

Abstract
Background and Purpose
Carbamazepine (CBZ), known for its anti-epileptic, analgesic and mood-stabilizing properties, is also known to induce weight gain but the pathophysiology of this adverse effect is still largely unknown. We tested the hypothesis that CBZ could have a direct effect on adipocyte development and metabolism.
Experimental Research
We studied the effects of CBZ on morphological biochemical and molecular markers of adipogenesis, using several pre-adipocyte murine cell lines (3T3-L1, 3T3-F442A and T37i cells) and primary cultures of human pre-adipocytes. To delineate the mechanisms underlying the effect of CBZ, clonal expansion of pre-adipocytes, pro-adipogenic transcription factors, glucose uptake and lipolysis were also examined.
Key Results
CBZ strongly inhibited pre-adipocyte differentiation and triglyceride accumulation in a time- and dose-dependent manner in all models. Pleiotropic mechanisms were at the basis of the inhibitory effects of CBZ on adipogenesis and cell lipid accumulation. They included suppression of both clonal expansion and major adipogenic transcription factors such as PPAR-γ and CCAAT/enhancer binding protein-α, activation of basal lipolysis and decrease in insulin-stimulated glucose transport.
Conclusions and Implications
The effect of CBZ on adipogenesis involves activation of the ERK1/2 pathway. Our results show that CBZ acts directly on pre-adipocytes and adipocytes to alter adipose tissue development and metabolism.
Keywords: adipogenesis, bipolar disorder, psychotropic drug, metabolism
Introduction
Mood disorders are today one of the main causes of disability in the world, because of their high incidence as well as the altered functioning that these patients encounter in both their professional and social lives. They were estimated to be the 4th leading cause of total disability-adjusted life years (DALYs) in 1990. According to the World Health Organization data, unipolar depression will be in 2030 the leading cause of DALYs in rich countries, and the second in the world (Mathers and Loncar, 2006).
Mood stabilizers such as lithium, valproic acid and carbamazepine (CBZ) remain the cornerstone of both treatment and prophylaxis of bipolar disorders despite recent evolutions in the past few years (American Psychiatric Association, 2002; Goodwin, 2009; Altamura et al., 2011; Frye, 2011). In major depressive disorder, various medications including several mood stabilizers are today associated with antidepressants in hope of improving response or preventing recurrences (Vigo and Baldessarini, 2009).
CBZ, a dibenzazepine, has been used as a treatment for epilepsy and bipolar mood disorders for decades. It has been suggested to induce a moderate weight gain in patients (Joffe et al., 1986; Lampl et al., 1991), but these observations remain controversial (Caksen et al., 2003; Uludag et al., 2009). However, very few data are available on the exact metabolic effect of CBZ: it has been reported to increase high-density lipoprotein- and low-density lipoprotein-cholesterol plasma levels (Hamed et al., 2009; Luef et al., 2009), while it does not modify insulin and leptin circulating levels (Hamed et al., 2009; Luef et al., 2009; Uludag et al., 2009). The mechanism of CBZ-induced weight gain remains to be elucidated. It probably proceeds at least from a central effect that intervenes in the regulation of appetite and food intake, but also through variations in resting energy expenditure (Jallon and Picard, 2001; Mintzer, 2010). At a molecular level, this central imbalance in energy homeostasis implies a dysregulation in neurotransmitters, neuropeptides and other modulators in brain neuronal circuits (Pijl and Meinders, 1996).
Alternatively, it is conceivable that in addition to its central effect, CBZ also exerts direct effects on peripheral tissues that are involved in the control of metabolic balance, including the pancreas, liver, skeletal muscle and adipose tissue. Accordingly, it has been reported that tricyclic antidepressants, which are structurally related to CBZ, can directly target hepatocytes to induce lipogenesis (Raeder et al., 2006). In addition, two reference mood stabilizers, lithium and valproic acid, are known to alter adipose tissue development. Lithium, through activation of the Wnt/β-catenin signalling pathway, can inhibit adipogenesis both in vitro and in vivo (Ross et al., 2000; Moldes et al., 2003; Longo et al., 2004; Prestwich and Macdougald, 2007). Valproic acid inhibits murine and human pre-adipocyte differentiation, and suppresses leptin and adiponectin gene expression, probably through its histone deacetylase properties (Lagace and Nachtigal, 2004; Lagace et al., 2004; Qiao et al., 2006; Catalioto et al., 2009). Peripheral effects of such drugs could thus represent a novel and underestimated mechanism, which modulates their undesirable metabolic properties.
The aim of the present study was to evaluate the potential direct effect of CBZ on pre-adipocyte differentiation. Using different pre-adipose murine cell lines and primary cultures of human pre-adipocytes, we showed that CBZ strongly inhibits adipogenesis through pleiotropic mechanisms. Our findings show that in addition to its central effect on food intake and energy expenditure, the mood stabilizer and anti-epileptic compound CBZ can directly act on pre-adipocytes to inhibit differentiation.
Methods
Cell culture and differentiation
Mouse 3T3-L1 and 3T3-F442A cells were maintained in high-glucose DMEM, with 10% heat-inactivated FCS and penicillin G/streptomycin sulfate.
For 3T3-L1 differentiation experiments, when pre-adipocytes reached confluence, they were induced to differentiate with DMEM, 10% FCS, 250 nM dexamethasone, 100 μM IBMX and 175 nM human insulin. After 2 days, pre-adipocytes were cultured in DMEM containing 10% FCS and 1 nM insulin. For 3T3-F442A differentiation experiments, confluent pre-adipocytes were cultured with DMEM, antibiotics, 10% FCS and 10 nM insulin. For T37i differentiation experiments, confluent cells were cultured in DMEM/Ham's F12, 10% FCS, 2 nM triiodothyronine (T3) and 20 nM insulin. Primary cultures of human subcutaneous pre-adipocytes were performed as previously described (Chiche et al., 2009). CBZ (Sigma Aldrich, Saint Quentin Fallavier, France) was dissolved in ethanol. The ethanol level was kept at 0.5% in all cells, and did not influence pre-adipocyte differentiation.
Morphological and biochemical determinations
For estimation of cell lipid content, cells were photographed under microscopy either without or with triglyceride staining by oil red O. At the indicated times, cells were washed with PBS, harvested in Tris 25 mM, pH 7.5, EDTA 1 mM, and homogenized. An aliquot of the homogenate was used to determine triglyceride content with a colorimetric kit (Biomerieux, Marcy l'Etoile, France). The remaining homogenate was centrifuged at 10 000× g for 15 min at 4°C, and glycerol-3-phosphate dehydrogenase (G3PDH) activity was measured in the cell supernatant by measuring the oxidation rate of NADH at 340 nm (Kozak and Jensen, 1974). Protein content was determined by a BCA assay (Interchim, Montluçon, France).
The proliferation of pre-adipocyte cells during the clonal expansion phase was measured by [3H]-thymidine incorporation into DNA, as previously reported (Viengchareun et al., 2008). Cell viability was measured with the [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] (MTT) assay.
[3H]-deoxyglucose ([3H]-DOG) uptake and lipolysis were determined in mature adipocytes as previously mentioned (Chiche et al., 2009).
To investigate the pattern of cytokine secretion, 3T3-L1 differentiated adipocytes were shifted in serum-free DMEM in the absence or presence of CBZ for 24 h. Thereafter, media were harvested and tested for cytokine concentration with a dedicated array (Proteome Profiler Mouse Cytokine array kit, panel A, R&D Systems, Lille, France).
Western blot analysis
Cell lysates were separated by SDS-PAGE (10% gel), then electroblotted on a nitrocellulose membrane (Bio-Rad, Marnes-La Coquette, France). After blockade in a TBS-T buffer (20 mM Tris, pH 7.6, 137 mM NaCl, 0.1% Tween-20) with 5% fat-free milk, the membrane was incubated overnight at 4°C with primary antibodies at the indicated dilutions (Table 1). After extensive washing in TBS-T, the membrane was then incubated with a horseradish peroxidase-conjugated secondary antibody (1:15 000) for 1 h at room temperature, washed, and proteins were visualized with an ECL+ detection kit (Perkin Elmer, Courtaboeuf, France).
Table 1.
Dilutions of primary antibodies used in Western blot analysis
| Name | Supplied by | Dilution | MW (kDa) |
|---|---|---|---|
| aP2 | Farmer SR | 1:40 000 | 15 |
| Adiponectin | Santa Cruz | 1:1 000 | 28 |
| C/EBP-α | Santa Cruz | 1:1 000 | 30–42 |
| UCP-1 | Calbiochem | 1:500 | 30 |
| Cyclin D1 | Cell Signaling | 1:500 | 36 |
| ERK 1/2 | Cell Signaling | 1:1 000 | 42–44 |
| C/EBP-β | Santa Cruz | 1:1 000 | 45 |
| PPAR-γ | Santa Cruz | 1:250 | 55 |
| LHS | Santa Cruz | 1:20 000 | 90 |
| SREBP1c | Thermo Scientific | 1:1 000 | 68–100 |
| β-Catenin | BD transduction Lab | 1:1 000 | 92 |
| FAS | Cell Signaling | 1:1 000 | 250 |
| ACC | Cell Signaling | 1:1 000 | 280 |
Quantitative real-time PCR
Total RNA was extracted by the method of Cathala et al. (1983), and quantitative real-time PCR was performed as described previously (Chiche et al., 2009). Sequences of the sense and antisense primers are listed in Table 2.
Table 2.
Sequence of forward and reverse primers used in RT-qPCR analysis
Statistical analysis
The results are represented as means ± SEM from three to eight independent experiments. The results were analysed for statistical significance in GraphPad Prism (GraphPad, San Diego, CA, USA) by using one-way ANOVA or two-way ANOVA followed by Bonferroni post-tests for multiple comparisons to compare means, and Student's t-test to evaluate differences between groups. Statistical significance was accepted when P < 0.05.
Results
CBZ inhibits adipose conversion in various murine pre-adipocyte cell lines and in primary cultures of human pre-adipocytes
We first examined the effect of CBZ on cell triglyceride content in various murine cell lines (3T3-L1, 3T3-F442A and T37i) during their differentiation. Sustained exposure to various CBZ concentrations induced a sharp dose-dependent decrease in lipid content in both white (Figure 1A and D) and brown pre-adipocytes (Figure 1G). The maximal effect was obtained at a concentration of 500 μM, where cell triglyceride content represented only 5–10% of control values. Half-maximal effect was estimated at 150 μM. This inhibition of adipocyte differentiation was confirmed by macroscopic (Figure 1B) and microscopic examination (Figure 1E and H) of red oil staining, showing a marked decrease in the number and size of lipid droplets. We estimated that 90% of the cells were differentiated in control conditions. This fell to 50% with 200 μM CBZ and approximately 5% with 500 μM of the drug. Interestingly, G3PDH activity, which reflects the magnitude of adipose conversion (Pairault and Green, 1979), decreased only at the highest CBZ concentration (Figure 1A). We also examined the expression of several proteins that are usually induced during the adipose conversion process. At 250 and/or 500 μM of the drug, there was a dramatic decrease in acetyl-CoA carboxylase (ACC), adipocyte lipid-binding protein 2 (aP2), fatty acid synthase (FAS), hormone-sensitive lipase (HSL) and adiponectin expression (Figure 1C, F and I). Furthermore, CBZ strongly down-regulated the expression of uncoupling protein 1, the key functional protein of thermogenesis (Figure 1I).
Figure 1.

Carbamazepine inhibits adipocyte differentiation in a dose-dependent manner. Confluent pre-adipocytes (day 0) were cultured for 10 days in the absence or in the presence of various concentrations of carbamazepine. At day 10, cell triglyceride content was tested in 3T3-L1 cells (A), 3T3-F442A cells (D) and T37i cells (G). Results represent the mean ± SEM of four to eight separate determinations and are expressed as % of the control value. G3PDH-specific activity was measured on 3T3-L1 cytosolic extracts prepared at the same time (A). Results represent the mean ± SEM of four separate determinations and are expressed as % of the control value. At day 10, 3T3-L1 cells were stained with red oil and photographed macroscopically (B), whereas 3T3-F442A cells (E), T37i cells (H) and human pre-adipocytes (J) were photographed microscopically, scale bar 50 μm. At day 10, cell extracts were prepared and FAS, ACC, HSL, adiponectin, aP2, uncoupling protein 1 (UCP-1), PPAR-γ and ERK 1/2 protein expression was studied using Western blot analysis on 3T3-L1 cells (C), 3T3-F442A cells (F), T37i cells (I) and human pre-adipocytes (K). **P < 0.01; ***P < 0.001, carbamazepine-treated versus control.
The effects of CBZ were also tested on primary cultures of human pre-adipocytes. Likewise, there was a clear decrease in cell lipid content as well as in several typical markers of adipogenesis (Figure 1J and K).
The time dependence of the inhibitory effect of CBZ was also tested during the conversion process. Confluent 3T3-L1 cells were cultured from confluence in the absence or in the presence of 500 μM CBZ, and cell extracts were prepared at intervals. At day 6 following confluence, the compound provoked a dramatic 20-fold decrease in cell triglyceride content, an effect that was maintained throughout the differentiation process (Figure 2A). The expression of FAS, HSL and PPAR-γ was decreased in parallel (Figure 2B).
Figure 2.
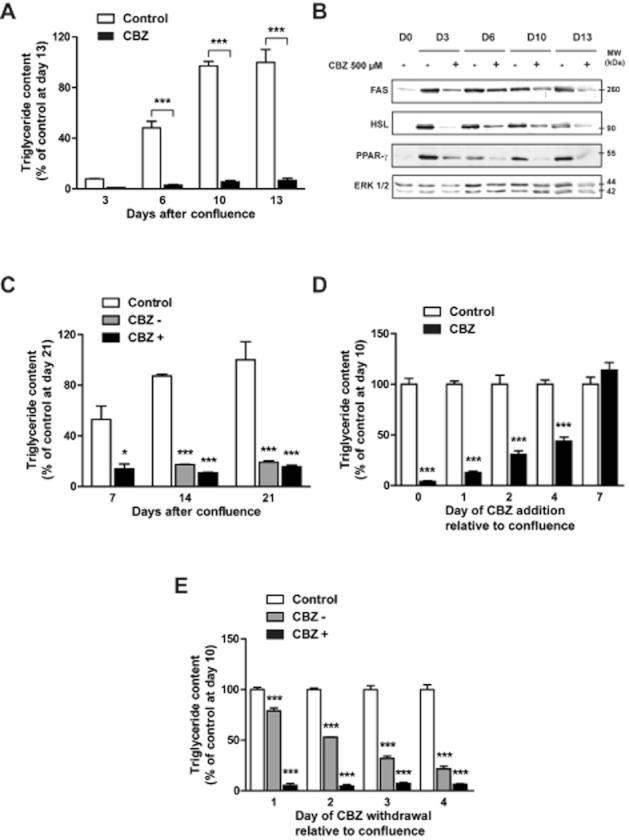
Carbamazepine inhibits adipocyte differentiation in a time-dependent and irreversible manner. Confluent 3T3-L1 cells (day 0) were cultured in the absence or in the presence of 500 μM carbamazepine. At days 3, 6, 10 and 13, cell triglyceride content was tested (A). Results represent the mean ± SEM of three separate determinations and are expressed as % of the control value at day 13. Cells extracts were prepared at the same intervals to study FAS, HSL and PPAR-γ protein expression (B). Confluent 3T3-L1 cells were cultured in the absence or in the presence of 500 μM carbamazepine. At day 7, in cells previously exposed to carbamazepine, the drug was then either omitted (CBZ –) or maintained (CBZ +). At days 7, 14 and 21, cell triglyceride content was tested (C). Results represent the mean ± SEM of four to six separate determinations and are expressed as % of the control value at day 21. Confluent 3T3-L1 preadipocytes (day 0) were cultured for 10 days. 500 μM carbamazepine was added to the medium at different times following confluence (days 1, 2, 4 and 7). At day 10, cell triglyceride content was measured (D). Confluent pre-adipocytes (day 0) were cultured for 10 days. 500 μM carbamazepine was added to the medium at day 0 and then omitted at different times following confluence (days 1, 2, 3 and 4). At day 10, cell triglyceride content was measured (E). Results represent the mean ± SEM of four separate determinations and are expressed as % of the control value. *P < 0.05; ***P < 0.001, carbamazepine-treated versus respective ethanol control.
The reversibility of the CBZ effect was also tested in 3T3-L1 cells (Figure 2C). From confluence (day 0) to day 7, cells were cultured in the absence or in the presence of 500 μM of the drug; thereafter, CBZ was either maintained or omitted from the culture medium. Strikingly, CBZ withdrawal from the culture medium at day 7 did not allow a partial or total recovery of cell lipid content, even at day 21.
The critical period during which the addition of CBZ is required to inhibit adipocyte differentiation was also evaluated. Even when the drug was added as late as day 4 after confluence, there was a marked decrease in cell lipid content at day 10 (Figure 2D). Interestingly, we noticed that the earlier the addition of CBZ, the strongest the suppression of triglyceride concentration.
In a second set of experiments, an exposure as short as 24 h after confluence was sufficient to induce a significant reduction in cell triglyceride content at day 10 (Figure 2E). A 4-day treatment induced a fall in cell triglyceride content close to the maximal effect previously obtained with a continuous exposure to the drug for 10 days.
Long-term exposure of mature adipocytes decreases cell lipid content and adiponectin production
We also evaluated the effects of CBZ on fully mature adipocytes. Day 8 post-confluent 3T3-L1 adipocytes were exposed for 3 days (Figure 3A) or for 7 days (Figure 3B) to various concentrations of the drug. While a 72 h treatment was not sufficient to suppress cell lipid content, a 1 week exposure induced a slight but significant 15–20% decrease in mature adipocyte triglyceride concentration. This was associated with a parallel slight decrease in FAS expression with 500 μM CBZ, while HSL levels remained unchanged (Figure 3C). Interestingly, PPAR-γ and adiponectin expressions were markedly decreased after a 7-day treatment with CBZ. The effect of the drug on adiponectin secretion was confirmed by an ELISA assay (Figure 3D). It showed a decreased hormone concentration in both cell homogenates and culture medium after a 48 h exposure to CBZ, although adipocyte lipid content was not affected.
Figure 3.
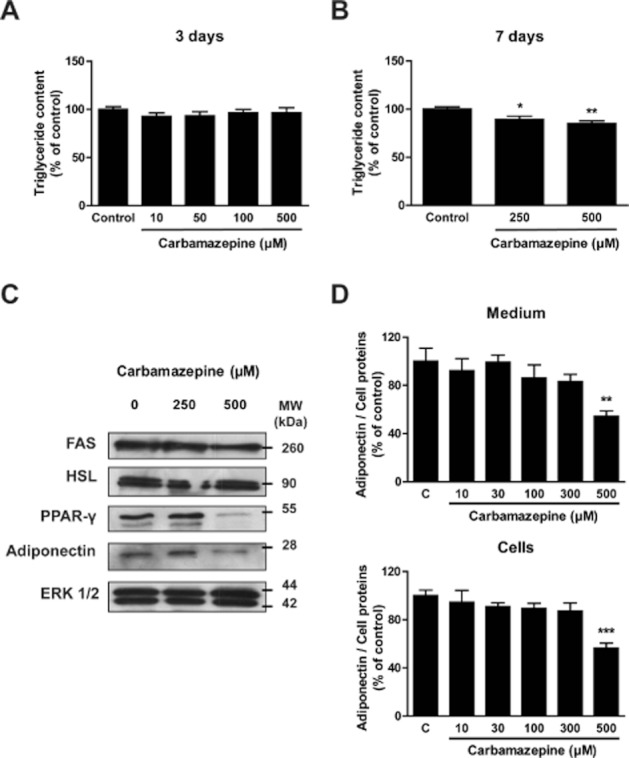
Carbamazepine has a significant effect on mature adipocytes. Mature 3T3-L1 adipocytes (day 7) were treated with concentrations of carbamazepine ranging from 10 to 500 μM for either 3 (A) or 7 days (B). Thereafter, cell triglyceride content was tested. Results represent the mean ± SEM of four separate determinations and are expressed as % of the control value. Mature 3T3-L1 cells were incubated with 250 or 500 μM carbamazepine for 7 days before cell extracts were prepared to determine FAS, HSL, adiponectin and PPAR-γ protein expression (C). Mature adipocytes were treated with 10–500 μM carbamazepine for 2 days. Adiponectin concentration was measured in the medium (upper panel) and cells (lower panel) and normalized to total cell protein concentration (D). Results represent the mean ± SEM of four separate determinations and are expressed as % of the control value. *P < 0.05; **P < 0.01; ***P < 0.001 carbamazepine-treated versus control.
Mechanisms involved in CBZ effects on lipid accumulation and adipose conversion
We then investigated the mechanisms by which CBZ exerts its inhibitory effects on adipose conversion and lipid accumulation. We first verified that CBZ did not exert a cytotoxic effect on pre-adipocytes. Cell viability, assessed in confluent 3T3-L1 cells by the MTT assay, was not altered following a 48 h CBZ exposure, even at the maximal concentration of 500 μM (Figure 4A).
Figure 4.
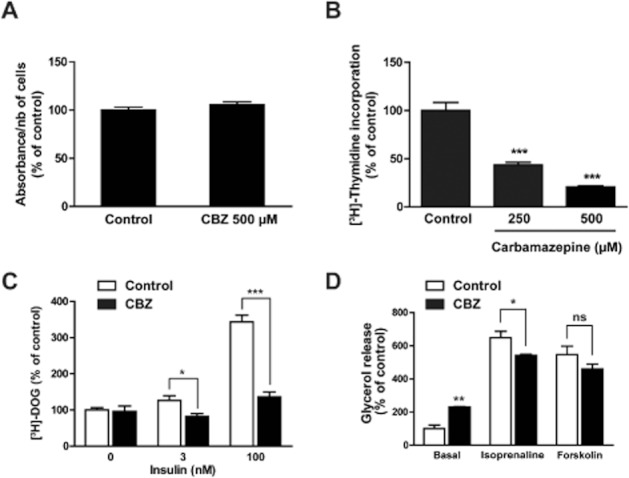
Carbamazepine alters clonal expansion, glucose uptake and lipolysis but not cell viability. Confluent 3T3-L1 cells (day 0) were treated with 500 μM carbamazepine for 2 days. Viability was then tested using a MTT assay (n = 42) (A). At the same time, clonal expansion was measured by the amount of [3H]-thymidine incorporated in cell DNA (n = 16) (B). Mature 3T3-L1 cells were treated with 500 μM carbamazepine for 2 days and [3H]-deoxyglucose uptake was measured in basal conditions and after insulin stimulation (n = 4) (C). Mature 3T3-L1 cells were treated with 500 μM carbamazepine for 2 days and lipolysis was measured in basal conditions and after stimulation by 10 μM (-)-isoprenaline or forskolin (n = 4) (D). All results represent the mean ± SEM and are expressed as % of the control value. *P < 0.05; **P < 0.01; ***P < 0.001, carbamazepine-treated versus respective control.
We also examined the possibility that CBZ could alter clonal expansion, i.e. the limited number of mitoses that follows growth arrest at confluence. [3H]-thymidine incorporation assays showed that a 48 h treatment with CBZ reduced cell proliferation by 60% and 80% at 250 μM and 500 μM, respectively (Figure 4B).
The inhibitory effect of CBZ on cell triglyceride content could also result from a reduction in glucose availability, with a subsequent decrease in fatty acid and triacylglycerol synthesis and storage. Thus, we tested whether in mature 3T3-L1 adipocytes, a 48 h exposure to CBZ could regulate basal and insulin-stimulated glucose transport. While CBZ had no effect on basal [3H]-DOG uptake, it markedly decreased insulin-stimulated [3H]-DOG transport, both at submaximal and maximal insulin concentrations (Figure 4C).
The decrease in cell fat stores induced by CBZ could also be related to an activation of triacylglycerol hydrolysis, i.e. lipolysis. This was assessed by measuring glycerol release under basal conditions or in response to an optimal concentration (10 μM) of the β-adrenoceptor agonist (-)-isoprenaline or of the adenylyl cyclase effector forskolin. CBZ exposure provoked an approximate doubling of basal lipolysis, while (-)-isoprenaline- and forskolin-stimulated glycerol release was only weakly or not significantly altered by the drug (Figure 4D).
We next tested whether CBZ could markedly alter the transcriptional mechanisms of adipogenesis. We studied the expression of key adipogenic transcription factors such as PPAR-γ, CCAAT/enhancer binding protein-α (C/EBP-α), C/EBP-β and sterol regulatory element-binding protein 1c (SREBP-1c). At a concentration of 500 μM, CBZ caused a sharp decrease in PPAR-γ and C/EPB-α protein levels (Figure 5A). By contrast, the drug induced only a slight decrease in C/EBP-β levels and no significant variation in SREBP-1c could be detected (data not shown). To examine whether this dramatic decrease in PPAR-γ and C/EPB-α protein expression was associated with a similar decrease in the expression of the related genes, C/EBP-α and PPAR-γ transcript levels were tested in parallel using RT-qPCR analysis (Figure 5B). A more than 10-fold decrease in PPAR-γ and C/EBP-α mRNA levels was observed with 250 and 500 μM CBZ. An approximate twofold reduction in PPAR-γ transcript was already detectable at 100 μM. The effect of CBZ on adipogenic transcription factors was independent of its effect on clonal expansion since the drug also reduced PPAR-γ expression once clonal expansion was achieved (data not shown).
Figure 5.
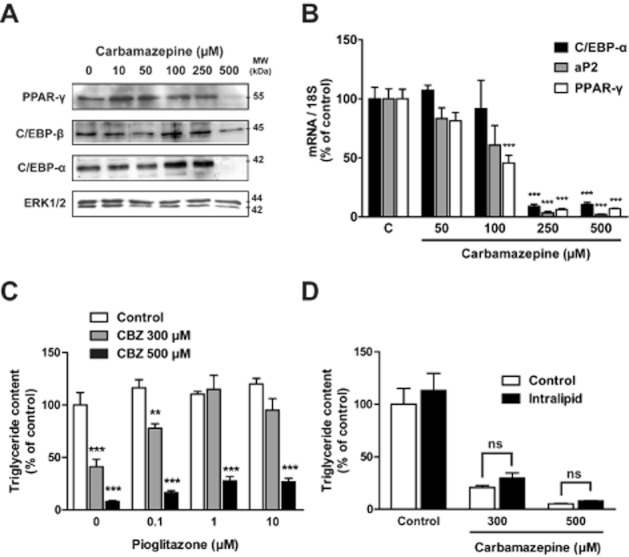
Carbamazepine alters the transcriptional programme of adipogenesis. Confluent 3T3-L1 pre-adipocytes (day 0) were cultured for 10 days in the absence or in the presence of various concentrations of carbamazepine. At day 10, total and nuclear cell extracts were prepared and PPAR-γ, C/EBP-α and C/EBP-β protein expression was studied (A). C/EBP-α, aP2 and PPAR-γ mRNA levels were studied using RT-qPCR (B). Results are expressed as % of the control value. Confluent 3T3-L1preadipocytes (day 0) were cultured for 7 days in the absence or in the presence of carbamazepine and/or pioglitazone (0.1–10 μM) before triglyceride measurement (C). Results represent the mean ± SEM of four determinations and are expressed as % of the control value without pioglitazone or carbamazepine. Confluent 3T3-L1 preadipocytes (day 0) were cultured for 7 days in the absence or in the presence of carbamazepine and/or intralipid (100 mg·L−1) before triglyceride measurement (D). Results represent the mean ± SEM of six determinations and are expressed as % of the basal control value. **P < 0.01; ***P < 0.001, carbamazepine-treated versus respective control.
Since PPAR-γ is considered to be the major adipogenic transcription factor and is particularly affected by CBZ exposure, we investigated whether the inhibitory effect of CBZ on differentiation could be prevented by a parallel treatment with the PPAR-γ agonist pioglitazone. At a submaximal concentration of CBZ (300 μM), pioglitazone completely prevented the inhibitory effect of the compound on cell triglyceride accumulation. However, only a partial effect was achieved at the maximal CBZ concentration of 500 μM (Figure 5C).
We also investigated whether the blockade of the lipogenic pathway could be involved in the inhibitory effect of CBZ on adipogenesis. We attempted to prevent the effect of CBZ by directly providing fatty acids to 3T3-L1 cells during adipose conversion. Whatever CBZ concentration used, intralipid addition did not change cell triglyceride content (Figure 5D).
Since most prescribed mood stabilizers, lithium and valproate, as well as CBZ, all exhibit an anti-adipogenic effect, we examined whether CBZ could share a common mechanism with lithium: activation of the Wnt/β-catenin pathway. As shown in Supporting Information Figure S2, β-catenin activation by CBZ was very weak. This was also confirmed by a slight induction of cyclin D1, a well-known target of the β-catenin/T-cell factor complex (Behrens, 2000).
CBZ effects on adipocytes and pre-adipocytes are reminiscent to those observed in the presence of pro-inflammatory cytokines such as TNF-α, IL-1 and IL-6, and include inhibition of adipogenesis and insulin-stimulated glucose uptake as well as activation of basal lipolysis. Thus, using a cytokine array, we tested the ability of CBZ to modulate 3T3-L1 adipocyte cytokine secretion. Several cytokines and chemokines were induced or down-regulated by CBZ in the culture medium (Supporting Information Figure S1), but no typical pro- or anti-inflammatory profile was observed. However, since cytokines are known to modulate the activity of several kinases, we investigated whether CBZ could target downstream effectors, such as ERK 1/2 and p38 MAPK, independently of cytokine mediation (Figure 6). There was a clear increase in ERK 1/2 phosphorylation after a 10 min exposure to CBZ (Figure 6A). CBZ treatment also induced increased p38 MAPK phosphorylation but did not modify JNK phosphorylation (data not shown).
Figure 6.
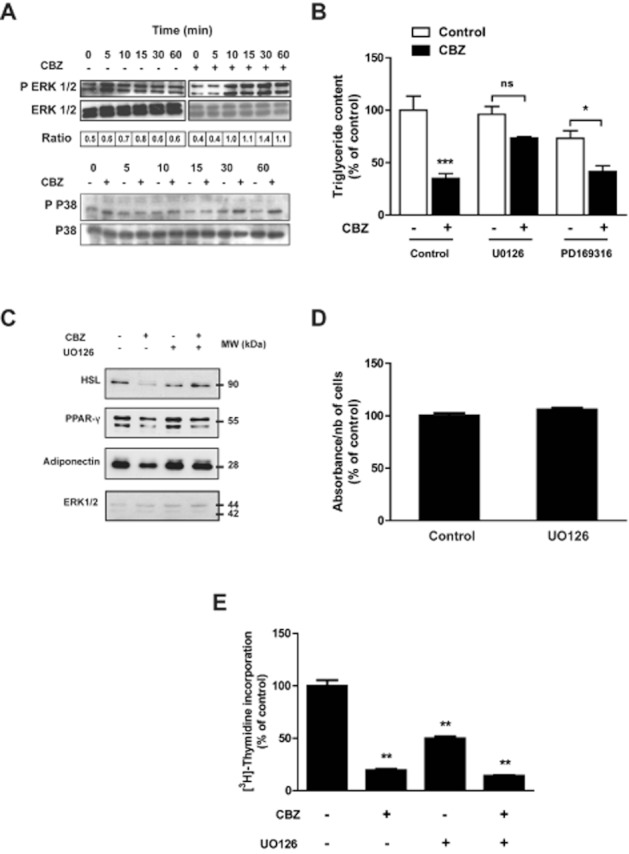
The anti-adipogenic effect of carbamazepine is mediated by ERK 1/2 activation. Confluent 3T3-L1 pre-adipocytes were exposed for the indicated times to 500 μM carbamazepine, then cell extracts were prepared and tested for expression of total and phosphorylated ERK and p38 MAPK (A). Confluent 3T3-L1 cells were exposed or not to 250 μM carbamazepine in the absence or in the presence of UO126 or PD169316 before cell triglyceride content (B) and protein expression (C) were measured at day 8 (n = 4). Confluent 3T3-L1 pre-adipocytes were treated or not for 48 h with 1 μM UO126, then cell viability was tested with a MTT assay (D). Confluent 3T3-L1 pre-adipocytes were exposed or not to 500 μM carbamazepine and/or 1 μM UO126 for 48 h, then [3H]-thymidine incorporation was measured (n = 6) (E). Results represent the mean ± SEM of six determinations and are expressed as % of the basal control value. *P < 0.05; **P < 0.01; ***P < 0.001, carbamazepine-treated versus respective control.
We then tested whether specific inhibitors could prevent the inhibitory effect of CBZ on adipocyte differentiation. While p38 MAPK inhibitor (PD169316) did not modify cell triglyceride content when added to CBZ, the MAPK/ERK kinase (MEK) inhibitor UO126 prevented the inhibitory effect of CBZ on adipose conversion (Figure 6B). We further demonstrated that UO126 could reverse the inhibitory effect of CBZ on several protein markers of adipocyte differentiation such as PPAR-γ, HSL and adiponectin (Figure 6C). To ensure that UO126 does not have a non-specific effect on cell viability, we performed a MTT assay that showed no significant effect of this compound (Figure 6D). Finally, we also examined whether UO126 could antagonize the effect of CBZ on clonal expansion. As shown in Figure 6E, UO126 induced a twofold decrease in [3H]-thymidine incorporation and did not modify CBZ effect on cell proliferation. Collectively, these results suggest that activation of the ERK 1/2 pathway is at least partly involved in the anti-adipogenic effect of CBZ.
Discussion
CBZ is today largely used in several fields, including pain management, epilepsy and mood regulation (Arain and Abou-Khalil, 2009; Grunze, 2010; Wiffen et al., 2011). However, long-term exposure to this drug is associated with the onset of several metabolic side effects, such as weight gain (Jallon and Picard, 2001; Mintzer, 2010). Unexpectedly, by using convergent morphological, biochemical and molecular approaches, we showed that in various models of pre-adipocyte differentiation, CBZ strongly inhibits adipogenesis through pleiotropic mechanisms.
The repression of the transcriptional programme of adipogenesis appears to be central in the inhibitory effect of CBZ. PPAR-γ and C/EBP-α, which are known to be the two main transcription factors of adipogenesis (Rosen et al., 2000), were strongly down-regulated by CBZ. The role of PPAR-γ repression as a key mechanism was strengthened by the finding that the CBZ effect was reversed in the presence of the PPAR-γ-selective agonist pioglitazone. By contrast, C/EBP-β and SREBP-1c expression was only weakly reduced or unaffected by CBZ. This absence of significant effect of CBZ on the SREBP-1c pathway has already been shown in cultured hepatocytes (Raeder et al., 2006). SREBP-1c is the main transcription factor of the lipogenic pathway since it transactivates the genes coding for lipogenic enzymes such as ACC, FAS and stearoyl CoA desaturase (Girard et al., 1997). In agreement with the absence of a significant effect of CBZ on the lipogenic pathway, the addition of intralipid to CBZ-treated cells, which would circumvents thi metabolic pathway, was unable to restore cell triglyceride stores. The potent inhibitory effect of CBZ on PPAR-γ and C/EBP-α expression could also be related to the blockade of clonal expansion (Figure 4). However, this effect of CBZ was retained, but to a lesser extent, when the drug was added following clonal expansion. Altogether, these findings strongly suggest that repression of PPAR-γ and of clonal expansion are independent mechanisms that both contribute to the anti-adipogenic effect of CBZ. Interestingly, we have recently shown that phenelzine, an antidepressant of the monoamine oxidase inhibitor family, also potently inhibits adipose conversion (Chiche et al., 2009). However, the blockade of the lipogenic pathway through SREBP-1c down-regulation is a central mechanism that accounts for this effect of phenelzine.
While less pronounced than the effect on differentiating pre-adipocytes, CBZ also triggered mature adipocytes to reduce their cell triglyceride content. Two additional mechanisms could contribute to the depletion of fat cell stores in differentiated adipocytes. Firstly, CBZ markedly reduces insulin-stimulated glucose uptake, while basal glucose transfer remains unaltered. Secondly, it increases basal lipolysis. Although the mechanisms underlying CBZ effect on glucose uptake and lipolysis remain to be investigated, this underlines the pleiotropy of the metabolic pathways altered by this drug. Both reduced insulin-stimulated glucose uptake and increased lipolysis represent key mechanisms of insulin resistance. Remarkably, CBZ decreases adiponectin synthesis and secretion, which could also contribute to insulin resistance (Kahn et al., 2006; Shetty et al., 2009). Whether CBZ influences insulin sensitivity in patients is another major issue.
CBZ mimics the effects of pro-inflammatory cytokine on adipocytes; it increases lipolysis and suppresses adipogenesis, insulin-stimulated glucose uptake and adiponectin secretion (Feve and Bastard, 2009). Since these pro-inflammatory cytokine effects are mediated by various signalling pathways, we tested the hypothesis that some of these could also be involved in the effect of CBZ on adipogenesis. ERK 1/2 appears to be a privileged target that mediates at least part of CBZ's effect on adipogenesis. It is known that MEK inhibitors such as UO126 alter clonal expansion but not terminal differentiation (Qiu et al., 2001; Bost et al., 2005). Thus, it was not surprising that in our experiments, UO126 inhibited [3H]-thymidine incorporation during clonal expansion. However, since UO126 was still able to reverse the anti-adipogenic effect of CBZ, this further indicates that inhibition of the transcriptional programme of adipogenesis is a central mechanism contributing to the anti-adipogenic effect of CBZ but not to its blockade of clonal expansion. To extend this observation, the effects of CBZ could be further examined in ERK 1/2-deficient pre-adipocytes or animals.
The CBZ concentration required (100 μM) to significantly decrease fat cell lipid content is slightly higher than the therapeutic plasma concentration of 17–50 μM (Bertilsson, 1978; Citrome et al., 2007). However, we believe that our results remain relevant for the following reasons: (i) our study was carried out within a limited time lag, and lower concentrations over a longer period could have similar effects; (ii) therapeutic plasma concentrations have been defined for epileptic patients, and CBZ can be prescribed at higher doses when used as a mood stabilizer (Grunze, 2010); and (iii) CBZ is a lipophilic compound that could accumulate in adipose tissue during chronic exposure.
To our knowledge, this work is the first to report a strong inhibitory effect of CBZ on adipogenesis. In addition, two other mood stabilizers, lithium and valproic acid, have also been shown to have similar properties. So far, two distinct anti-adipogenic mechanisms have been identified: while lithium activates the Wnt/β-catenin pathway (Ross et al., 2000), valproic acid's inhibitory effect is thought to be mediated by its histone deacetylase properties (Lagace and Nachtigal, 2004; Ebmeier et al., 2006) After excluding a major involvement of the Wnt/β-catenin signalling, we suggest that a third distinct mechanism is the basis of CBZ effect on adipocytes. Although we think that these three mood stabilizers each use an independent mechanism, it is tempting to speculate that these three drugs share some common pathways to block adipogenesis.
Like other mood stabilizers, CBZ can promote weight gain in patients (Joffe et al., 1986; Lampl et al., 1991). It seems paradoxical that in vitro CBZ inhibits adipogenesis yet in vivo it induces adiposity. Weight homeostasis depends on both central and peripheral inputs. CBZ is known to increase appetite and this central effect may overcome its direct inhibitory effect on adipose tissue development. From a clinical point of view, we propose that the development of CBZ derivatives with less central effect but higher anti-adipogenic potency could represent a relevant approach to limit its metabolic side effects, particularly for epileptic or bipolar patients who often suffer from a disturbed energy homeostasis. Further to the effect of CBZ on weight regulation, it would also be of major interest to examine whether CBZ has specific effects on glucose and lipid metabolism.
In conclusion, we demonstrated that CBZ, through ERK 1/2 activation, has a potent inhibitory effect on the differentiation of fat cells by altering the transcriptional programme of adipogenesis. This illustrates a potential direct effect of CBZ on a key metabolic tissue and suggests that closer metabolic monitoring is needed when treating patients with mood stabilizers.
Acknowledgments
This work was supported by grants from INSERM and Universities Paris 6 and 11. E Turpin received a personal grant from the Société Française d'Endocrinologie.
Glossary
- ACC
acetyl-CoA carboxylase
- C/EBP-α
CCAAT/enhancer binding protein-α
- C/EBP-β
CCAAT/enhancer binding protein-β
- CBZ
carbamazepine
- DOG
deoxyglucose
- FAS
fatty acid synthase
- G3PDH
glycerol-3-phosphate dehydrogenase
- HSL
hormone-sensitive lipase
- MEK
MAPK/ERK kinase
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- SREBP-1c
sterol regulatory element-binding protein 1c
Conflict of interest
None of the authors declare any conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Carbamazepine-induced changes in cytokines secreted by 3T3-L1 adipocytes. Differentiated 3T3-L1 adipocytes were exposed for 24 hours to 500 μM carbamazepine in a serum-free medium, then media were collected and tested for cytokine expression with a specific array. Results are expressed as the percentage of cytokine level measured in control conditions. Only repressed (A) and induced (B) cytokines are shown in the graph.
Figure S2 Effect of carbamazepine on the Wnt/β-catenin pathway. 3T3-L1 pre-adipocytes were exposed or not to carbamazepine (300 μM) from confluence to day 8 following confluence. Cell extracts were prepared and tested in Western blot analysis for FAS, total β-catenin, cyclin D1 and ERK 1/2 expression. The blot is representative of 4 independent experiments.
References
- Altamura AC, Lietti L, Dobrea C, Benatti B, Arici C, Dell'Osso B. Mood stabilizers for patients with bipolar disorder: the state of the art. Expert Rev Neurother. 2011;11:85–99. doi: 10.1586/ern.10.181. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Practice guideline for the treatment of patients with bipolar disorder (revision) Am J Psychiatry. 2002;159:1–50. [PubMed] [Google Scholar]
- Arain AM, Abou-Khalil BW. Management of new-onset epilepsy in the elderly. Nat Rev Neurol. 2009;5:363–371. doi: 10.1038/nrneurol.2009.74. [DOI] [PubMed] [Google Scholar]
- Behrens J. Control of beta-catenin signaling in tumor development. Ann N Y Acad Sci. 2000;910:21–33. doi: 10.1111/j.1749-6632.2000.tb06698.x. discussion 33–25. [DOI] [PubMed] [Google Scholar]
- Bertilsson L. Clinical pharmacokinetics of carbamazepine. Clin Pharmacokinet. 1978;3:128–143. doi: 10.2165/00003088-197803020-00003. [DOI] [PubMed] [Google Scholar]
- Bost F, Aouadi M, Caron L, Even P, Belmonte N, Prot M, et al. The extracellular signal-regulated kinase isoform erk1 is specifically required for in vitro and in vivo adipogenesis. Diabetes. 2005;54:402–411. doi: 10.2337/diabetes.54.2.402. [DOI] [PubMed] [Google Scholar]
- Caksen H, Deda G, Berberoglu M, Icagasioglu D, Turan EB. Serum leptin levels in children receiving long-term carbamazepine. Acta Paediatr Taiwan. 2003;44:82–83. [PubMed] [Google Scholar]
- Catalioto RM, Maggi CA, Giuliani S. Chemically distinct hdac inhibitors prevent adipose conversion of subcutaneous human white preadipocytes at an early stage of the differentiation program. Exp Cell Res. 2009;315:3267–3280. doi: 10.1016/j.yexcr.2009.09.012. [DOI] [PubMed] [Google Scholar]
- Cathala G, Savouret JF, Mendez B, West BL, Karin M, Martial JA, et al. A method for isolation of intact, translationally active ribonucleic acid. DNA. 1983;2:329–335. doi: 10.1089/dna.1983.2.329. [DOI] [PubMed] [Google Scholar]
- Chiche F, Le Guillou M, Chetrite G, Lasnier F, Dugail I, Carpene C, et al. Antidepressant phenelzine alters differentiation of cultured human and mouse preadipocytes. Mol Pharmacol. 2009;75:1052–1061. doi: 10.1124/mol.108.052563. [DOI] [PubMed] [Google Scholar]
- Citrome L, Macher JP, Salazar DE, Mallikaarjun S, Boulton DW. Pharmacokinetics of aripiprazole and concomitant carbamazepine. J Clin Psychopharmacol. 2007;27:279–283. doi: 10.1097/jcp.0b013e318056f309. [DOI] [PubMed] [Google Scholar]
- Ebmeier KP, Donaghey C, Steele JD. Recent developments and current controversies in depression. Lancet. 2006;367:153–167. doi: 10.1016/S0140-6736(06)67964-6. [DOI] [PubMed] [Google Scholar]
- Feve B, Bastard JP. The role of interleukins in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol. 2009;5:305–311. doi: 10.1038/nrendo.2009.62. [DOI] [PubMed] [Google Scholar]
- Frye MA. Clinical practice. Bipolar disorder – a focus on depression. N Engl J Med. 2011;364:51–59. doi: 10.1056/NEJMcp1000402. [DOI] [PubMed] [Google Scholar]
- Girard J, Ferre P, Foufelle F. Mechanisms by which carbohydrates regulate expression of genes for glycolytic and lipogenic enzymes. Annu Rev Nutr. 1997;17:325–352. doi: 10.1146/annurev.nutr.17.1.325. [DOI] [PubMed] [Google Scholar]
- Goodwin GM. Evidence-based guidelines for treating bipolar disorder: revised second edition-recommendations from the British Association for Psychopharmacology. J. Psychopharmacol. 2009;23:346–388. doi: 10.1177/0269881109102919. [DOI] [PubMed] [Google Scholar]
- Grunze HC. Anticonvulsants in bipolar disorder. J Ment Health. 2010;19:127–141. doi: 10.3109/09638230903469186. [DOI] [PubMed] [Google Scholar]
- Hamed SA, Fida NM, Hamed EA. States of serum leptin and insulin in children with epilepsy: risk predictors of weight gain. Eur J Paediatr Neurol. 2009;13:261–268. doi: 10.1016/j.ejpn.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Jallon P, Picard F. Bodyweight gain and anticonvulsants: a comparative review. Drug Saf. 2001;24:969–978. doi: 10.2165/00002018-200124130-00004. [DOI] [PubMed] [Google Scholar]
- Joffe RT, Post RM, Uhde TW. Effect of carbamazepine on body weight in affectively ill patients. J Clin Psychiatry. 1986;47:313–314. [PubMed] [Google Scholar]
- Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- Kozak LP, Jensen JT. Genetic and developmental control of multiple forms of l-glycerol 3-phosphate dehydrogenase. J Biol Chem. 1974;249:7775–7781. [PubMed] [Google Scholar]
- Lagace DC, Nachtigal MW. Inhibition of histone deacetylase activity by valproic acid blocks adipogenesis. J Biol Chem. 2004;279:18851–18860. doi: 10.1074/jbc.M312795200. [DOI] [PubMed] [Google Scholar]
- Lagace DC, McLeod RS, Nachtigal MW. Valproic acid inhibits leptin secretion and reduces leptin messenger ribonucleic acid levels in adipocytes. Endocrinology. 2004;145:5493–5503. doi: 10.1210/en.2004-0877. [DOI] [PubMed] [Google Scholar]
- Lampl Y, Eshel Y, Rapaport A, Sarova-Pinhas I. Weight gain, increased appetite, and excessive food intake induced by carbamazepine. Clin Neuropharmacol. 1991;14:251–255. doi: 10.1097/00002826-199106000-00009. [DOI] [PubMed] [Google Scholar]
- Longo KA, Wright WS, Kang S, Gerin I, Chiang SH, Lucas PC, et al. Wnt10b inhibits development of white and brown adipose tissues. J Biol Chem. 2004;279:35503–35509. doi: 10.1074/jbc.M402937200. [DOI] [PubMed] [Google Scholar]
- Luef G, Rauchenzauner M, Waldmann M, Sturm W, Sandhofer A, Seppi K, et al. Non-alcoholic fatty liver disease (NAFLD), insulin resistance and lipid profile in antiepileptic drug treatment. Epilepsy Res. 2009;86:42–47. doi: 10.1016/j.eplepsyres.2009.04.004. [DOI] [PubMed] [Google Scholar]
- Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3:e442. doi: 10.1371/journal.pmed.0030442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintzer S. Metabolic consequences of antiepileptic drugs. Curr Opin Neurol. 2010;23:164–169. doi: 10.1097/WCO.0b013e32833735e7. [DOI] [PubMed] [Google Scholar]
- Moldes M, Zuo Y, Morrison RF, Silva D, Park BH, Liu J, et al. Peroxisome-proliferator-activated receptor gamma suppresses wnt/beta-catenin signalling during adipogenesis. Biochem J. 2003;376:607–613. doi: 10.1042/BJ20030426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pairault J, Green H. A study of the adipose conversion of suspended 3t3 cells by using glycerophosphate dehydrogenase as differentiation marker. Proc Natl Acad Sci U S A. 1979;76:5138–5142. doi: 10.1073/pnas.76.10.5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijl H, Meinders AE. Bodyweight change as an adverse effect of drug treatment. Mechanisms and management. Drug Saf. 1996;14:329–342. doi: 10.2165/00002018-199614050-00005. [DOI] [PubMed] [Google Scholar]
- Prestwich TC, Macdougald OA. Wnt/beta-catenin signaling in adipogenesis and metabolism. Curr Opin Cell Biol. 2007;19:612–617. doi: 10.1016/j.ceb.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao L, Schaack J, Shao J. Suppression of adiponectin gene expression by histone deacetylase inhibitor valproic acid. Endocrinology. 2006;147:865–874. doi: 10.1210/en.2005-1030. [DOI] [PubMed] [Google Scholar]
- Qiu Z, Wei Y, Chen N, Jiang M, Wu J, Liao K. DNA synthesis and mitotic clonal expansion is not a required step for 3t3-l1 preadipocyte differentiation into adipocytes. J Biol Chem. 2001;276:11988–11995. doi: 10.1074/jbc.M011729200. [DOI] [PubMed] [Google Scholar]
- Raeder MB, Ferno J, Vik-Mo AO, Steen VM. Srebp activation by antipsychotic- and antidepressant-drugs in cultured human liver cells: relevance for metabolic side-effects? Mol Cell Biochem. 2006;289:167–173. doi: 10.1007/s11010-006-9160-4. [DOI] [PubMed] [Google Scholar]
- Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM. Transcriptional regulation of adipogenesis. Genes Dev. 2000;14:1293–1307. [PubMed] [Google Scholar]
- Ross SE, Hemati N, Longo KA, Bennett CN, Lucas PC, Erickson RL, et al. Inhibition of adipogenesis by wnt signaling. Science. 2000;289:950–953. doi: 10.1126/science.289.5481.950. [DOI] [PubMed] [Google Scholar]
- Shetty S, Kusminski CM, Scherer PE. Adiponectin in health and disease: evaluation of adiponectin-targeted drug development strategies. Trends Pharmacol Sci. 2009;30:234–239. doi: 10.1016/j.tips.2009.02.004. [DOI] [PubMed] [Google Scholar]
- Uludag IF, Kulu U, Sener U, Kose S, Zorlu Y. The effect of carbamazepine treatment on serum leptin levels. Epilepsy Res. 2009;86:48–53. doi: 10.1016/j.eplepsyres.2009.04.005. [DOI] [PubMed] [Google Scholar]
- Viengchareun S, Servel N, Feve B, Freemark M, Lombes M, Binart N. Prolactin receptor signaling is essential for perinatal brown adipocyte function: a role for insulin-like growth factor-2. PLoS ONE. 2008;3:e1535. doi: 10.1371/journal.pone.0001535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigo DV, Baldessarini RJ. Anticonvulsivants in the treatment of major depressive disorder: an overview. Harv Rev Psychiatry. 2009;17:231–241. doi: 10.1080/10673220903129814. [DOI] [PubMed] [Google Scholar]
- Wiffen PJ, Derry S, Moore RA, McQuay HJ. Carbamazepine for acute and chronic pain in adults. Cochrane Database Syst Rev. 2011;19(1) doi: 10.1002/14651858.CD005451.pub2. January: CD005451. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.