 cells in a dose-responsive manner as compared to cells in the vehicle-treated control group (Figure 1(a)). Similarly, treatment with 4–8 μM γ-tocotrienol, 6.4–25 μM rosiglitazone, 3.2–50 μM troglitazone, 3.2–12 μM GW9662, and 12–50 μM T0070907 significantly inhibited MDA-MB-231 cell growth in a dose-responsive manner as compared to cells in the vehicle-treated control group (Figure 1(b)).
Figure 1.
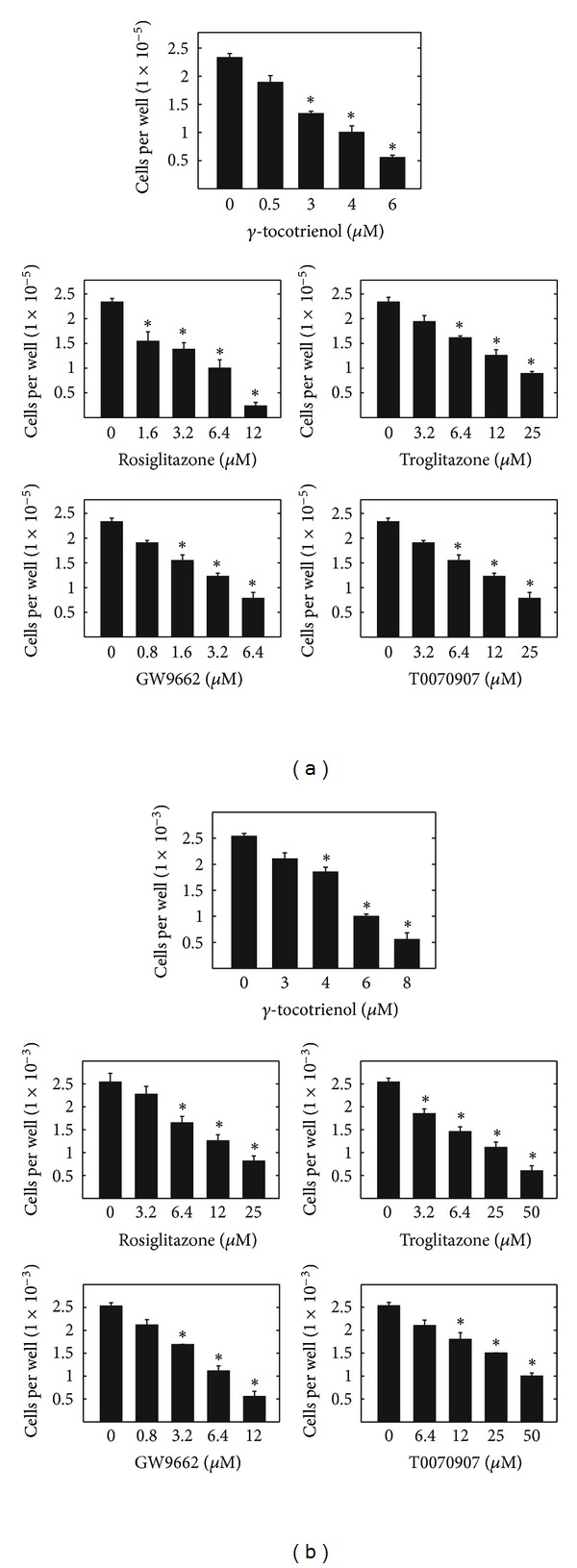
Antiproliferative effects of γ-tocotrienol, PPARγ agonists (rosiglitazone and troglitazone), and PPARγ antagonists (GW9662 and T0070907) on (a) MCF-7 and (b) MDA-MB-231 human breast cancer cells. MCF-7 cells were plated at a density of 5 × 104 (6 wells per group) in 24-well culture plates and exposed to treatment media for a 4-day period. Afterwards viable cell number was determined using MTT colorimetric assay. MDA-MB-231 cells were plated at a density of 1 × 104 (6 wells per group) in 96-well culture plates and exposed to treatment media for a 4-day period. Afterwards viable cell number was determined using MTT colorimetric assay. Vertical bars indicate mean cell count ± SEM in each treatment group. *P < 0.05 as compared with vehicle-treated controls.
3.2. Antagonistic Effects of PPARγ Agonist Rosiglitazone and Troglitazone on the Antiproliferative Effects of γ-Tocotrienol
Treatment with 1–6 μM γ-tocotrienol alone significantly inhibited growth of MCF-7 (Figure 2(a)) and MDA-MB-231 (Figure 2(b)) breast cancer cells after a 4-day treatment period. However, the growth inhibitory effects of 1–4 μM γ-tocotrienol on MCF-7 cells were reversed when given in combination with 3.2 μM rosiglitazone or troglitazone (Figure 2(a), Top and Bottom). A similar, but less pronounced, reversal in 3–6 μM γ-tocotrienol-induced growth inhibitory effects on MDA-MB-231 breast cancer cells was observed when used in combination with 6.4 μM rosiglitazone or troglitazone (Figure 2(b), Top and Bottom).
Figure 2.
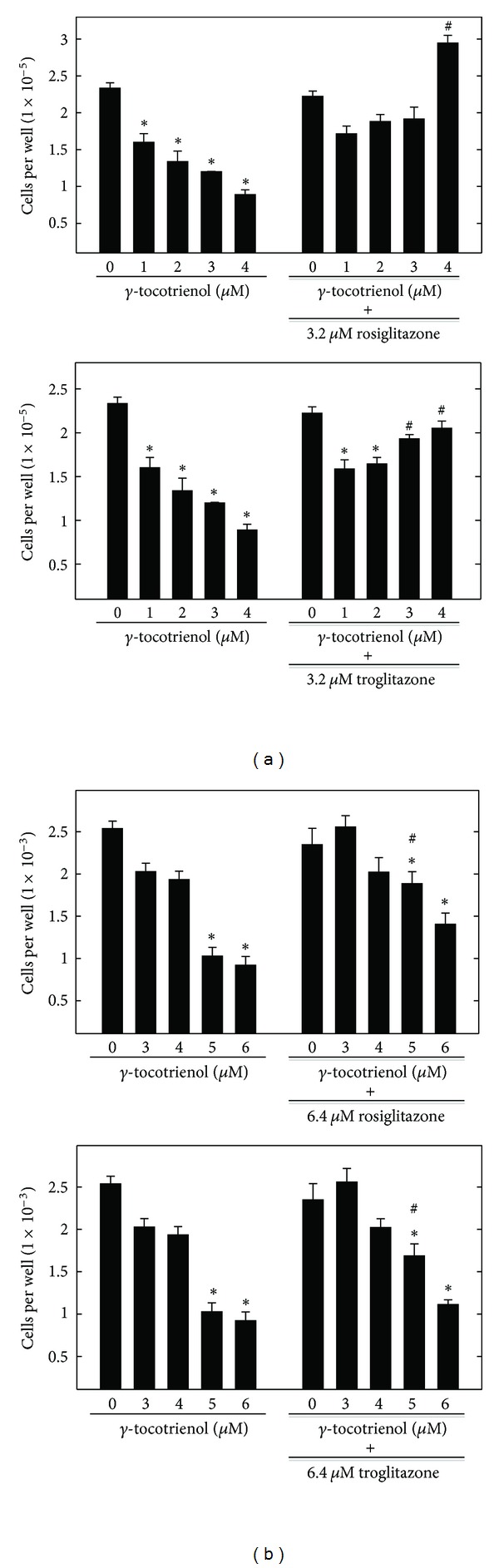
Effects of γ-tocotrienol, PPARγ agonist rosiglitazone and troglitazone treatment alone or in combination on growth of (a) MCF-7 and (b) MDA-MB-231 human breast cancer cells. MCF-7 cells were initially plated at a density of 5 × 104 (6 wells per group) in 24-well plates and (b) MDA-MB-231 were initially plated at a density of 1 × 104 (6 wells per group) in 96-well culture plates and exposed to treatment media for a 4-day period. Afterwards, viable cell number was determined using MTT colorimetric assay. Vertical bars indicate the mean cell count ± SEM in each treatment group. *P < 0.05 as compared with vehicle-treated controls and # P < 0.05 as compared to their corresponding control treated with γ-tocotrienol alone.
3.3. Enhancement of γ-Tocotrienol-Induced Antiproliferative Effects When Given in Combination with PPARγ Antagonist GW9662 or T0070907
The growth inhibitory effects of 1–4 μM γ-tocotrienol was significantly enhanced when given in combination with a subeffective dose (3.2 μM) of the PPARγ antagonist, GW9662, in MCF-7 breast cancer cells (Figure 3(a), Top). A slight, but insignificant enhancement of the growth inhibitory effects 1–4 μM γ-tocotrienol was observed when combined with a subeffective dose (3.2 μM) of the PPARγ antagonist, T0070907, in MCF-7 breast cancer cells (Figure 3(a), Bottom). In MDA-MB-231 cells, 0.5–3 μMγ-tocotrienol was used in combination with 6.4 μM of the PPARγ antagonists, GW9662 (Figure 3(b), Top) or T0070907 (Figure 3(b), Bottom) and was found to significantly enhanced the growth inhibitory effects of these agents. Higher dose ranges of γ-tocotrienol in combination with these same doses of PPARγ antagonists resulted in a complete suppression in breast cancer cell growth such that viable cell number was undetectable using the MTT assay (data not shown).
Figure 3.
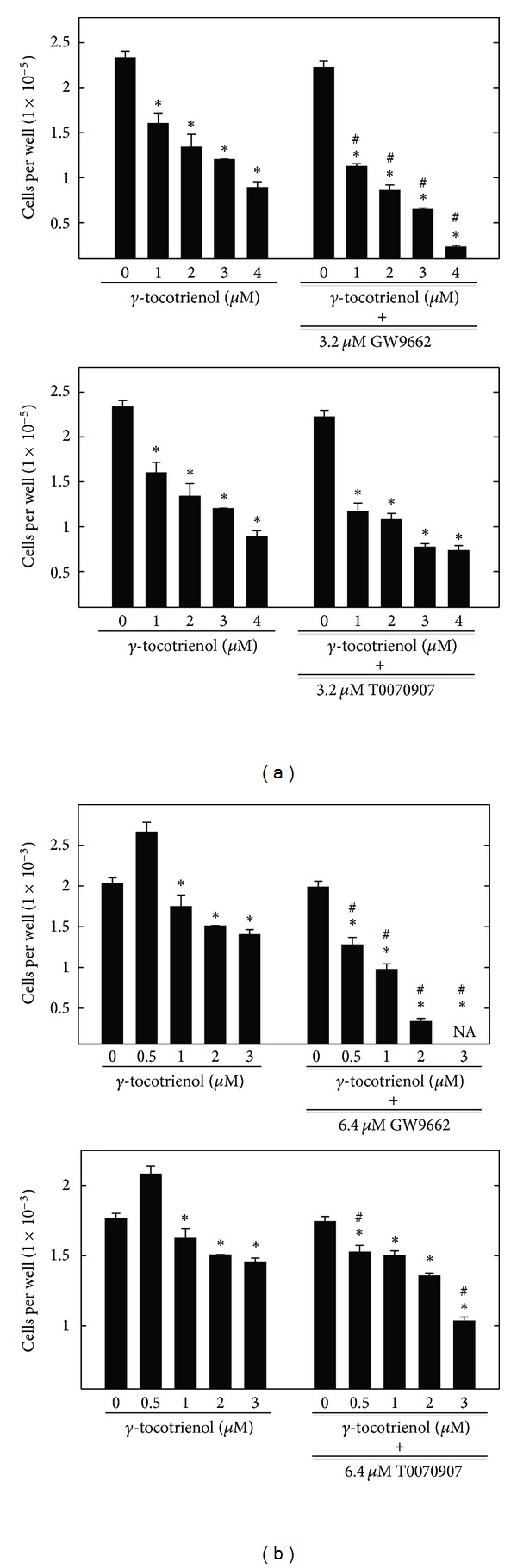
Effects of γ-tocotrienol, PPARγ antagonists GW9662 and T0070907 treatment alone or in combination on growth of (a) MCF-7 and (b) MDA-MB-231 human breast cancer cells. MCF-7 cells were initially plated at a density of 5 × 104 (6 wells per group) in 24-well plates and (b) MDA-MB-231 were initially plated at a density of 1 × 104 (6 wells per group) in 96-well culture plates and exposed to treatment media for a 4-day period. Afterwards, viable cell number was determined using MTT colorimetric assay. Vertical bars indicate the mean cell count ± SEM in each treatment group. *P < 0.05 as compared with vehicle-treated controls and # P < 0.05 as compared to their corresponding control treated with γ-tocotrienol alone.
3.4. Isobologram Analysis of Combined Treatment Effects of γ-Tocotrienol with PPARγ Agonists and Antagonists
Combined treatment of γ-tocotrienol with PPARγ agonists, rosiglitazone, and troglitazone was found to be statistically antagonistic on MCF-7 (Figure 4(a)) and MDA-MB-231 (Figure 4(b)) breast cancer cell growth, as evidenced by the location of the data point in the isobologram being well above the line defining additive effect. In contrast, the growth inhibitory effect of combined treatment of γ-tocotrienol with PPARγ antagonists, GW9662, and T0070907 were found to be statistically synergistic in both MCF-7 (Figure 4(a)) and MDA-MB-231 (Figure 4(b)) breast cancer cells, as evidenced by the location of the data point in the isobologram being well below the line defining additive effect.
Figure 4.
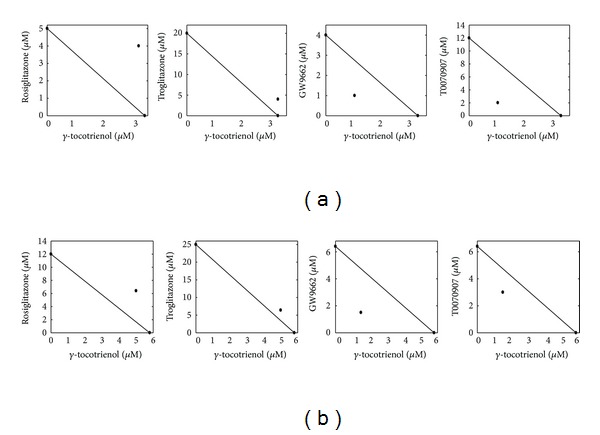
Isobologram analysis of combined treatment of γ-tocotrienol and PPARγ ligands on (a) MCF-7 and (b) MDA-MB-231 human breast cancer cells. Individual IC50 doses for γ-tocotrienol, PPARγ agonists (rosiglitazone and troglitazone), and PPARγ antagonists (GW9662 and T0070907) were calculated and then plotted on the x-axes and y-axes, respectively. The data point on the isobologram represents the actual doses of combined γ-tocotrienol and PPARγ ligands. Combined treatment of PPARγ agonists rosiglitazone and troglitazone with γ-tocotrienol was found to be antagonistic, as evidenced by the location of the data point in the isobologram being well above the line defining additive effect. In contrast, the growth inhibitory effect of combined treatment of γ-tocotrienol with PPARγ antagonists GW9662 and T0070907 was found to be synergistic, as evidenced by the location of the data point in the isobologram being well below the line defining additive effect for both cell lines.
3.5. Effects of γ-Tocotrienol and PPARγ Agonist Rosiglitazone and Troglitazone Given Alone or in Combination on PPARγ and RXR Levels
Western blot analysis shows that treatment with 2 μM (MCF-7 cells) or 3 μM (MDA-MB-231 cells) γ-tocotrienol alone induced a decreased expression of PPARγ and RXR as compared to the vehicle-treated controls (Figures 5(a) and 5(b)). Treatment with 3.2 μM rosiglitazone or troglitazone alone in MCF-7 cells or 6.4 μM rosiglitazone or troglitazone alone in MDA-MB-231 cells had little or no effect on PPARγ or RXR levels (Figures 5(a) and 5(b)). However, combined treatment with similar doses of γ-tocotrienol and rosiglitazone or troglitazone resulted in a significant increase in PPARγ and RXR expression in both MCF-7 and MDA-MB-231 breast cancer cell lines (Figures 5(a) and 5(b)).
Figure 5.
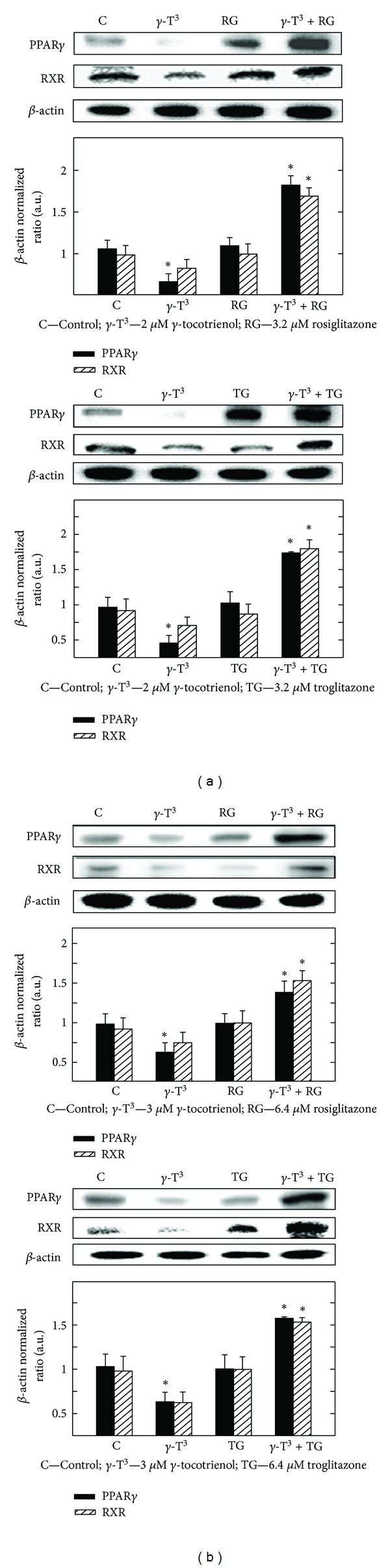
Western blot analysis of γ-tocotrienol and PPARγ agonists (rosiglitazone and troglitazone) given alone or in combination on the levels of PPARγ and RXR after a 4-day incubation period in (a) MCF-7 and (b) MDA-MB-231 human breast cancer cells. MCF-7 cells were initially plated at 1 × 106 cells/100 mm culture dish and treated with control or treatment media containing 2 μM γ-tocotrienol, 3.2 μM rosiglitazone, or troglitazone alone or in combination. MDA-MB-231 cells were plated in a similar manner and treated with control or treatment media containing either 3 μM γ-tocotrienol, 6.4 μM rosiglitazone, or 6.4 μM troglitazone alone or in combination. All cells were fed fresh treatment media every other day for 4-day incubation period. Afterwards, whole cell lysates were prepared for subsequent separation by polyacrylamide gel electrophoresis (50 μg/lane) followed by Western blot analysis. Scanning densitometric analysis was performed on all blots done in triplicate and the integrated optical density of each band was normalized with corresponding β-actin, as shown in bar graphs below their respective Western blot images. Vertical bars in the graph indicate the normalized integrated optical density of bands visualized in each lane ± SEM. *P < 0.05 as compared with vehicle-treated controls.
3.6. Effects of γ-Tocotrienol and PPARγ Antagonist GW9662 and T0070907 Given Alone or in Combination on PPARγ and RXR Levels
Western blot analysis shows that treatment with 2 μM (MCF-7 cells) or 3 μM (MDA-MB-231 cells) γ-tocotrienol alone induced decrease expression of PPARγ and RXR as compared to the vehicle-treated controls (Figures 6(a) and 6(b)). Treatment with 3.2 μM (MCF-7 cells) or 6.4 μM (MDA-MB-231 cells) of the PPARγ antagonists, GW9662 or T0070907 alone had only slight effects on PPARγ and RXR expression (Figures 6(a) and 6(b)). However, combined treatment with these same doses of γ-tocotrienol and GW9662 or T0070907 caused a significant reduction in PPARγ and its heterodimer partner, RXR, in both MCF-7 and MDA-MB-231 cells as compared to vehicle treated controls (Figures 6(a) and 6(b)).
Figure 6.
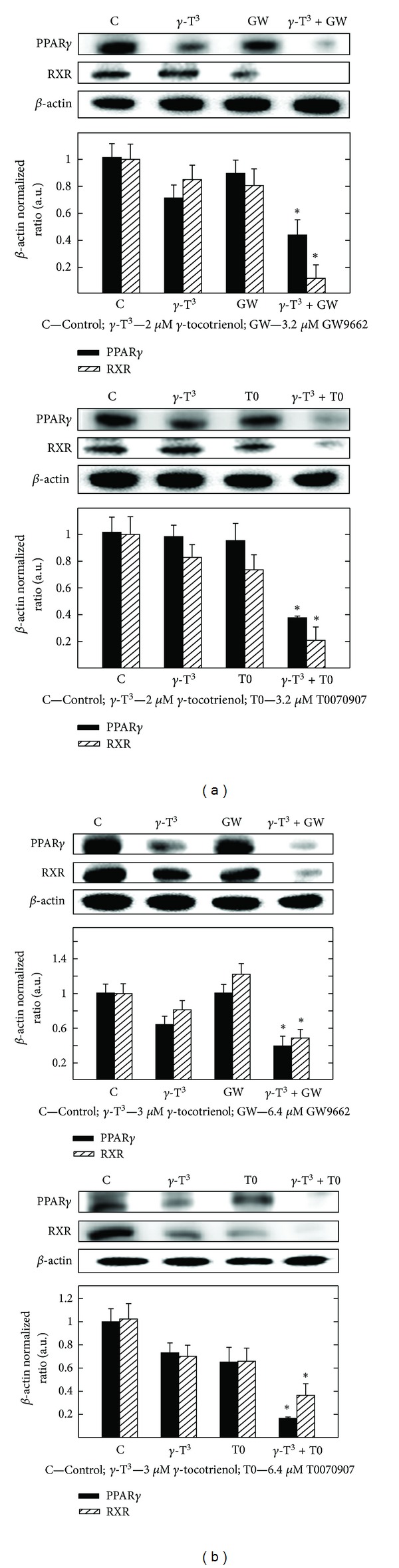
Western blot analysis of γ-tocotrienol and PPARγ antagonists (GW9662 and T0070907) given alone or in combination on the levels of PPARγ and RXR after a 4-day incubation period in (a) MCF-7 and (b) MDA-MB-231 cells. MCF-7 cells were initially plated at 1 × 106 cells/100 mm culture dish and treated with control or treatment media containing either 2 μM γ-tocotrienol, 3.2 μM GW9662, or T0070907 alone or in combination. MDA-MB-231 cells were plated in a similar manner and treated with control or treatment media containing either 3 μM γ-tocotrienol, 6.4 μM GW9662, or 6.4 μM T0070907 alone or in combination. All cells were fed fresh treatment media every other day for 4-day incubation period. Afterwards, whole cell lysates were prepared from each treatment group for subsequent separation by polyacrylamide gel electrophoresis (50 μg/lane) followed by Western blot analysis. Scanning densitometric analysis was performed on all blots done in triplicate and the integrated optical density of each band was normalized with corresponding β-actin, as shown in bar graphs below their respective Western blot images. Vertical bars in the graph indicate the normalized integrated optical density of bands visualized in each lane ± SEM. *P < 0.05 as compared with vehicle-treated controls.
3.7. Effects of γ-Tocotrienol and PPARγ Agonist Rosiglitazone and Troglitazone Given Alone or in Combination on PPRE Mediated Reporter Activity
Luciferase assay shows that the treatment with 2 μM (MCF-7 cells) or 3 μM (MDA-MB-231 cells) γ-tocotrienol alone induced only slight effects in the PPRE mediated reporter activity as compared to vehicle treated controls (Figures 7(a) and 7(b), Top and Bottom). Treatment with 3.2 μM (MCF-7 cells) or 6.4 μM (MDA-MB-231 cells) with the PPARγ agonists, rosiglitazone, and troglitazone, or PPARγ antagonists, GW9662 and T0070907, alone, caused a slight, but insignificant decrease in PPRE mediated reporter activity (Figures 7(a) and 7(b), Top and Bottom). However, combined treatment with these same doses of γ-tocotrienol and rosiglitazone or troglitazone caused an increase in transcription activity of PPARγ in both MCF-7 and MDA-MB-231 cells as compared to vehicle-treated controls (Figures 7(a) and 7(b), Top). In contrast, combined treatment with these same doses of γ-tocotrienol and GW9662 or T0070907 caused a significant decrease PPRE mediated reporter activity in both MCF-7 and MDA-MB-231 cells as compared to vehicle-treated controls (Figures 7(a) and 7(b), Bottom).
Figure 7.
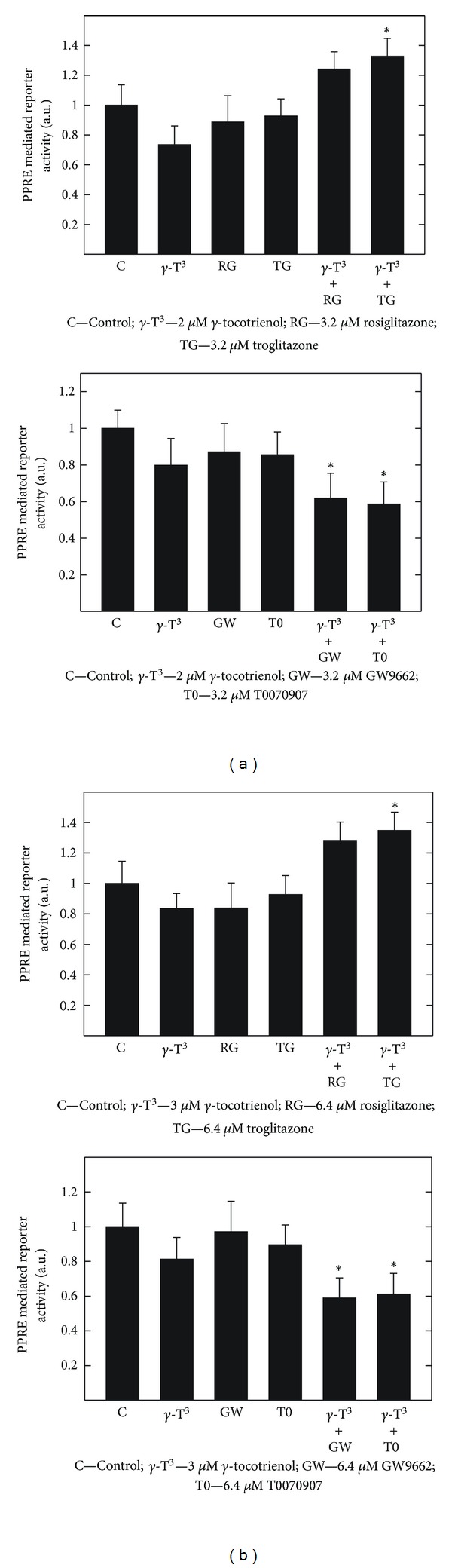
Luciferase assay was performed on (a) MCF-7 and (b) MDA-MB-231 human breast cancer cells. The cells were initially plated at a density of 2 × 104 cells/well in 96-well plates. Cells were then transfected by adding 32 ng of PPRE X3-TK-luc and 3.2 ng of renilla luciferase plasmid in 0.8 μL of lipofectamine 2000 transfection reagent. Following a 6-h incubation period, MCF-7 cells were treated with control or treatment media containing 0–2 μM γ-tocotrienol, 0–3.2 μM rosiglitazone, 0–3.2 μM troglitazone, 0–3.2 μM GW9662, or 0–6.4 μM T0070907 alone or in combination. MDA-MB-231 cells were initially plated in a similar manner and treated with control or treatment media containing 0–3 μM γ-tocotrienol, 0–6.4 μM rosiglitazone, 0–6.4 μM troglitazone, 0–6.4 μM GW9662, or 0–6.4 μM T0070907 alone or in combination. All cells were fed fresh treatment media every other day for 4-day incubation period. Afterwards, cells were lysed with 75 μL of passive lysis buffer and treated according to manufacturer's instructions using the dual-glo luciferase assay system. Results were calculated as raw luciferase units divided by raw renilla units. Vertical bars indicate PPRE mediated reporter activity ± SEM (arbitrary units) in each treatment group. *P < 0.05 as compared with vehicle-treated controls.
3.8. Effects of γ-Tocotrienol and PPARγ Agonist Rosiglitazone and Troglitazone Given Alone or in Combination on Coactivator Expression
Western blot analysis shows that treatment with 2 μM (MCF-7 cells) or 3 μM (MDA-MB-231 cells) γ-tocotrienol alone induced only slight, but insignificant effects in the expression of CBP p/300, CBP C-20, or SRC-1 as compared to the vehicle-treated controls (Figures 8(a) and 8(b)). Treatment with 3.2 μM (MCF-7 cells) or 6.4 μM (MDA-MB-231 cells) with the PPARγ agonists, rosiglitazone and troglitazone alone caused a slight decrease in CBP p/300 and SRC-1, but not CBP C-20, expression (Figures 8(a) and 8(b)). However, combined treatment with these same doses of γ-tocotrienol and rosiglitazone and troglitazone cause a significant decrease in CBP p/300, CBP C-20, or SRC-1 expression in both MCF-7 and MDA-MB-231 cells as compared to vehicle treated controls (Figures 8(a) and 8(b)).
Figure 8.
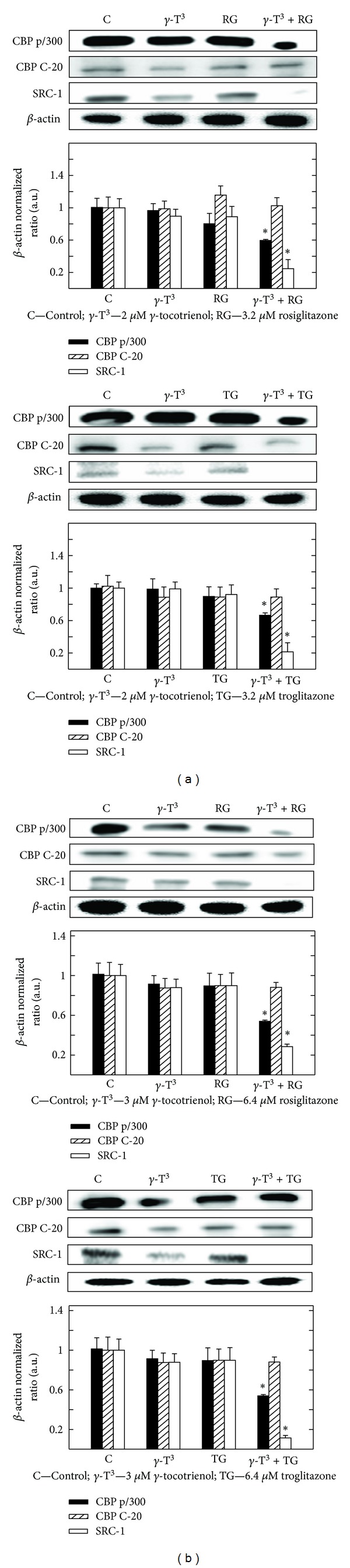
Western blot analysis of γ-tocotrienol and PPARγ agonists (rosiglitazone and troglitazone) when used alone or in combination on the levels of CBP p/300, CBP C-20, and SRC-1 in (a) MCF-7 and (b) MDA-MB-231 human breast cancer cells. MCF-7 cells were initially plated at 1 × 106 cells/100 mm culture dish and treated with control or treatment media containing 2 μM γ-tocotrienol, 3.2 μM rosiglitazone, or 3.2 μM troglitazone alone or in combination. MDA-MB-231 cells were plated in a similar manner and treated with control or treatment media containing either 3 μM γ-tocotrienol or 6.4 μM rosiglitazone or 6.4 μM troglitazone alone or in combination. All cells were fed fresh treatment media every other day for a 4-day incubation period. Afterwards, whole cell lysates were from cell in each treatment group and prepared for subsequent separation by polyacrylamide gel electrophoresis (50 μg/lane) followed by Western blot analysis. Scanning densitometric analysis was performed on all blots done in triplicate and the integrated optical density of each band was normalized with corresponding β-actin, as shown in bar graphs below their respective Western blot images. Vertical bars in the graph indicate the normalized integrated optical density of bands visualized in each lane ± SEM. *P < 0.05 as compared with vehicle-treated controls.
3.9. Effects of γ-Tocotrienol and PPARγ Antagonist GW9662 and T0070907 Given Alone or in Combination on Coactivator Expression
Western blot analysis shows that treatment with 2 μM (MCF-7 cells) or 3 μM (MDA-MB-231 cells) γ-tocotrienol alone induced only slight effects in the expression of CBP p/300, CBP C-20, or SRC-1 as compared to the vehicle-treated controls (Figures 9(a) and 9(b)). Treatment with 3.2 μM (MCF-7 cells) or 6.4 μM (MDA-MB-231 cells) of the PPARγ antagonists, GW9662 and T0070907, alone had only slight effects on CBP p/300, CBP C-20, or SRC-1 expression (Figures 9(a) and 9(b)). However, combined treatment with these same doses of γ-tocotrienol and rosiglitazone and troglitazone cause a significant increase in CBP p/300, CBP C-20, or SRC-1 expression in both MCF-7 and MDA-MB-231 cells as compared to vehicle-treated controls (Figures 9(a) and 9(b)).
Figure 9.
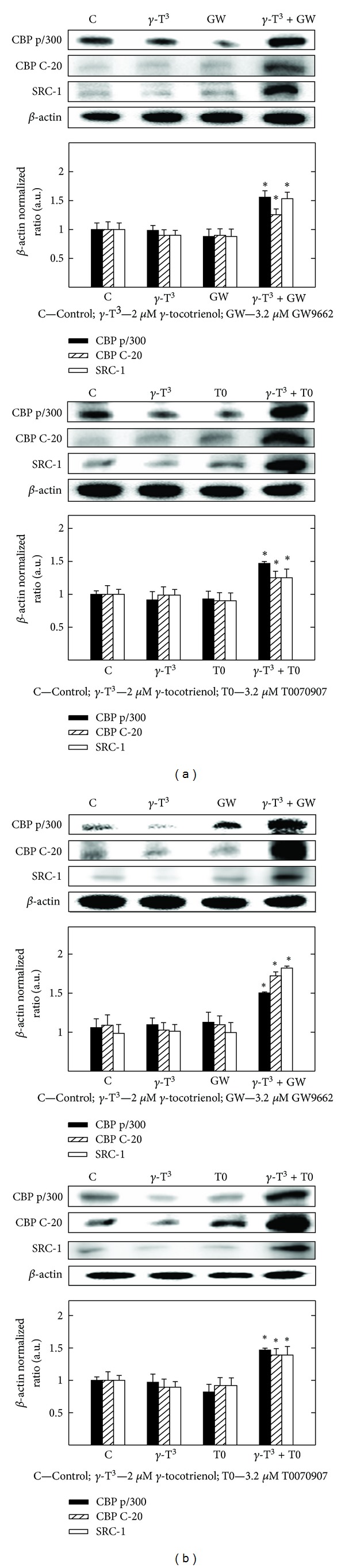
Western blot analysis of γ-tocotrienol and PPARγ antagonists (GW9662 and T0070907) when used alone or in combination with each other to determine protein levels of CBP p/H300, CBP C-20, and SRC-1 in (a) MCF-7 and (b) MDA-MB-231 cells. MCF-7 cells were initially plated at 1 × 106 cells/100 mm culture plate and treated with control or treatment media containing either 2 μM γ-tocotrienol, 3.2 μM GW9662, or 3.2 μM T0070907 alone and in combination. MDA-MB-231 cells were plated in a similar manner and cells were treated with control or treatment media containing 3 μM γ-tocotrienol, 6.4 μM GW9662, or 6.4 μM T0070907 alone or in combination. All cells were fed fresh treatment media every other day for a 4-day incubation period. Afterwards, whole cell lysates were prepared from cells in each treatment group for subsequent separation by polyacrylamide gel electrophoresis (50 μg/lane) followed by Western blot analysis. Scanning densitometric analysis was performed on all blots done in triplicate and the integrated optical density of each band was normalized with corresponding β-actin, as shown in bar graphs below their respective Western blot images. Vertical bars in the graph indicate the normalized integrated optical density of bands visualized in each lane ± SEM. *P < 0.05 as compared with vehicle-treated controls.
3.10. Effects of γ-Tocotrienol and PPARγ Antagonist GW9662 and T0070907 Given Alone or in Combination on PI3K/Akt Mitogenic Signaling
Treatment of 2 μM γ-tocotrienol with 3.2 μM of the PPARγ antagonists GW9662 or T0070907 alone had little or no effects on intracellular levels of Akt, phospho-Akt, PTEN, phospho-PTEN, PI3K, and PDK-1 in MCF-7 cells after a 4-day treatment period (Figure 10(a)). However, combined treatment with the same doses of these agents caused a significant decrease in levels of phospho-Akt, PDK-1, and PI3K, but had little or no effect on total Akt and PTEN, and phospho-PTEN levels as compared to MCF-7 cells in the vehicle-treated control groups (Figure 10(a)). Similarly, treatment of 3 μM γ-tocotrienol, 6.4 μM GW9662 or 6.4 μM T0070907 alone had little or no effect on intracellular levels of phospho-Akt (activated), PDK-1, PI3K, Akt, PTEN, and phospho-PTEN in MDA-MB-231 breast cancer cells, as compared to vehicle-treated controls (Figure 10(b)). Combined treatment with the same doses of these agents resulted in a significant decrease in phospho-Akt, PDK-1, and PI3K levels as compared to MDA-MB-231 breast cancer cells in the vehicle-treated control group (Figure 10(b)).
Figure 10.
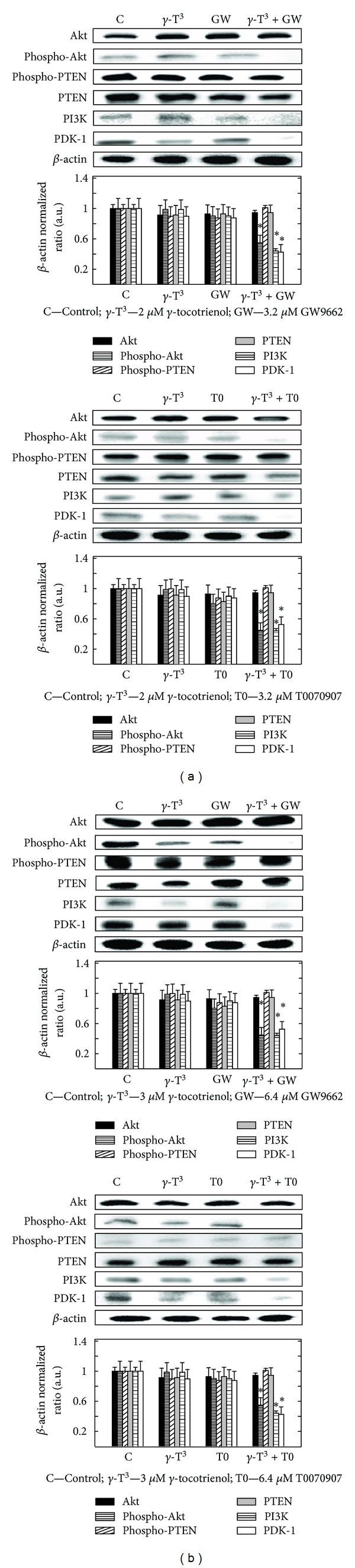
Western blot analysis of γ-tocotrienol and PPARγ antagonists (GW9662 or T0070907) alone or in combination on Akt, phospho-Akt, PTEN, phospho-PTEN, PI3K, and PDK-1 levels on (a) MCF-7 and (b) MDA-MB-231 cells. MCF-7 cells were initially plated at 1 × 106 cells/100 mm culture dish and treated with control or treatment media containing 2 μM γ-tocotrienol, 3.2 μM GW9662, or 3.2 μM T0070907 alone or in combination. MDA-MB-231 cells were also plated in a similar manner and cells were treated with control or treatment media containing 3 μM γ-tocotrienol, 6.4 μM GW9662, or 6.4 μM T0070907 alone or in combination. All cells were fed fresh treatment media every other day for a 4-day incubation period. Afterwards, whole cell lysates were prepared from cells in each treatment group for subsequent separation by polyacrylamide gel electrophoresis (50 μg/lane) followed by Western blot analysis. Scanning densitometric analysis was performed on all blots done in triplicate and the integrated optical density of each band was normalized with corresponding β-actin, as shown in bar graphs below their respective Western blot images. Vertical bars in the graph indicate the normalized integrated optical density of bands visualized in each lane ± SEM. *P < 0.05 as compared with vehicle-treated controls.
Similar studies were conducted to determine the effects of combined γ-tocotrienol treatment with PPARγ agonist rosiglitazone and troglitazone on PI3K/Akt mitogenic signaling in MCF-7 and MDA-MB-231 breast cancer cells. However, little or no differences in the relative levels of these mitogenic proteins were observed among the different treatment groups (data not shown), apparently because cells in the various treatment groups were actively proliferating at a near maximal growth rate.
3.11. Apoptotic Effects of γ-Tocotrienol and PPARγ Antagonist GW9662 and T0070907 Given Alone or in Combination
In order to determine if the growth inhibitory effects resulting from combined treatment with subeffective doses of γ-tocotrienol and PPARγ antagonists might result from a reduction in viable cell number, studies were conducted to determine the acute effects (24-h) and chronic effects (96-h) of these treatment on the initiation of apoptosis and cell viability. Western blot analysis shows that treatment with 2 μM (MCF-7 cells) or 3 μM (MDA-MB-231 cells) γ-tocotrienol alone had no effect on the expression of cleaved PARP, cleaved caspase-3 or viable cell number after a 24-h and 96-h treatment exposure (Figures 11(a) and 11(b)). Treatment with 3.2 μM (MCF-7 cells) or 6.4 μM (MDA-MB-231 cells) of the PPARγ antagonists, GW9662 and T0070907, alone, or in combination with their respective treatment dose of γ-tocotrienol was also found to have no effect on the expression of cleaved PARP, cleaved caspase-3 or viable cell number 24-h after treatment exposure (Figures 11(a) and 11(b)). However, treatment with 20 μM γ-tocotrienol, a dose previously shown to induce apoptosis in mammary cancer cells [13, 14] and used as an apoptosis-inducing positive control in this experiments was found to induce a large increase in cleaved PARP and cleaved caspase-3 levels, and corresponding decrease in viable cell number in both MCF-7 and MDA-MB-231 breast cancer cells 24 h following treatment exposure (Figures 11(a) and 11(b)). The positive apoptosis control treatment of 20 μM γ-tocotrienol was not included in the 96 h treatment exposure experiment, because by the end of this experiment there are no viable cells remaining in this treatment group.
Figure 11.
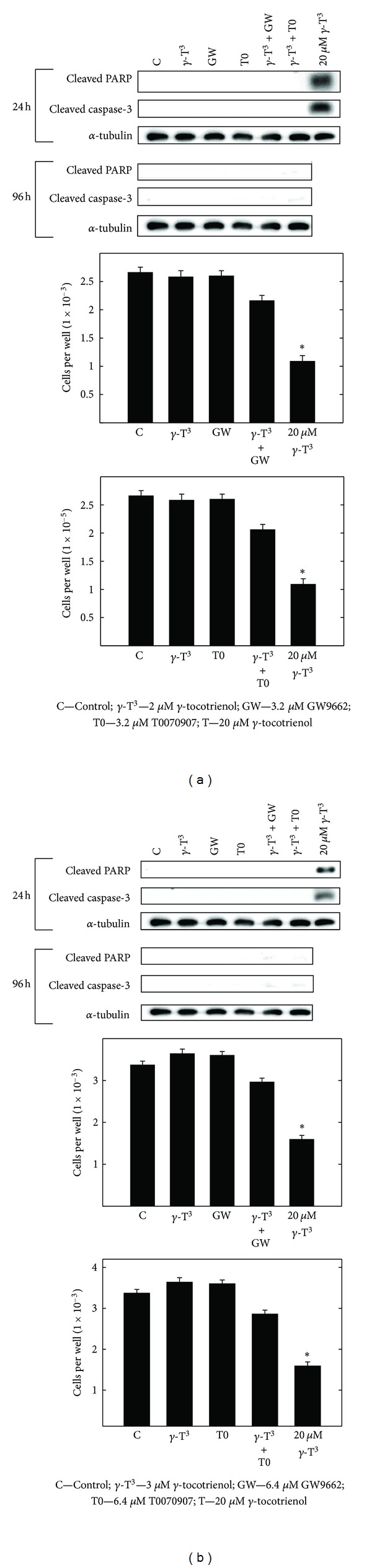
Apoptotic effects of γ-tocotrienol and PPARγ antagonists (GW9662 or T0070907) alone or in combination on caspase-3 and cleaved PARP levels on (a) MCF-7 and (b) MDA-MB-231 human breast cancer cells. For Western blot studies, MCF-7 and MDA-MB-231 cells were initially plated at 1 × 106 cells/100 mm culture dish and maintained on control media for a 3-day culture period. Afterwards, cells were divided into the various treatment groups, media was removed, and cells were exposed to their respective treatment media for a 24-h treatment period. In addition, cells were exposed to their respective treatment media for a 96-h treatment period, where fresh media was added every other day. MCF-7 cells were exposed to treatment media containing 0–2 μM γ-tocotrienol, 0–3.2 μM GW9662, or 0–3.2 μM T0070907 alone or in combination, whereas MDA-MB-231 cells exposed to treatment media containing 0–3 μM γ-tocotrienol, 0–6.4 μM GW9662, or 0–6.4 μM T0070907 alone or in combination. Afterwards, whole cell lysates were prepared from cells in each treatment group for subsequent separation by polyacrylamide gel electrophoresis (50 μg/lane) followed by western blot analysis. In parallel studies, (a) MCF-7 cells were plated at a density of 5 × 104 (6 wells per group) in 24-well culture plates, whereas (b) MDA-MB-231 cells were plated at a density of 1 × 104 (6 wells per group) in 96-well culture plates and exposed to the same treatments as described above. After a 24-h treatment exposure, viable cell number in all treatment groups was determined using MTT assay. Vertical bars indicate the mean cell count ± SEM in each treatment group. *P < 0.05 as compared with vehicle-treated controls.
4. Discussion
Results in these studies demonstrate that when given alone, treatment with γ-tocotrienol, PPARγ agonists (rosiglitazone and troglitazone), or PPARγ antagonists (GW9662 and T0070907), all induce a significant dose-responsive inhibition in the growth of MCF-7 and MDA-MB-231 human breast cancer cells in culture. However, when used in combination, treatment with low doses of PPARγ agonists were found to reverse, whereas treatment with low doses of PPARγ antagonists were found to synergistically enhance the antiproliferative effects of γ-tocotrienol. Additional studies determined that the synergistic inhibition of MCF-7 and MDA-MB-231 tumor cell growth resulting from combined low dose treatment of γ-tocotrienol with PPARγ antagonists was associated with a reduction in PPARγ, PPRE mediated reporter activity, and RXR, an increase in PPARγ coactivator expression, and a corresponding suppression in PI3K/Akt mitogenic-signaling. Conversely, enhancement in MCF-7 and MDA-MB-231 tumor cell growth resulting from combined low dose treatment of γ-tocotrienol with PPARγ agonists was associated with an increase in PPARγ, PPRE mediated reporter activity, and RXR, a decrease in PPARγ coactivator expression, and a corresponding restoration in EGF-dependent PI3K/Akt mitogenic-signaling as compared to their vehicle-treated control group. Taken together, these finding demonstrate that combined treatment of γ-tocotrienol with PPARγ antagonists display synergistic anticancer activity and may provide some benefit in the treatment of human breast cancer. These finding also demonstrate the importance of matching complimentary anticancer agents for use in combination therapy because a mismatch may result in an antagonistic and undesirable therapeutic response.
Previous investigations have shown that both PPARγ agonists and antagonists act as effective anticancer agents [28, 29]. The role of PPARγ agonists as anticancer agents has been well characterized in treatment of colon, gastric, and lung cancer [3, 11], whereas, PPARγ antagonists have been shown to induce potent antiproliferative effects in many hematopoietic and epithelial cancer cell lines [11, 28]. Results in the present study confirm and extend these previous findings. Dose-response studies showed that treatment with either PPARγ agonist or antagonist significantly inhibited the growth of human MCF-7 and MDA-MB-231 breast cancer cells in culture. Furthermore, treatment-induced antiproliferative effects were found to be more pronounce in MDA-MB-231 as compared to MCF-7 breast cancer cells, and these results are similar to those previously reported [28].
Numerous investigations have established that γ-tocotrienol acts as a potent anticancer agent that inhibits the growth of mouse [16, 30] and human [31, 32] breast cancer cells. Furthermore, studies have also shown that combined treatment of γ-tocotrienol with other traditional chemotherapies often results in an additive or synergistic inhibition in cancer cell growth and viability [16, 30]. The rationale for using tocotrienols in combination therapy is based on the principle that resistance to a single agent can be overcome with the use of multiple agents that display complimentary anticancer mechanisms of action. Initial studies showed the additive anticancer effects of mixed tocotrienols and tamoxifen on growth of the estrogen receptor positive MCF-7 and the estrogen receptor negative MDA-MB-435 cells [33] and these findings were later confirmed in other reports [34]. Recent studies have also shown synergistic anticancer effects of combined use γ-tocotrienol with statins [35–37], tyrosine kinase inhibitors [18, 38], COX-2 inhibitors [39, 40], and cMet inhibitors [41]. These studies concluded that combination therapy is most effective when the anticancer mechanism of action of γ-tocotrienol compliments the mechanism of action of the other drug, and may provide significant health benefits in the prevention and/or treatment of breast cancer in women, while at the same time avoiding tumor resistance or toxic effects that is commonly associated with high-dose monotherapy.
The exact role of PPARγ in breast cancer cell proliferation and survival is not clearly understood. Previous studies have suggested that PPARγ activation results in extensive accumulation of lipids and changes in mammary epithelial cell gene expression that promotes a more differentiated and less malignant phenotype, and attenuates breast cancer cell growth and progression [42, 43]. Other studies have shown that γ-tocotrienol enhances the expression of multiple forms of PPARs by selectively regulating PPAR target genes [21]. The antiproliferative effects of γ-tocotrienol have been previously hypothesized to be mediated by the action of γ-tocotrienol to stimulate PPARγ activation by increasing the production of the PPARγ ligand, 15-lipoxygenase-2, in human prostate cancer cells [22]. However, findings in the present study using two distinct types of human breast cancer cell lines showed that low-dose treatment with γ-tocotrienol decreased PPARγ levels, whereas combined treatment of γ-tocotrienol with PPARγ agonists resulted in an elevation in PPARγ levels and a corresponding increase in breast cancer cell growth. These contradictory findings might be explained by differences in the cancer cell types and experimental models used to examine combination treatment effects in these different studies. Nevertheless, the present finding clearly demonstrate an antagonistic effect on breast cancer cell proliferation when treated with the combination of γ-tocotrienol and PPARγ agonists, and provides strong evidence that increased expression of PPARγ is a negative indicator for breast cancer responsiveness to anticancer therapy. This hypothesis is further evidence by the finding that PPARγ expression is elevated in breast cancer cells as compared to normal mammary epithelial cells [9, 44], and mice genetically predisposed to developing mammary tumors constitutively express high levels of activated PPARγ as compared to control mice [9, 44]. It is also possible that the anticancer effects of high-dose treatment with PPARγ agonists may be mediated through PPARγ-independent mechanisms.
The present study also confirms and extends previous findings showing that treatment with PPARγ antagonists significantly inhibits growth of breast cancer cells. Experimental results showed that PPARγ antagonist downregulate PPARγ activation and expression and these effects were associated with enhanced responsiveness to anticancer therapy [45, 46]. However, the present study also shows that combined treatment of γ-tocotrienol with PPARγ antagonist induced a relative large decrease in transcription activity of PPARγ. This treatment was also shown to result in decreased expression of PPARγ and RXR, and these effects were associated with a significant decrease in breast cancer cell growth. PPARγ functions as a heterodimer with its obligate heterodimer partner-RXR. Like other nuclear hormone receptors, the PPARγ-RXR heterodimer recruits cofactor complexes, either coactivators or corepressors to modulate their transcriptional activity [45]. Upon binding of a ligand to the heterodimer complex, corepressors are displaced and the receptor then associates with a coactivator molecule. These coactivators include SRC-1, CBP C-20, and the CBP homologue p/300 [47, 48]. Combined treatment of γ-tocotrienol and PPARγ antagonists-induced suppression of transcription of PPARγ, appears to also decrease the recruitment of coactivator molecules to available PPARγ-RXR heterodimers for translocation into the nucleus, and ultimately resulting in an elevation of free coactivator levels in the cytoplasm. Taken together these results suggest that breast cancer cells require PPARγ activation for their survival, and that treatments designed to reduce or inhibition of PPARγ levels and/or activation and may provide an effective strategy in treatment of breast cancer.
PPARγ activity can be modulated by phosphorylation at multiple sites [49]. In addition, PPARγ ligands can reduce the activity of PI3K and its downstream target Akt [50]. Combined treatment of γ-tocotrienol with PPARγ antagonists was found to reduced PI3K, phosphorylated PDK-1 (active), and phosphorylated-Akt (active) levels in MCF-7 and MDA-MB-231 breast cancer cells. Furthermore, these effects were not associated with an increase in PTEN activity, the phosphatase involved in the inactivation of PDK and Akt. These findings indicate that the antiproliferative effects of combined γ-tocotrienol and PPARγ antagonists treatment is mediated through a suppression in PI3K/Akt mitogenic signaling. These effects were found to be cytostatic in nature, and not associated with a decrease in cell viability resulting from the initiation of apoptosis. Previous findings have also shown that treatment with PPARγ antagonists can cause a decrease in PI3K/Akt mitogenic signaling [51].
5. Conclusion
Result in these studies demonstrate that combined low-dose treatment of γ-tocotrienol and PPARγ antagonists act synergistically to inhibit human breast cancer cell proliferation, and this effect appears to be mediated by a large reduction in PPARγ expression and corresponding reduction in PI3K/Akt mitogenic signaling. Although high dose treatment with PPARγ agonist also was also found to inhibit human breast cancer cells growth, it is most likely that these effects are mediated through PPARγ-independent mechanisms because the preponderance of experimental evidence strongly suggest that elevations in PPARγ expression is an indicator of robust breast cancer cell growth and resistance to anticancer therapy, whereas a reduction in PPARγ expression is an indicator of decreased breast cancer proliferation and increased responsiveness to chemotherapeutic agents. These findings also show that combination anticancer therapy does not always result in an additive or synergistic anticancer response, but can result in a paradoxical/antagonistic response as was observed with the combined treatment of γ-tocotrienol with PPARγ agonist in MCF-7 and MDA-MB-231 human breast cancer cells. The importance of understanding the intracellular mechanism of action of anticancer agents is critical for optimizing therapeutic response. It is also clearly evident that use of γ-tocotrienol in combination with PPARγ antagonist may have potential therapeutic value in treatment of breast cancer in women.
Acknowledgments
This work was supported in part by Grants from First Tec International Ltd. (Hong Kong), the Malaysian Palm Oil Council (MPOC), and the Louisiana Cancer Foundation. The authors would like to thank the First Tech International Ltd. for generously providing γ-tocotrienol for use in these studies.
References
- 1.Badawi AF, Badr MZ. Chemoprevention of breast cancer by targeting cyclooxygenase-2 and peroxisome proliferator-activated receptor-gamma. International Journal of Oncology. 2002;20(6):1109–1122. [PubMed] [Google Scholar]
- 2.Krishnan A, Nair SA, Pillai MR. Biology of PPARγ in cancer: a critical review on existing lacunae. Current Molecular Medicine. 2007;7(6):532–540. doi: 10.2174/156652407781695765. [DOI] [PubMed] [Google Scholar]
- 3.Tachibana K, Yamasaki D, Ishimoto K, Doi T. The role of PPARs in cancer. PPAR Research. 2008;2008 doi: 10.1155/2008/102737.102737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang X, Southard RC, Kilgore MW. The increased expression of peroxisome proliferator-activated receptor-γ1 in human breast cancer is mediated by selective promoter usage. Cancer Research. 2004;64(16):5592–5596. doi: 10.1158/0008-5472.CAN-04-0043. [DOI] [PubMed] [Google Scholar]
- 5.Zaytseva YY, Wallis NK, Southard RC, Kilgore MW. The PPARγ antagonist T0070907 suppresses breast cancer cell proliferation and motility via both PPARγ-dependent and -independent mechanisms. Anticancer Research. 2011;31(3):813–823. [PubMed] [Google Scholar]
- 6.Zaytseva YY, Wang X, Southard RC, Wallis NK, Kilgore MW. Down-regulation of PPARγ1 suppresses cell growth and induces apoptosis in MCF-7 breast cancer cells. Molecular Cancer. 2008;7, article 90 doi: 10.1186/1476-4598-7-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Subbarayan V, Xu XC, Kim J, et al. Inverse relationship between 15-lipoxygenase-2 and PPAR-γ gene expression in normal epithelia compared with tumor epithelia. Neoplasia. 2005;7(3):280–293. doi: 10.1593/neo.04457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saez E, Rosenfeld J, Livolsi A, et al. PPARγ signaling exacerbates mammary gland tumor development. Genes and Development. 2004;18(5):528–540. doi: 10.1101/gad.1167804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cui Y, Miyoshi K, Claudio E, et al. Loss of the peroxisome proliferation-activated receptor gamma (PPARγ) does not affect mammary development and propensity for tumor formation but leads to reduced fertility. Journal of Biological Chemistry. 2002;277(20):17830–17835. doi: 10.1074/jbc.M200186200. [DOI] [PubMed] [Google Scholar]
- 10.Burton JD, Goldenberg DM, Blumenthal RD. Potential of peroxisome proliferator-activated receptor gamma antagonist compounds as therapeutic agents for a wide range of cancer types. PPAR Research. 2008;2008 doi: 10.1155/2008/494161.494161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lea MA, Sura M, Desbordes C. Inhibition of cell proliferation by potential peroxisome proliferator-activated receptor (PPAR) gamma agonists and antagonists. Anticancer Research. 2004;24(5):2765–2771. [PubMed] [Google Scholar]
- 12.Gronemeyer H, Gustafsson JÅ, Laudet V. Principles for modulation of the nuclear receptor superfamily. Nature Reviews Drug Discovery. 2004;3(11):950–964. doi: 10.1038/nrd1551. [DOI] [PubMed] [Google Scholar]
- 13.McIntyre BS, Briski KP, Tirmenstein MA, Fariss MW, Gapor A, Sylvester PW. Antiproliferative and apoptotic effects of tocopherols and tocotrienols on normal mouse mammary epithelial cells. Lipids. 2000;35(2):171–180. doi: 10.1007/BF02664767. [DOI] [PubMed] [Google Scholar]
- 14.Mcintyre BS, Briski KP, Gapor A, Sylvester PW. Antiproliferative and apoptotic effects of tocopherols and tocotrienols on preneoplastic and neoplastic mouse mammary epithelial cells. Experimental Biology and Medicine. 2000;224(4):292–301. doi: 10.1046/j.1525-1373.2000.22434.x. [DOI] [PubMed] [Google Scholar]
- 15.Shah S, Sylvester PW. Tocotrienol-induced caspase-8 activation is unrelated to death receptor apoptotic signaling in neoplastic mammary epithelial cells. Experimental Biology and Medicine. 2004;229(8):745–755. doi: 10.1177/153537020422900806. [DOI] [PubMed] [Google Scholar]
- 16.Sylvester PW, Shah SJ. Mechanisms mediating the antiproliferative and apoptotic effects of vitamin E in mammary cancer cells. Frontiers in Bioscience. 2005;10:699–709. doi: 10.2741/1565. [DOI] [PubMed] [Google Scholar]
- 17.Samant GV, Sylvester PW. γ-tocotrienol inhibits ErbB3-dependent PI3K/Akt mitogenic signalling in neoplastic mammary epithelial cells. Cell Proliferation. 2006;39(6):563–574. doi: 10.1111/j.1365-2184.2006.00412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bachawal SV, Wali VB, Sylvester PW. Enhanced antiproliferative and apoptotic response to combined treatment of γ-tocotrienol with erlotinib or gefitinib in mammary tumor cells. BMC Cancer. 2010;10, article 84 doi: 10.1186/1471-2407-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toker A. Protein kinases as mediators of phosphoinositide 3-kinase signaling. Molecular Pharmacology. 2000;57(4):652–658. [PubMed] [Google Scholar]
- 20.Baldwin AS., Jr. The NF-κB and IκB proteins: new discoveries and insights. Annual Review of Immunology. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 21.Fang F, Kang Z, Wong C. Vitamin E tocotrienols improve insulin sensitivity through activating peroxisome proliferator-activated receptors. Molecular Nutrition and Food Research. 2010;54(3):345–352. doi: 10.1002/mnfr.200900119. [DOI] [PubMed] [Google Scholar]
- 22.Campbell SE, Rudder B, Phillips RB, et al. γ-tocotrienol induces growth arrest through a novel pathway with TGFβ2 in prostate cancer. Free Radical Biology and Medicine. 2011;50(10):1344–1354. doi: 10.1016/j.freeradbiomed.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 23.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 24.Sylvester PW, Birkenfeld HP, Hosick HL, Briski KP. Fatty acid modulation of epidermal growth factor-induced mouse mammary epithelial cell proliferation in vitro. Experimental Cell Research. 1994;214(1):145–153. doi: 10.1006/excr.1994.1243. [DOI] [PubMed] [Google Scholar]
- 25.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proceedings of the National Academy of Sciences of the United States of America. 1979;76(9):4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim JB, Wright HM, Wright M, Spiegelman BM. ADD1/SREBP1 activates PPARγ through the production of endogenous ligand. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(8):4333–4337. doi: 10.1073/pnas.95.8.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tallarida RJ. Drug synergism: its detection and applications. Journal of Pharmacology and Experimental Therapeutics. 2001;298(3):865–872. [PubMed] [Google Scholar]
- 28.Burton JD, Castillo ME, Goldenberg DM, Blumenthal RD. Peroxisome proliferator-activated receptor-γ antagonists exhibit potent antiproliferative effects versus many hematopoietic and epithelial cancer cell lines. Anti-Cancer Drugs. 2007;18(5):525–534. doi: 10.1097/CAD.0b013e3280200414. [DOI] [PubMed] [Google Scholar]
- 29.Kim KY, Kim SS, Cheon HG. Differential anti-proliferative actions of peroxisome proliferator-activated receptor-γ agonists in MCF-7 breast cancer cells. Biochemical Pharmacology. 2006;72(5):530–540. doi: 10.1016/j.bcp.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 30.Sylvester PW. Vitamin E and apoptosis. Vitamins and Hormones. 2007;76:329–356. doi: 10.1016/S0083-6729(07)76012-0. [DOI] [PubMed] [Google Scholar]
- 31.Nesaretnam K, Guthrie N, Chambers AF, Carroll KK. Effect of tocotrienols on the growth of a human breast cancer cell line in culture. Lipids. 1995;30(12):1139–1143. doi: 10.1007/BF02536615. [DOI] [PubMed] [Google Scholar]
- 32.Nesaretnam K, Stephen R, Dils R, Darbre P. Tocotrienols inhibit the growth of human breast cancer cells irrespective of estrogen receptor status. Lipids. 1998;33(5):461–469. doi: 10.1007/s11745-998-0229-3. [DOI] [PubMed] [Google Scholar]
- 33.Guthrie N, Gapor A, Chambers AF, Carroll KK. Inhibition of proliferation of estrogen receptor-negative MDA-MB-435 and -positive MCF-7 human breast cancer cells by palm oil tocotrienols and tamoxifen, alone and in combination. Journal of Nutrition. 1997;127(3):544S–548S. doi: 10.1093/jn/127.3.544S. [DOI] [PubMed] [Google Scholar]
- 34.Nesaretnam K, Dorasamy S, Darbre PD. Tocotrienols inhibit growth of ZR-75-1 breast cancer cells. International Journal of Food Sciences and Nutrition. 2000;51:S95–S103. [PubMed] [Google Scholar]
- 35.Wali VB, Bachawal SV, Sylvester PW. Combined treatment of γ-tocotrienol with statins induce mammary tumor cell cycle arrest in G1. Experimental Biology and Medicine. 2009;234(6):639–650. doi: 10.3181/0810-RM-300. [DOI] [PubMed] [Google Scholar]
- 36.Wali VB, Bachawal SV, Sylvester PW. Suppression in mevalonate synthesis mediates antitumor effects of combined statin and γ-tocotrienol treatment. Lipids. 2009;44(10):925–934. doi: 10.1007/s11745-009-3344-0. [DOI] [PubMed] [Google Scholar]
- 37.Wali VB, Sylvester PW. Synergistic antiproliferative effects of γ-tocotrienol and statin treatment on mammary tumor cells. Lipids. 2007;42(12):1113–1123. doi: 10.1007/s11745-007-3102-0. [DOI] [PubMed] [Google Scholar]
- 38.Bachawal SV, Wali VB, Sylvester PW. Combined γ-tocotrienol and erlotinib/gefitinib treatment suppresses Stat and Akt signaling in murine mammary tumor cells. Anticancer Research. 2010;30(2):429–437. [PubMed] [Google Scholar]
- 39.Shirode AB, Sylvester PW. Synergistic anticancer effects of combined γ-tocotrienol and celecoxib treatment are associated with suppression in Akt and NFκB signaling. Biomedicine and Pharmacotherapy. 2010;64(5):327–332. doi: 10.1016/j.biopha.2009.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shirode AB, Sylvester PW. Mechanisms mediating the synergistic anticancer effects of combined γ-tocotrienol and celecoxib treatment. Journal of Bioanalysis and Biomedicine. 2011;3(1):1–7. doi: 10.4172/1948-593X.1000036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ayoub NM, Bachawal SV, Sylvester PW. γ-tocotrienol inhibits HGF-dependent mitogenesis and Met activation in highly malignant mammary tumour cells. Cell Prolif. 2011;44:516–526. doi: 10.1111/j.1365-2184.2011.00785.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mueller E, Sarraf P, Tontonoz P, et al. Terminal differentiation of human breast cancer through PPARγ . Molecular Cell. 1998;1(3):465–470. doi: 10.1016/s1097-2765(00)80047-7. [DOI] [PubMed] [Google Scholar]
- 43.Kilgore MW, Tate PL, Rai S, Sengoku E, Price TM. MCF-7 and T47D human breast cancer cells contain a functional peroxisomal response. Molecular and Cellular Endocrinology. 1997;129(2):229–235. doi: 10.1016/s0303-7207(97)04057-4. [DOI] [PubMed] [Google Scholar]
- 44.Lee G, Elwood F, McNally J, et al. T0070907, a selective ligand for peroxisome proliferator-activated receptor γ, functions as an antagonist of biochemical and cellular activities. Journal of Biological Chemistry. 2002;277(22):19649–19657. doi: 10.1074/jbc.M200743200. [DOI] [PubMed] [Google Scholar]
- 45.Michalik L, Desvergne B, Wahli W. Peroxisome-proliferator-activated receptors and cancers: complex stories. Nature Reviews Cancer. 2004;4(1):61–70. doi: 10.1038/nrc1254. [DOI] [PubMed] [Google Scholar]
- 46.Panigrahy D, Huang S, Kieran MW, Kaipainen A. PPARγ as a therapeutic target for tumor angiogenesis and metastasis. Cancer Biology and Therapy. 2005;4(7):687–693. doi: 10.4161/cbt.4.7.2014. [DOI] [PubMed] [Google Scholar]
- 47.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocrine Reviews. 1999;20(5):649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 48.Gelman L, Zhou G, Fajas L, Raspé E, Fruchart JC, Auwerx J. p300 Interacts with the N- and C-terminal part of PPARγ2 in a ligand- independent and -dependent manner, respectively. Journal of Biological Chemistry. 1999;274(12):7681–7688. doi: 10.1074/jbc.274.12.7681. [DOI] [PubMed] [Google Scholar]
- 49.Burns KA, Vanden Heuvel JP. Modulation of PPAR activity via phosphorylation. Biochimica et Biophysica Acta. 2007;1771(8):952–960. doi: 10.1016/j.bbalip.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goetze S, Bungenstock A, Czupalla C, et al. Leptin induces endothelial cell migration through Akt, which is inhibited by PPARγ-ligands. Hypertension. 2002;40(5):748–754. doi: 10.1161/01.hyp.0000035522.63647.d3. [DOI] [PubMed] [Google Scholar]
- 51.Shimada T, Kojima K, Yoshiura K, Hiraishi H, Terano A. Characteristics of the peroxisome proliferator activated receptor γ (PPARγ) ligand induced apoptosis in colon cancer cells. Gut. 2002;50(5):658–664. doi: 10.1136/gut.50.5.658. [DOI] [PMC free article] [PubMed] [Google Scholar]