

Abstract
BACKGROUND AND PURPOSE
Sirtuin 6 (SIRT6) is involved in regulation of glucose and fat metabolism. However, its possible contribution to cardiac dysfunction remains to be determined. In the present study, the effect of SIRT6 on cardiac hypertrophy induced by angiotensin II (AngII) and the underlying molecular mechanisms were investigated.
EXPERIMENTAL APPROACH
The expression and deacetylase activity of SIRT6 were measured in hypertrophic cardiomyocytes induced by AngII. After SIRT6 overexpression by transfection, or depletion by RNA interference in neonatal rat cardiomyocytes, cellular hypertrophy was monitored by measuring cell surface area and the mRNA levels of hypertrophic biomarkers. Further, the interaction between SIRT6 and the transcription factor NF-κB was investigated by co-immunoprecipitation, confocal immunofluorescence microscopy and luciferase reporter gene assay. The expression and deacetylase activity of SIRT6 were measured in vivo, using the abdominal aortic constriction (AAC) model of cardiac hypertrophy in rats.
KEY RESULTS
In AngII-induced hypertrophic cardiomyocytes and also in AAC-induced hypertrophic hearts, the expression of SIRT6 protein was upregulated, while its deacetylase activity was decreased. Overexpression of wild-type SIRT6 but not its catalytically inactive mutant, attenuated AngII-induced cardiomyocyte hypertrophy. We further demonstrated a physical interaction between SIRT6 and NF-κB catalytic subunit p65, whose transcriptional activity could be repressed by SIRT6 overexpression.
CONCLUSIONS AND IMPLICATIONS
Our findings suggest that SIRT6 suppressed cardiomyocyte hypertrophy in vitro via inhibition of NF-κB-dependent transcriptional activity and that this effect was dependent on its deacetylase activity.
Keywords: sirtuin 6, cardiac hypertrophy, nicotinamide adenine dinucleotide, deacetylase activity, nuclear factor kappa B
Introduction
Cardiac hypertrophy is a response of the heart to a variety of physiological or pathological stimuli. It may initially be adaptive, but prolonged hypertrophy leads to deterioration in heart function and eventually results in heart failure (Ma et al., 2010). The incidence of cardiac hypertrophy increases dramatically with age (Lakatta and Levy, 2003), implying that aging-associated mechanisms might be critical for the molecular regulation of cardiac hypertrophy.
Sirtuins are evolutionarily conserved NAD-dependent class III histone deacetylases that play pivotal roles in the process of aging. Up to now, seven sirtuins have been found in mammals, named SIRT1–7. These mammalian sirtuins are involved in the molecular regulation of gene silencing, DNA damage repair, life span extension, metabolic homeostasis, tumorigenesis, neurodegeneration, and other disease pathophysiologies (Haigis and Sinclair, 2010).
Recently, the roles of sirtuins in age-related cardiovascular diseases have been the focus of numerous studies. SIRT1 has been implicated in the control of physiological and pathological cardiac growth. The expression of SIRT1 was found to be significantly up-regulated in the hypertrophic or failing hearts of various models of hypertensive, ischemic, diabetic and inflammatory cardiomyopathies (Alcendor et al., 2007; Li et al., 2009; Vahtola et al., 2010). SIRT1 overexpression prevented the age-dependent increase in cardiac hypertrophy, apoptosis/fibrosis and cardiac dysfunction (Alcendor et al., 2007). In addition to SIRT1, SIRT3 and SIRT7 were also identified as modulators in heart diseases. Two recent studies using gene knockout and transgenic mouse models of SIRT3 demonstrated that SIRT3 protected hearts from cardiac hypertrophy by suppressing reactive oxygen species (ROS) (Sundaresan et al., 2009; Pillai et al., 2010). SIRT7 deficiency resulted in cardiac hypertrophy and inflammatory cardiomyopathy, suggesting an important role of SIRT7 in the regulation of stress responses in heart (Vakhrusheva et al., 2008). However, the roles of other sirtuin isoforms in cardiopathy remain to be explored.
Sirtuin 6 (SIRT6), a NAD-dependent deacetylase and ADP-ribosyltransferase, is predominantly expressed in the nucleus (Michishita et al., 2005). It can promote proper chromatin function in several physiological contexts, including telomere and genome stabilization, and DNA repair (Tennen and Chua, 2011). SIRT6 has also been implicated in glucose homeostasis (Xiao et al., 2010; Zhong et al., 2010) and fat metabolism (Kanfi et al., 2010; Kim et al., 2010), suggesting a protective role in metabolic diseases. The metabolic syndrome, characterized by disturbances of glucose and fat metabolism, is widely accepted to be an independent risk factor for cardiovascular diseases (Mottillo et al., 2010).
In the current study, we hypothesized that SIRT6 played a role in the regulation of cardiac hypertrophy. This hypothesis was tested in vitro using gain- and loss-of-function approaches coupled with an in vivo model of cardiac hypertrophy. To the best of our knowledge, these findings uncover, for the first time, a role for SIRT6 in protecting cardiomyocytes from developing hypertrophy.
Methods
Cell culture
All animal care and experimental procedures conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication no. 85-23, revised 1996) and were approved by the Institutional Ethics Review Board of Sun Yat-Sen University. The results of all studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (McGrath et al., 2010). The total number of animals used to provide the cell cultures was 300 and the total number used in the AAC model was 40. Neonatal rat cardiomyocytes were isolated from the hearts of 1- to 3-day-old Sprague-Dawley (SD) rats and cultured as previously described (Li et al., 2007). Briefly, rats were killed by exposing to a rising concentration of CO2. The hearts were removed from animals immediately, and then ventricles were excised and chopped into pieces. The minced tissues were digested with repetitive trypsinization (0.08% trypsin solution). Cardiomyocytes were purified with differential attachment and seeded at a density of 1 × 106 cells·per well into six-well plates and cultured in DMEM supplemented with 10% fetal bovine serum and 5-bromodeoxyuridine (0.1 mM).
Quantitative real-time (RT) PCR
Total RNA from cultured cells or tissues was extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. One microgram of total RNA was reverse transcripted using one-step RT Kit (Takara Biotechnology, Dalian, China) and the resulting cDNA was used as a PCR template for determining the mRNA expression level using SYBR-Green Quantitative PCR kit (Takara Biotechnology) by iCycler iQ system (Bio-Rad, Hercules, CA, USA). Rat-specific primers for SIRT1-SIRT7, atrial natriuretic factor (ANF), brain natriuretic polypeptide (BNP), myosin heavy chain β (β-MHC) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used (see Supporting Information Table S1).
Western blotting and co-immunoprecipitation (co-IP)
Rabbit anti-SIRT6 polyclonal antibody and mouse anti-α-tubulin monoclonal antibody were purchased from Sigma (St Louis, MO, USA). Mouse anti-p65 monoclonal antibody was purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Western blotting analyses were performed as previously described (Zhou et al., 2006). For co-IP, nuclear protein was extracted with the CelLytic NuCLEAR Extraction Kit (Sigma). Nuclear proteins (100 µg) were incubated with 1 µg anti-SIRT6 or anti-p65 antibodies for 2 h (rabbit normal IgG was used as a control), followed by overnight incubation with protein G-agarose beads (Pierce, Rockford, IL, USA) at 4°C. The immunoprecipitated proteins were detected by Western blotting.
Deacetylase activity assay
The deacetylase activity of SIRT6 was determined with a fluorometric assay kit (CycLex, Ina, Nagano, Japan). Nuclear extracts (100 µg) from cardiomyocytes or heart tissues were immunoprecipitated with rabbit anti-SIRT6 polyclonal antibody (Sigma). Precipitates (15 µL) were mixed with a reaction mixture (35 µL) containing 50 mM Tris-HCl (pH 8.8), 0.25 mAU·mL−1 lysyl-endopeptidase, 1 µM trichostatin A, 800 µM NAD, 20 µM fluorosubstrate peptide, followed by incubation for 30 min at room temperature. The fluorescence intensity was measured using a microtiter plate fluorimeter with excitation at 490 ± 10 nm and emission at 530 ± 10 nm, and normalized to the protein concentration. All tests were performed in triplicate.
NAD determination
The NAD levels were measured as described previously (Jacobson and Jacobson, 1976) with a slight modification. An average of 1 × 105 cells or 50 mg of frozen crushed tissue was suspended in 200 µL of 0.5 M perchloric acid. The cell extract was centrifuged at 10000×g for 10 min.after neutralization with an equal volume of 1 M KOH and 0.33 M KH2PO4/K2HPO4 (pH 7.5). The supernatant (50 µL, or NAD standard) was added to 200 µL of NAD reaction mixture (600 mM ethanol, 0.5 mM 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, 2 mM phenazine ethosulfate, 5 mM EDTA, 1 mg·mL−1 of bovine serum albumin, and 120 mM bicine, pH 7.8) and incubated for 5 min at 37°C. The reaction was initiated by the addition of 25 µL of alcohol dehydrogenase (0.5 mg·mL−1 in 100 mM bicine, pH 7.8) and stopped by addition of 250 µL of 12 mM iodoacetate after incubation for 20 min at 37°C. The absorbance of the reaction mixture was read at a wavelength of 570 nm. The NAD content was calculated from the standard curve and normalized to the protein content of the sample.
Plasmid transfection
Cardiomyocytes were transiently transfected with FLAG-SIRT6 or FLAG-H133Y or empty vector using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. After incubation for 24 h, the cells were treated with or without AngII for indicated time.
RNA interference
Three different duplex siRNAs for SIRT6 (S1, S2, S3), p65 (si01, si02, si03) and negative control siRNA were purchased from Invitrogen. The sequences of siRNAs are shown in Supporting Information Tables S2 and S3. Rat cardiomyocytes were transfected either with SIRT6 siRNAs or with negative control siRNA using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Quantitative RT-PCR and Western blotting were performed at 48 h after transfection to compare silencing efficiency of different duplex siRNAs (see Supporting Information Figure S3).
Measurement of cell surface area
Rhodamine-phalloidin (Invitrogen) was used to visualize actin filaments by fluorescence microscopy. Cardiomyocytes grown on coverslips were fixed with 4% paraformaldehyde in PBS for 15 min at room temperature, and further incubated with 0.1% rhodamine-phalloidin and 0.1% saponin for 1 h. After washing with PBS, coverslips were mounted in prolong Gold anti-fade reagent with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen) and inspected with a confocal microscope (Zeiss 710). Cells from randomly selected fields in three culture plates were examined and the surface area of cells from each group was measured by using image analysis software.
Luciferase reporter gene assay
The promega Renilla Luciferase vector contains the herpes simplex virus thymidine kinase promoter (pRL-TK) and NF-κB reporter plasmids pGL4.32, and dual-luciferase reporter assay system were all purchased from Promega (Madison, WI, USA). Cells were seeded at 5 × 104 cells·well−1 into 96-well plates, and cotransfected with NF-κB reporter plasmid (100 ng per well) and pRL-TK (10 ng per well) as an internal control. Total amounts of transfected DNA were equalized by the addition of empty vector. After 8 h of incubation, the cells were serum-deprived for 12 h and subjected to AngII stimulation, RNA interference, or transfection with FLAG-SIRT6 or FLAG-H133Y. The cell lysates were prepared with passive lysis buffer, and the luciferase activity was measured by the dual-luciferase reporter assay system (Promega) and normalized by the transfection efficiency calculated by the Renilla luciferase activity from pRL-TK.
Abdominal aortic constriction (AAC) surgery and echocardiography
Male SD rats (180–200 g; total number) were housed under controlled environmental conditions (12:12-h light/dark cycle and room temperature 21–23°C) and had free access to standard laboratory food pellets and water. Pressure overload was induced by suprarenal AAC, as previously described (Phrommintikul et al., 2008). Briefly, rats were randomized into two groups and anaesthetized with sodium pentobarbital (30 mg·kg−1, i.p.). Fentanyl (0.16 mg·kg−1, s.c.) was given as an analgesic agent. The adequacy of anaesthesia was monitored by evaluating and recording body temperature, cardiac and respiratory rates and pattern, muscle relaxation, and lash reflex. Under sterile conditions, the abdominal aorta above the kidneys was exposed through a midline abdominal incision and constricted at the suprarenal level with a 4-0 silk suture tied around both the aorta and a blunted 22-gauge needle. The needle was promptly removed after constriction. Sham-operated group underwent a similar procedure without banding the aorta.
Two-dimensionally guided M-mode echocardiography was performed using a Technos MPX ultrasound system (ESAOTE, Italy) equipped with a 8.5-MHz imaging transducer as previously described (Zhou et al., 2006). After assessment of echocardiography, rats were killed by exposing to a rising concentration of CO2. The hearts were carefully excised, and heart weight was determined. The left ventricle (LV) was then carefully trimmed away from the right ventricle and atria and weighed. The weight of heart or LV was expressed as a ratio relative to body weight.
Statistical analysis
Data are presented as mean ± standard error. Statistical analyses between two groups were performed by unpaired Student's t-test. Differences among groups were tested by one-way anova with Tukey's post hoc test. In all cases, differences were considered statistically significant with P < 0.05.
Materials
Angiotensin II, sodium pentobarbital, fentanyl, alcohol dehydrogenase, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, phenazine ethosulfate, bicine and iodoacetate were supplied by Sigma, St Louis, MO, USA and NAD by Roche, Basel, Switzerland).
Results
SIRT6 expression was increased in AngII-induced hypertrophic neonatal rat cardiomyocytes
AngII has been known to induce hypertrophic responses in cardiomyocytes (Wollert and Drexler, 1999). Here, quantitative RT-PCR was performed to detect the effect of AngII on mRNA levels of all seven sirtuins in primary neonatal rat cardiomyocytes. The results revealed that the mRNA levels of SIRT1 and SIRT6 were increased while that of SIRT7 was decreased in response to AngII stimulation. However, the other SIRT isoforms were not significantly changed (Figure 1A). Furthermore, the AngII-induced elevation of SIRT6 mRNA and protein expression levels was concentration- and time-dependent (Figure 1B and C).
Figure 1.
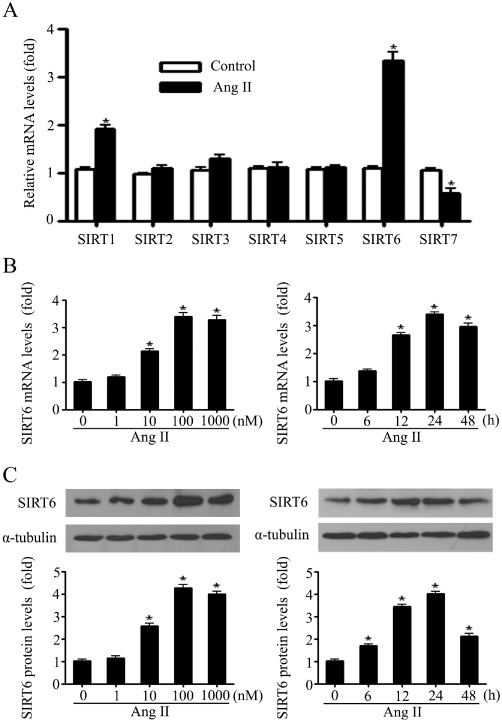
SIRT6 mRNA and protein levels were upregulated in the in vitro model of cardiomyocyte hypertrophy. (A) Primary neonatal rat cardiomyocytes were treated with 100 nM of AngII for 24 h, and quantitative RT-PCR was performed to screen the mRNA changes of all seven sirtuins. (B and C) Cardiomyocytes were treated with indicated concentrations of AngII for 24 h (left panel) or with 100 nM of AngII for indicated time (right panel). The levels of mRNA (B) and protein (C) of SIRT6 were measured and normalized (mRNA by GAPDH and protein by α-tubulin), and then presented as the fold of control levels. Data are presented as means ± SE. *P < 0.05, significantly different from. control group, n= 5–7.
Deacetylase activity of SIRT6 and NAD levels were decreased in response to AngII
Because the deacetylase activity of SIRT6 plays a critical role under physiological conditions (Du et al., 2009), it was measured in AngII-treated primary neonatal rat cardiomyocytes. Interestingly, the results revealed that although the mRNA and protein levels of SIRT6 were increased, its deacetylase activity was markedly decreased (Figure 2A).
Figure 2.
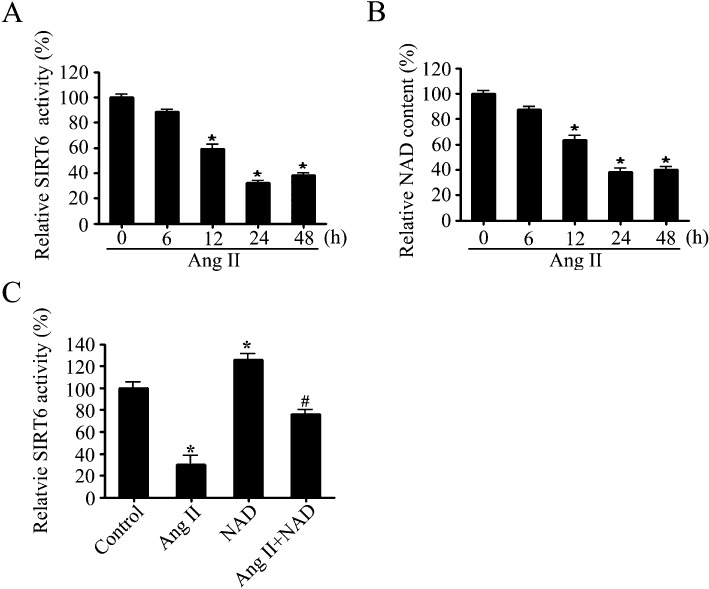
The deacetylase activity of SIRT6 and the intracellular NAD contents were both decreased in cardiomyocyte hypertrophy. (A and B) The SIRT6 deacetylase activity (A) and intracellular NAD contents (B) were measured, in primary neonatal rat cardiomyocytes stimulated with AngII (100 nM) for the indicated times. (C) The SIRT6 deacetylase activity was measured in primary neonatal rat cardiomyocytes pretreated with NAD (50 µM) for 1 h followed by treatment with AngII (100 nM) for 24 h. Data are presented as means ± SE. *P < 0.05, significantly different from. control group, n= 5).
In addition, it is well known that the deacetylase activity of SIRT6 is NAD-dependent. Therefore, we also investigated the cellular NAD levels in our experiments. As shown in Figure 2B, AngII treatment significantly reduced the NAD content of cardiomyocytes in a time-dependent manner.
To explore the potential effect of NAD on deacetylase activity of SIRT6, primary neonatal rat cardiomyocytes were preincubated with 50 µM NAD followed by treatment with or without 100 nM AngII for 24 h. The results showed that AngII-induced decrease in SIRT6 deacetylase activity was partially blocked by NAD supplementation (Figure 2C).
SIRT6 protected cardiomyocytes against hypertrophic response induced by AngII
SIRT6 was transiently overexpressed in primary neonatal rat cardiomyocytes by using a FLAG-tagged wild-type SIRT6 plasmid. The data revealed that both protein level and deacetylase activity of SIRT6 were increased in wild-type SIRT6 transfected cells. However, when the cells were transfected with a catalytically inactive SIRT6 mutant plasmid (FLAG-H133Y), which abolished the deacetylase activity, the protein expression of SIRT6 was increased, but its deacetylase activity remained at control level (Figure 3A and B).
Figure 3.
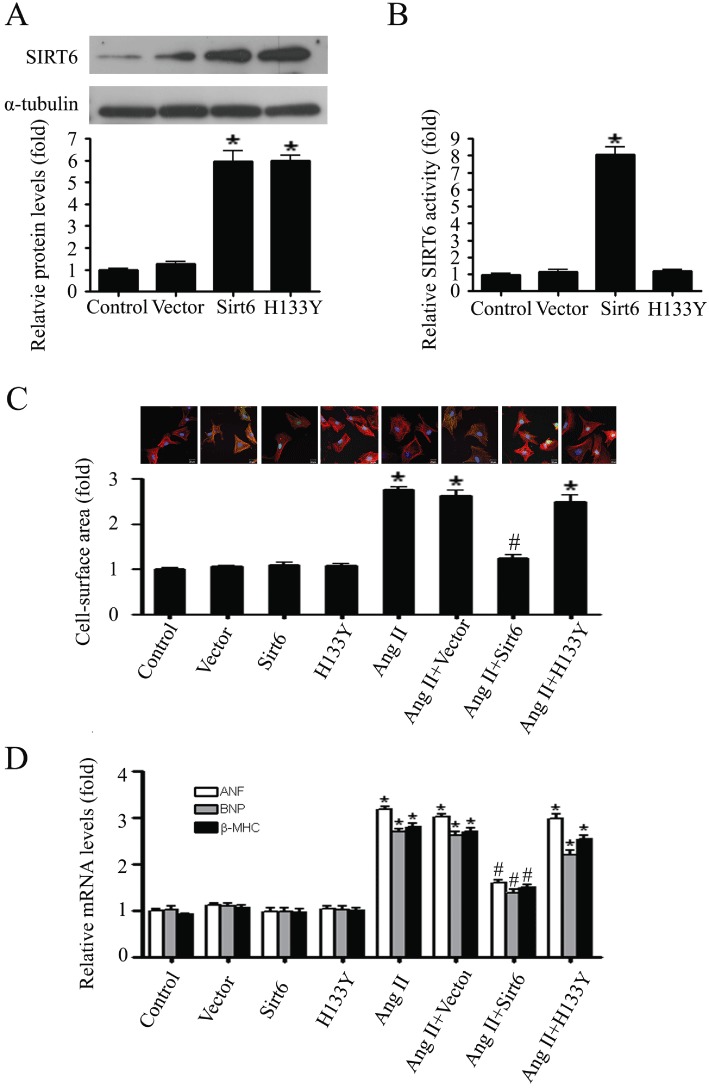
SIRT6 blocked cardiomyocyte hypertrophy through its deacetylase activity. (A) Western blotting analysis of SIRT6 protein in primary neonatal rat cardiomyocytes transiently transfected with SIRT6 WT (Sirt6) or mutant plasmid (H133Y). (B) The deacetylase activity of SIRT6 was measured in primary neonatal rat cardiomyocytes transfected with Sirt6 or H133Y. (C and D) Primary neonatal rat cardiomyocytes were tansfected with Vector, Sirt6 or H133Y and then treated with or without AngII (100 nM, for 24 h). Cardiomyocyte hypertrophic responses were demonstrated by changes of cell surface areas (C) and mRNA levels of hypertrophic biomarkers ANF, BNP, and β-MHC (D). Data are presented as means ± SE. *P < 0.05, significantly different from. control group, #P < 0.05 significantly different from AngII treatment, n= 5.
Following that, the transfected cells were treated with AngII. The cardiomyocyte hypertrophy was monitored by measuring cell surface area and the mRNA levels of ANF, BNP, and β-MHC, which are biomarkers of the hypertrophic response (Du, 2007). As shown in Figure 3C and D, AngII increased the surface area of cardiomyocytes and upregulated the expression of the hypertrophic marker genes. This hypertrophic response in the cells overexpressed with wild-type SIRT6 was significantly attenuated, while in the cells overexpressed with the inactive H133Y mutant was not affected.
To further determine whether endogenous SIRT6 is involved in cardiomyocyte hypertrophy, its expression was knocked down in neonatal rat cardiomyocytes by using three independent siRNAs, S1, S2, and S3. As shown in Figure 4A and B, S1 was the most effective in SIRT6 knockdown. Then the cell surface area and mRNA levels of ANF, BNP and β-MHC were measured. The results showed that down-regulation of SIRT6 by S1 could mimic the effects of AngII by increasing both the cell surface area and the expression of hypertrophic biomarkers (Figure 4C and D).
Figure 4.
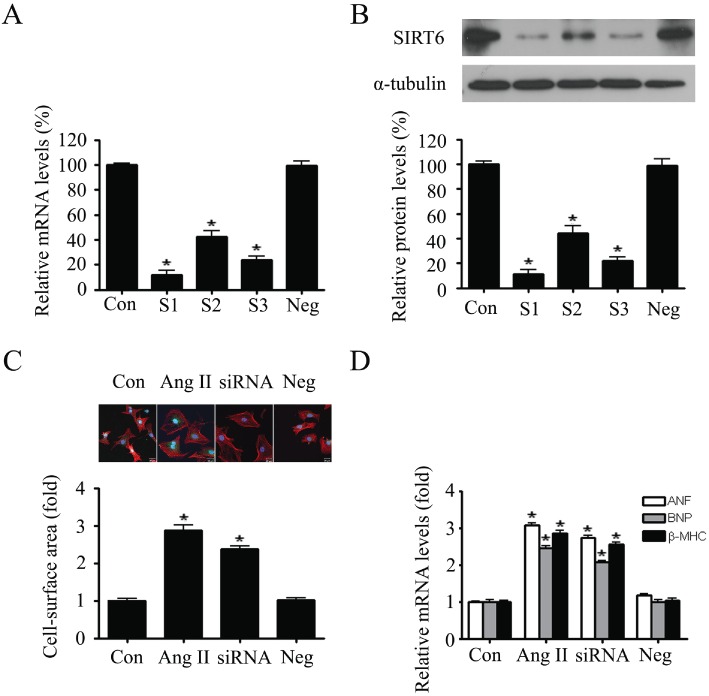
SIRT6 knockdown mimicked the hypertrophic responses in primary neonatal rat cardiomyocytes. (A and B) Three independent siRNAs (marked as S1, S2 and S3), as well as the negative control (Neg), were transfected into cardiomyocytes. The mRNA and protein expression levels of SIRT6 were measured by quantitative RT-PCR (A) and Western blotting (B). (C) Cardiomyocytes were treated with AngII (100 nM, for 24 h) or subjected to SIRT6 knockdown (siSIRT6). Cell surface area was measured to demonstrate the hypertrophic responses in cardiomyocytes. (D) The mRNA levels of hypertrophic biomarkers ANF, BNP, and β-MHC were detected by quantitative RT-PCR. Data are presented as means ± SE. *P < 0.05, significantly different from. control group n= 5.
SIRT6 interacted physically with NF-κB subunit p65 in cardiomyocytes
Previous studies have demonstrated that NF-κB activation is required for the hypertrophic growth of primary neonatal rat cardiomyocytes (Purcell et al., 2001; Van der Heiden et al., 2010) and our earlier results identified a protective role of SIRT6 in cardiac hypertrophy. Thus, it was possible that the protective function of SIRT6 might be exerted through regulation of the NF-κB signalling pathway during the process of cardiac hypertrophy. To test this hypothesis, co-IP was performed in nuclear extracts of cardiomyocytes. The results showed that the NF-κB catalytic subunit p65 was precipitated by anti-SIRT6 antibody, suggesting an interaction between p65 and SIRT6 (Figure 5A). Furthermore, this SIRT6-p65 interaction was detected at different time points of AngII stimulation, with a peak at 60 min (Figure 5C).
Figure 5.
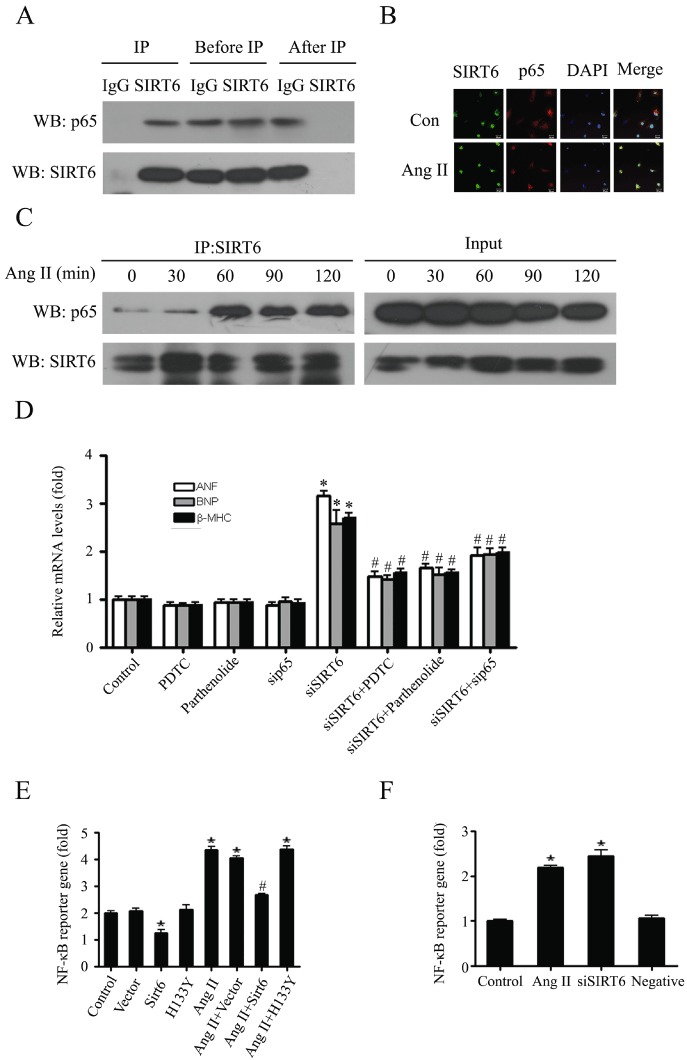
SIRT6 physically interacted with the NF-κB catalytic subunit p65 and repressed NF-κB-dependent transcriptional activity. (A) After immunoprecipitation of cardiomyocyte nuclear extracts with anti-SIRT6 or control IgG, samples of precipitated proteins (IP), of lysate (Before IP) and supernatant (After IP), were separated by 10% SDS-PAGE gels and immunoblotted with anti-SIRT6 and anti-p65 antibodies. (B) Cardiomyocytes were treated with 100 mM AngII for 60 min, and then the co-localization of SIRT6 (green) and p65 (red) were examined by confocal immunofluorescence microscopy. DAPI was used to mark the position of nuclei. (C) Cardiomyocytes were treated with 100 nM AngII for indicated time. Co-IP assay was performed to show the effect of AngII on SIRT6-p65 interaction. The samples of lysate before co-immunoprecipitation are lebelled as ‘Input’. Images representative of three independent experiments are shown. (D) SIRT6 depleted cardiomyocytes (siSIRT6) were treated with or without PDTC, parthenolide or transfected with sip65. The mRNA levels of hypertrophic biomarkers were measured by quantitative RT-PCR. Data are presented as means ± SE. *P < 0.05, significantly different from. control group, #P < 0.05 vs. siSIRT6, n= 3. (E) Cardiomyocytes transfected with FLAG-SIRT6 or FLAG-H133Y were treated with or without 100 nM AngII for 24 h. Dual luciferase reporter assays were performed to evaluate the NF-κB-dependent transcriptional activity. (F) Dual luciferase reporter assays were performed to evaluate the influence of SIRT6 knockdown (siSIRT6) or AngII (100 nM, for 24 h) treatment on NF-κB-dependent transcriptional activity. Data are presented as means ± SE. *P < 0.05, significantly different from. control group, #P < 0.05 vs. AngII treatment, n= 3.
The intracellular co-localization of SIRT6 and p65 in primary neonatal rat cardiomyocytes was also identified by confocal immunofluorescence microscopy. As shown in Figure 5B, SIRT6 was predominantly localized in the nucleus. NF-κB subunit p65 was localized in both the nucleus and the cytoplasm under basal conditions, and AngII induced its nuclear translocation, which might facilitate its interaction with SIRT6.
SIRT6 protected cardiomyocytes from hypertrophy via inhibition of NF-κB-dependent transcriptional activity
To further elucidate the role of SIRT6-p65 interaction during cardiac hypertrophy, cardiomyocytes were treated with pyrrolidine dithiocarbamate (PDTC) or parthenolide, which are well-characterized inhibitors of NF-κB activity (Mahmoodzadeh et al., 2009). As shown in Figure 5D, SIRT6 knockdown increased the expression of hypertrophic markers ANF, BNP and β-MHC. This effect could be reversed by PDTC or parthenolide treatment. Similar results were observed when p65 was knocked down with a specific siRNA (sip65), indicating that anti-hypertrophic effect of SIRT6 required the participation of NF-κB.
Subsequently, we tested the effect of SIRT6 on NF-κB-dependent transcriptional activity by using the luciferase reporter gene assay. In these assays, overexpression of wild-type SIRT6 repressed both basal and AngII-induced NF-κB-Luc reporter gene activity, but overexpression of the catalytically inactive H133Y mutant failed to do so (Figure 5E). Moreover, SIRT6 knock-down by siRNAs led to increased NF-κB-Luc reporter gene activity, mimicking the effect of AngII stimulation (Figure 5F). Taken together, these results suggested that SIRT6 protected cardiomyocytes from hypertrophy through inhibition of NF-κB-dependent transcriptional activity, and this effect was dependent on the deacetylase activity of SIRT6.
SIRT6 expression and deacetylase activity in pressure overload-induced cardiac hypertrophy of rats
Because the expression of SIRT6 was up-regulated but its deacetylase activity was decreased in AngII-induced cardiomyocyte hypertrophy, we further investigated whether there was a similar alteration in vivo, in AAC rats. As shown in Figure 6A and B, SIRT6 mRNA and protein levels increased gradually in rat hearts after AAC. However, the deacetylase activity of SIRT6 decreased along with a decrease of the cardiac NAD content, following AAC (Figure 6C and D).
Figure 6.
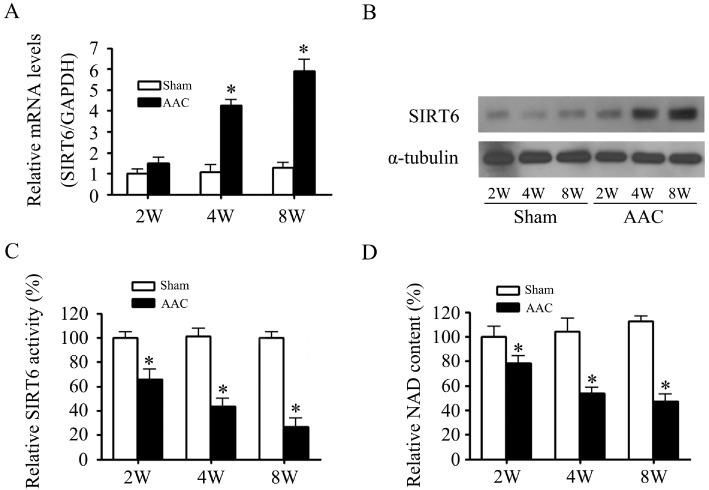
SIRT6 expression was increased and deacetylase activity were decreased in AAC rats. Rats were sampled at different times (2–8 weeks, as shown) after induction of AAC. The mRNA expression (A), protein expression (B), deacetylase activity (C) of SIRT6 and NAD content (D) were measured. Data are presented as means ± SE. *P < 0.05, significantly different from sham group, n= 6.
Discussion and conclusions
Although major advances have been made in our knowledge of the fundamental molecular activity of SIRT6, relatively little is known regarding its function and cellular pathways in cardiovascular diseases (Finkel et al., 2009). Here we demonstrated for the first time that SIRT6 negatively regulated cardiomyocyte hypertrophy. In the present study, we screened the mRNA expressions of all sirtuin isoforms in AngII-induced hypertrophic cardiomyocytes and found that changes in SIRT6 were the most significant. In addition, our data revealed that the expression of SIRT1 was also up-regulated. This is consistent with recent studies in which expression of SIRT1 protein was elevated during pressure overload, left ventricular hypertrophy and heart failure (Alcendor et al., 2007; Li et al., 2009). On the other hand, the deacetylase activity of SIRT1 was decreased during heart failure due to the depletion of NAD (Pillai et al., 2005). Together with reports that SIRT3 (Sundaresan et al., 2009; Pillai et al., 2010) and SIRT7 (Vakhrusheva et al., 2008) also play important roles in the regulation of stress responses in the heart, all these findings have opened up a new line of investigation into the relationship between sirtuins and cardiovascular disease. Through gain- and loss-of-function approaches in cardiomyocytes, we further showed that SIRT6 exhibited cardioprotective effects against hypertrophic stimuli and these effects were dependent on its deacetylase activity. Taken together, these results suggest the potential feasibility of treatments targeted to SIRT6.
In recent years, there has been an increasing interest in the modulation of cardiomyopathy-related pathways in mammals, by the NF-κB family of transcription factors. Several lines of evidences confirmed a strong link between NF-κB and cardiac hypertrophy. Activation of NF-κB was required for the hypertrophic response of primary neonatal rat cardiomyocytes, whereas its inhibition blocked cardiomyocyte hypertrophy and the expression of fetal genes such as ANF and BNP (Purcell et al., 2001; Hirotani et al., 2002). In myotrophin-transgenic mice, inhibition of NF-κB, using a short hairpin RNA against p65, resulted in regression of cardiac hypertrophy (Gupta et al., 2008). Moreover, NF-κB was activated in the hypertrophic and failing human heart (Purcell and Molkentin, 2003). Although the precise role of NF-κB pathway in the pathogenesis of cardiac hypertrophy remains to be elucidated, the activation of NF-κB is now believed to be critical for the development of cardiac hypertrophy. In line with these previous studies, we also found significant activation of NF-κB in the in vitro model of cardiac hypertrophy. Given the intriguing links of both SIRT6 and NF-κB to cardiomyocyte hypertrophy, we hypothesized that these two pathways might functionally overlap. Therefore, we explored whether the protective role of SIRT6 against hypertrophic stimuli was achieved through NF-κB signalling. Our data indicated a physical interaction between NF-κB and SIRT6 in cardiomyocytes, and this interaction could be significantly enhanced by AngII. In another experimental system, 293T cells, Kawahara et al. (2009) had already identified an interaction between SIRT6 and the NF-κB catalytic subunit p65. More importantly, we found that SIRT6 markedly repressed NF-κB-dependent transcriptional activity and that inhibition of NF-κB signalling was essential for the anti-hypertrophic effect of SIRT6. Thus, we propose that the protective role of SIRT6 in heart against hypertrophic stimuli may be attributed to its inhibitory effect on NF-κB-dependent transcriptional activity. In addition, a growing body of evidence has indicated the participation of sirtuins in cellular processes such as apoptosis, fibrosis and inflammation (Alcendor et al., 2007; Vakhrusheva et al., 2008; Sundaresan et al., 2009). Resveratrol-induced SIRT1 up-regulation promoted tumour cell apoptosis through negative regulation of NF-κB (Singh et al., 2011), and overexpression of SIRT1 is reported to suppress inflammation via blocking activation of NF-κB in cardiomyocytes (Planavila et al., 2011). Based on the modulatory function of SIRT6 on NF-κB activity (Kawahara et al., 2009), it may be that SIRT6 is also involved in these processes that are all implicated in the pathogenesis of cardiac hypertrophy. Certainly, further experiments will be needed to address this point.
Because sirtuins fulfil their functions by acting as deacetylases as well as ADP-ribosyltransferases (Du et al., 2009; Tennen and Chua, 2011), it was also necessary to determine whether the cardioprotective effect of SIRT6 was dependent on its enzymic activities. Here, we focused largely on the deacetylase activity of SIRT6, because the ADP-ribosyltransferase activity of sirtuins was considered a low-efficiency side-reaction and might not be physiologically relevant (Du et al., 2009). In the present study, the deacetylase activity of SIRT6 was decreased in response to hypertrophic stimuli both in vitro and in vivo. Overexpression of wild-type SIRT6, but not its catalytically inactive mutant H133Y, was able to block the decease in SIRT6 activity (Supporting Information Figure S2) and diminish the hypertrophic responses induced by AngII (Figure 3), indicating that the deacetylase activity is critical for the SIRT6-mediated cardioprotection. These observations are consistent with reports from other researchers. Kawahara et al. (2009) have described a model in which SIRT6 was recruited to NF-κB target gene promoters via a physical interaction with the NF-κB catalytic subunit p65. Deacetylation of histone H3-Lys9 (H3K9) on target gene promoters led to p65 destabilization from chromatin and thereby contributed to NF-κB signal termination. Those results suggested that SIRT6 could negatively regulate NF-κB-dependent transcription via histone H3K9 deacetylation. Another interesting finding in our study is that the expression of SIRT6 was increased during the development of cardiac hypertrophy. Although the underlying molecular mechanisms need to be determined in future studies, we speculate that upregulated expression of SIRT6 might be an insufficient compensatory mechanism against hypertrophic stimuli.
Furthermore, the deacetylase activity of sirtuins is known to depend on NAD (Michishita et al., 2005). Sirtuins perform deacetylation at modified lysine residues via a unique enzymatic mechanism that requires NAD cleavage with each reaction cycle (Imai et al., 2000; Sauve et al., 2001). In our experiments, the cellular NAD contents were decreased following AngII stimulation or pressure overload. Consistent with our finding, Pillai et al. (2006) reported that AngII-mediated cardiomyocyte hypertrophy was accompanied with depletion of cellular NAD content. NAD is an endogenous molecule that plays important roles in metabolism. Thus, NAD repressed agonist-induced cardiac hypertrophy and this effect was mediated through the activation of SIRT3 but not SIRT1 (Pillai et al., 2010). Here, our results revealed that the deacetylase activity of SIRT6 decreased along with the intracellular NAD level, indicating a close relationship between NAD and SIRT6 in cardiac hypertrophy. Furthermore, exogenous supplement of NAD elevated the SIRT6 deacetylase activity reduced by AngII stimulation, thus suggesting the suppression of SIRT6 deacetylase activity was partially due to decreased NAD level. It will be of great interest to explore whether NAD can also exert its anti-hypertrophic effect through SIRT6. Investigation now in progress in our laboratory may clarify this question.
Previous studies have indicated that oxidative stress plays an important role in the pathogenesis of cardiac hypertrophy (Takimoto and Kass, 2007). In cultured cardiomyocytes, AngII induced ROS generation and pretreatment with antioxidants abolished AngII-induced cardiac hypertrophy (Nakamura et al., 1998). Shih et al. (2001) also revealed that AngII-induced expression of the hypertrophic marker β-MHC was mediated by ROS-dependent activation of the MAPK pathway. In accordance with these studies, we found that vitamin E almost completely eliminated the increase in ROS generation and malondialdehyde level, but only partially inhibited the increased expression of hypertrophic biomarkers, induced by AngII (Supporting Information Figure S3A–E). Moreover, vitamin E had no effects on AngII-induced SIRT6 protein expression and deacetylase activity, thus suggesting that oxidative stress was not involved in the regulation of SIRT6 in cardiac hypertrophy (Supporting Information Figure S3F and G).
The physiological function of SIRT6 in vivo has been partially elucidated by using SIRT6 knockout mice, which developed striking degenerative phenotypes and experienced dramatic metabolic defects, and died by 4 weeks of age (Mostoslavsky et al., 2006). Moreover, liver-specific deletion of SIRT6 in mice caused the altered expression of genes involved in glycolysis and lipid metabolism, leading to increased glycolysis, triglyceride synthesis and fatty liver formation (Kim et al., 2010). In contrast, SIRT6 overexpression in mice could alleviate metabolic damage induced by high-fat diets (Kanfi et al., 2010). These findings suggest that SIRT6 is required for the molecular regulation of aging and metabolism. In the present study, our in vitro work further indicated a novel protective effect of SIRT6 against cardiac hypertrophy. However, further in vivo experiments are necessary to confirm the anti-hypertrophic effect of SIRT6. Overexpression of SIRT6 in vivo by adenoviral transfection or heart-specific knockout of SIRT6, offer convincing and feasible approaches to clarify the function of SIRT6 in cardiac hypertrophy. Besides this, AngII was used in our study as the hypertrophic stimulator in vitro. Although it has been widely reported that AngII, the main effector peptide of the renin–angiotensin system, can induce hypertrophic response of cardiomyocytes (Hu et al., 2004; Schluter and Wenzel, 2008; Yang et al., 2011), cardiac hypertrophy is in fact a multifactorial disease involving both haemodynamic and neurohumoral abnormalities (Heineke and Molkentin, 2006; McKinsey and Kass, 2007). Therefore, the model of cell hypertrophy induced by AngII can only partially represent the pathogenesis of cardiac hypertrophy, and further investigations on other animal models are necessary to confirm our conclusions.
In conclusion, our study suggests that SIRT6 protected cardiomyocytes from hypertrophy via inhibition of NF-κB-dependent transcriptional activity, which was dependent on its deacetylase activity (Figure 7). In light of this finding, there is an exciting possibility of managing cardiac hypertrophy by modulating SIRT6 deacetylase activity. Development of specific activators of SIRT6 may be beneficial for the prevention and/or therapeutic treatment of cardiac hypertrophy and heart failure.
Figure 7.
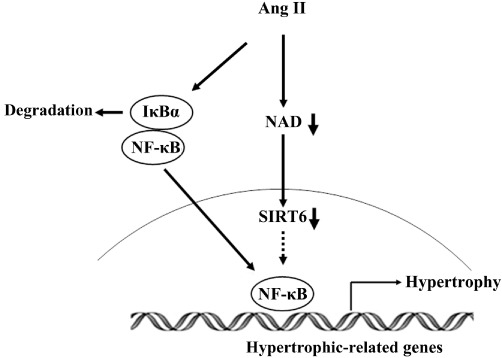
Scheme of the involvement of SIRT6 in AngII-induced cardiomyocyte hypertrophy. Stimulation of cardiomyocytes with AngII decreased intracellular NAD content, resulting in inhibition of the deacetylase activity of SIRT6. SIRT6 with lower deacetylase activity could increase the transcription of NF-κB-dependent hypertrophic genes through its interaction with the NF-κB subunit p65, and eventually lead to cardiac hypertrophy.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No. 81072641); NSFC-CIHR CHINA-CANADA Joint Health Research Initiative Proposal (No. 30811120434); and the National Science and Technology Major Project of China ‘Key New Drug Creation and Manufacturing Program’ (No. 2009ZX09102-152, 2011ZX09401-307).
Glossary
- AAC
abdominal aortic constriction
- ANF
atrial natriuretic factor
- AngII
angiotensin II
- β-MHC
myosin heavy chain β
- BNP
brain natriuretic polypeptide
- LVW
left ventricle weight
- PDTC
pyrrolidine dithiocarbamate
- ROS
reactive oxygen species
- SIRT6
sirtuin 6
Conflicts of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Three independent siRNAs targeted p65 (marked as si01, si02, and si03), as well as the negative control (Neg), were transfected into primary neonatal rat cardiomyocytes. The protein expression of p65 was measured by Western blotting.
Figure S2 The deacetylase activity of SIRT6 was measured in primary neonatal rat cardiomyocytes transfected with Sirt6 or H133Y followed by treatment with 100 nM AngII for 24 h.
Figure S3 Effect of vitamin E on SIRT6 proteinexpression and deacetylase activity in primary neonatal ratcardiomyocytes treated with Ang II. (A, B and C) Cardiomyocyteswere pretreated with vitamin E for 1 h followed by stimulation with100 nM AngII for 24 h. MDA contents (A), SOD activity (B) andGSH/GSSG ratio (C) were detected respectively. (D and E)Cardiomyocytes were pretreated with vitamin E for 1 h followed bystimulation with 100 nM AngII for 24 h. The cells were stained withdihydroethidium (DHE) to visualize the intracellular generation ofROS (D). Cardiomyocyte hypertrophic responses were demonstrated bychanges of mRNA levels of hypertrophic biomarkers ANF, BNP, andβ-MHC (E). (F and G) Cardiomyocytes were pretreated withvitamin E for 1 h followed by stimulation with 100 nM AngII for 24h. Deacetylase activity (F) and protein expression (G) of SIRT6were measured. Data are presented as means ± SE. *P< 0.05, significantly different from. control group;#P < 0.05, significantly different from AngIItreatment, n = 5.
Table S1 Primer sequences for quantitative RT-PCR
Table S2 Sequences of siRNAs for SIRT6
Table S3 Sequences of siRNAs for p65
References
- Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, et al. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res. 2007;100:1512–1521. doi: 10.1161/01.RES.0000267723.65696.4a. [DOI] [PubMed] [Google Scholar]
- Du J, Jiang H, Lin H. Investigating the ADP-ribosyltransferase activity of sirtuins with NAD analogues and 32P-NAD. Biochemistry. 2009;48:2878–2890. doi: 10.1021/bi802093g. [DOI] [PubMed] [Google Scholar]
- Du XJ. Divergence of hypertrophic growth and fetal gene profile: the influence of beta-blockers. Br J Pharmacol. 2007;152:169–171. doi: 10.1038/sj.bjp.0707353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Young D, Maitra RK, Gupta A, Popovic ZB, Yong SL, et al. Prevention of cardiac hypertrophy and heart failure by silencing of NF-kappaB. J Mol Biol. 2008;375:637–649. doi: 10.1016/j.jmb.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- Hirotani S, Otsu K, Nishida K, Higuchi Y, Morita T, Nakayama H, et al. Involvement of nuclear factor-kappaB and apoptosis signal-regulating kinase 1 in G-protein-coupled receptor agonist-induced cardiomyocyte hypertrophy. Circulation. 2002;105:509–515. doi: 10.1161/hc0402.102863. [DOI] [PubMed] [Google Scholar]
- Hu CM, Chen YH, Chiang MT, Chau LY. Heme oxygenase-1 inhibits angiotensin II-induced cardiac hypertrophy in vitro and in vivo. Circulation. 2004;110:309–316. doi: 10.1161/01.CIR.0000135475.35758.23. [DOI] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Jacobson EL, Jacobson MK. Pyridine nucleotide levels as a function of growth in normal and transformed 3T3 cells. Arch Biochem Biophys. 1976;175:627–634. doi: 10.1016/0003-9861(76)90553-1. [DOI] [PubMed] [Google Scholar]
- Kanfi Y, Peshti V, Gil R, Naiman S, Nahum L, Levin E, et al. SIRT6 protects against pathological damage caused by diet-induced obesity. Aging Cell. 2010;9:162–173. doi: 10.1111/j.1474-9726.2009.00544.x. [DOI] [PubMed] [Google Scholar]
- Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell. 2009;136:62–74. doi: 10.1016/j.cell.2008.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Xiao C, Wang RH, Lahusen T, Xu X, Vassilopoulos A, et al. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 2010;12:224–236. doi: 10.1016/j.cmet.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part II: the aging heart in health: links to heart disease. Circulation. 2003;107:346–354. doi: 10.1161/01.cir.0000048893.62841.f7. [DOI] [PubMed] [Google Scholar]
- Li L, Zhao L, Yi-Ming W, Yu YS, Xia CY, Duan JL, et al. Sirt1 hyperexpression in SHR heart related to left ventricular hypertrophy. Can J Physiol Pharmacol. 2009;87:56–62. doi: 10.1139/Y08-099. [DOI] [PubMed] [Google Scholar]
- Li R, Zheng W, Pi R, Gao J, Zhang H, Wang P, et al. Activation of peroxisome proliferator-activated receptor-alpha prevents glycogen synthase 3beta phosphorylation and inhibits cardiac hypertrophy. FEBS Lett. 2007;581:3311–3316. doi: 10.1016/j.febslet.2007.06.017. [DOI] [PubMed] [Google Scholar]
- Ma TK, Kam KK, Yan BP, Lam YY. Renin-angiotensin-aldosterone system blockade for cardiovascular diseases: current status. Br J Pharmacol. 2010;160:1273–1292. doi: 10.1111/j.1476-5381.2010.00750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoodzadeh S, Fritschka S, Dworatzek E, Pham TH, Becher E, Kuehne A, et al. Nuclear factor-kappaB regulates estrogen receptor-alpha transcription in the human heart. J Biol Chem. 2009;284:24705–24714. doi: 10.1074/jbc.M109.000463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA, Kass DA. Small-molecule therapies for cardiac hypertrophy: moving beneath the cell surface. Nat Rev Drug Discov. 2007;6:617–635. doi: 10.1038/nrd2193. [DOI] [PubMed] [Google Scholar]
- Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell. 2005;16:4623–4635. doi: 10.1091/mbc.E05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–329. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- Mottillo S, Filion KB, Genest J, Joseph L, Pilote L, Poirier P, et al. The metabolic syndrome and cardiovascular risk a systematic review and meta-analysis. J Am Coll Cardiol. 2010;56:1113–1132. doi: 10.1016/j.jacc.2010.05.034. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Fushimi K, Kouchi H, Mihara K, Miyazaki M, Ohe T, et al. Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor-alpha and angiotensin II. Circulation. 1998;98:794–799. doi: 10.1161/01.cir.98.8.794. [DOI] [PubMed] [Google Scholar]
- Phrommintikul A, Tran L, Kompa A, Wang B, Adrahtas A, Cantwell D, et al. Effects of a Rho kinase inhibitor on pressure overload induced cardiac hypertrophy and associated diastolic dysfunction. Am J Physiol Heart Circ Physiol. 2008;294:H1804–H1814. doi: 10.1152/ajpheart.01078.2007. [DOI] [PubMed] [Google Scholar]
- Pillai JB, Isbatan A, Imai S, Gupta MP. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J Biol Chem. 2005;280:43121–43130. doi: 10.1074/jbc.M506162200. [DOI] [PubMed] [Google Scholar]
- Pillai JB, Gupta M, Rajamohan SB, Lang R, Raman J, Gupta MP. Poly(ADP-ribose) polymerase-1-deficient mice are protected from angiotensin II-induced cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2006;291:H1545–H1553. doi: 10.1152/ajpheart.01124.2005. [DOI] [PubMed] [Google Scholar]
- Pillai VB, Sundaresan NR, Kim G, Gupta M, Rajamohan SB, Pillai JB, et al. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J Biol Chem. 2010;285:3133–3144. doi: 10.1074/jbc.M109.077271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planavila A, Iglesias R, Giralt M, Villarroya F. Sirt1 acts in association with PPARalpha to protect the heart from hypertrophy, metabolic dysregulation, and inflammation. Cardiovasc Res. 2011;90:276–284. doi: 10.1093/cvr/cvq376. [DOI] [PubMed] [Google Scholar]
- Purcell NH, Molkentin JD. Is nuclear factor kappaB an attractive therapeutic target for treating cardiac hypertrophy? Circulation. 2003;108:638–640. doi: 10.1161/01.CIR.0000085362.40608.DD. [DOI] [PubMed] [Google Scholar]
- Purcell NH, Tang G, Yu C, Mercurio F, DiDonato JA, Lin A. Activation of NF-kappa B is required for hypertrophic growth of primary rat neonatal ventricular cardiomyocytes. Proc Natl Acad Sci U S A. 2001;98:6668–6673. doi: 10.1073/pnas.111155798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve AA, Celic I, Avalos J, Deng H, Boeke JD, Schramm VL. Chemistry of gene silencing: the mechanism of NAD+-dependent deacetylation reactions. Biochemistry. 2001;40:15456–15463. doi: 10.1021/bi011858j. [DOI] [PubMed] [Google Scholar]
- Schluter KD, Wenzel S. Angiotensin II: a hormone involved in and contributing to pro-hypertrophic cardiac networks and target of anti-hypertrophic cross-talks. Pharmacol Ther. 2008;119:311–325. doi: 10.1016/j.pharmthera.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Shih NL, Cheng TH, Loh SH, Cheng PY, Wang DL, Chen YS, et al. Reactive oxygen species modulate angiotensin II-induced beta-myosin heavy chain gene expression via Ras/Raf/extracellular signal-regulated kinase pathway in neonatal rat cardiomyocytes. Biochem Biophys Res Commun. 2001;283:143–148. doi: 10.1006/bbrc.2001.4744. [DOI] [PubMed] [Google Scholar]
- Singh NP, Singh UP, Hegde VL, Guan H, Hofseth L, Nagarkatti M, et al. Resveratrol (trans-3,5,4′-trihydroxystilbene) suppresses EL4 tumor growth by induction of apoptosis involving reciprocal regulation of SIRT1 and NF-kappaB. Mol Nutr Food Res. 2011;55:1207–1218. doi: 10.1002/mnfr.201000576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest. 2009;119:2758–2771. doi: 10.1172/JCI39162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension. 2007;49:241–248. doi: 10.1161/01.HYP.0000254415.31362.a7. [DOI] [PubMed] [Google Scholar]
- Tennen RI, Chua KF. Chromatin regulation and genome maintenance by mammalian SIRT6. Trends Biochem Sci. 2011;36:39–46. doi: 10.1016/j.tibs.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahtola E, Louhelainen M, Forsten H, Merasto S, Raivio J, Kaheinen P, et al. Sirtuin1-p53, forkhead box O3a, p38 and post-infarct cardiac remodeling in the spontaneously diabetic Goto-Kakizaki rat. Cardiovasc Diabetol. 2010;9:5. doi: 10.1186/1475-2840-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakhrusheva O, Smolka C, Gajawada P, Kostin S, Boettger T, Kubin T, et al. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ Res. 2008;102:703–710. doi: 10.1161/CIRCRESAHA.107.164558. [DOI] [PubMed] [Google Scholar]
- Van der Heiden K, Cuhlmann S, Luong le A, Zakkar M, Evans PC. Role of nuclear factor kappaB in cardiovascular health and disease. Clin Sci (Lond) 2010;118:593–605. doi: 10.1042/CS20090557. [DOI] [PubMed] [Google Scholar]
- Wollert KC, Drexler H. The renin-angiotensin system and experimental heart failure. Cardiovasc Res. 1999;43:838–849. doi: 10.1016/s0008-6363(99)00145-5. [DOI] [PubMed] [Google Scholar]
- Xiao C, Kim HS, Lahusen T, Wang RH, Xu X, Gavrilova O, et al. SIRT6 deficiency results in severe hypoglycemia by enhancing both basal and insulin-stimulated glucose uptake in mice. J Biol Chem. 2010;285:36776–36784. doi: 10.1074/jbc.M110.168039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Ago T, Zhai P, Abdellatif M, Sadoshima J. Thioredoxin 1 negatively regulates angiotensin II-induced cardiac hypertrophy through upregulation of miR-98/let-7. Circ Res. 2011;108:305–313. doi: 10.1161/CIRCRESAHA.110.228437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, D'Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell. 2010;140:280–293. doi: 10.1016/j.cell.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou SG, Zhou SF, Huang HQ, Chen JW, Huang M, Liu PQ. Proteomic analysis of hypertrophied myocardial protein patterns in renovascularly hypertensive and spontaneously hypertensive rats. J Proteome Res. 2006;5:2901–2908. doi: 10.1021/pr050456l. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.