

Abstract
Background and Purpose
Intrathecally (i.t.) administered nociceptin/orphanin FQ (N/OFQ) evokes antinociceptive effects in rodents. Recent studies in monkeys demonstrated that i.t. co-application of N/OFQ and morphine elicits synergistic antinociceptive actions suggesting mixed N/OFQ peptide (NOP) and μ opioid receptor agonists as innovative spinal analgesics. Thus, novel N/OFQ related peptides were synthesized in order to identify and pharmacologically characterize a mixed NOP/ μ opioid receptor agonist.
Experimental Approach
The following in vitro assays were used: calcium mobilization in cells expressing the human NOP or classical opioid receptors and chimeric G proteins, receptor and [35S]-GTPγS binding, [35S]-GTPγS binding in rat spinal cord membranes, guinea pig ileum bioassay. In vivo experiments were performed in monkeys using the tail withdrawal assay.
Key Results
From calcium mobilization studies [Dmt1]N/OFQ(1–13)-NH2 was selected as the most potent and least selective compound. The mixed NOP/opioid full agonist activity and high affinity of [Dmt1]N/OFQ(1–13)-NH2 was confirmed at human recombinant receptors in receptor binding, calcium mobilization and/or [35S]-GTPγS binding studies, at rat spinal cord receptors in [35S]-GTPγS binding experiments, and at guinea pig receptors inhibiting neurogenic contractions in the ileum. In vivo in the tail withdrawal assay in monkeys i.t. [Dmt1]N/OFQ(1–13)-NH2 was able to elicit robust and long-lasting antinociceptive effects.
Conclusions and Implications
Collectively, these results demonstrate that [Dmt1]N/OFQ(1–13)-NH2 behaves as NOP/opioid receptor universal agonist and substantiate the suggestion that such mixed ligands are worthy of development as innovative spinal analgesics.
Keywords: [Dmt1]N/OFQ(1–13)-NH2, NOP receptor, opioid receptors, calcium mobilization, receptor and [35S]-GTPγS binding, guinea pig ileum, spinal cord, tail withdrawal assay, monkey
Introduction
The heptadecapeptide nociceptin/orphanin FQ (N/OFQ) modulates various biological functions including locomotor activity, anxiety and mood, memory, food intake, immunity, heart rate and blood pressure, diuresis, gastrointestinal motility, micturition and cough reflexes, and nociception (Lambert, 2008) via selective activation of the N/OFQ peptide (NOP) receptor (Meunier et al., 1995; Reinscheid et al., 1995). In particular, pain transmission is modulated by N/OFQ in an opposite manner depending on the site of administration. In fact, rodent studies demonstrated that N/OFQ given supraspinally produces pronociceptive effects and counteracts opioid-induced analgesia while, when given spinally, the peptide evokes, similar to opioids, antinociceptive effects (Zeilhofer and Calo, 2003). The spinal antinociceptive action of N/OFQ is dose-dependent, naloxone-insensitive, no longer evident in mice knockout for the NOP receptor gene [NOP(–/–)], mimicked by the NOP selective agonist UFP-112, and prevented by selective NOP antagonists (UFP-101, SB-612111, Compound 24) (Nazzaro et al., 2007; Rizzi et al., 2007a,b; Fischetti et al., 2009). This effect of N/OFQ has been confirmed in non-human primates (Ko et al., 2006). The spinal antinociceptive action of N/OFQ in monkeys is mimicked by the intrathecal (i.t.) injection of the peptide UFP-112 (Hu et al., 2009) as well as by the systemic administration of the NOP selective non-peptide agonist Ro 64–6198 (Ko et al., 2009). Moreover, the spinal antinociceptive effects of N/OFQ and NOP agonists in monkeys are sensitive to the NOP selective antagonist J-113397 (Ko et al., 2006; 2009; Hu et al., 2009). Interestingly, N/OFQ in combination with i.t. morphine dose-dependently potentiated alkaloid-induced analgesia in monkeys (Ko and Naughton, 2009). Thus, it seems that the simultaneous activation of spinal NOP and opioid (particularly the μ opioid receptor) receptors is able to produce a synergistic antinociceptive effect. Based on these findings we may anticipate that molecules able to activate with similar potencies NOP and opioid receptors (i.e. mixed NOP/opioid agonists) represent an innovative and interesting class of spinal analgesics.
Small molecules acting as mixed NOP/opioid agonists have been previously described by industrial (Gruenenthal and Purdue Pharma, see patent literature reviewed by Mustazza and Bastanzio, 2011) and academic (Khroyan et al., 2007; 2011; Spagnolo et al., 2008; Toll et al., 2009) laboratories. The opposite action on nociception exerted by N/OFQ at spinal versus supraspinal levels makes it difficult to predict the analgesic potential of these compounds after systemic administration; however, it is worth noting that several mixed NOP/opioid receptor agonists have been demonstrated to elicit robust antinociceptive effects in rodents after peripheral administration.
The aim of the present study was the design, synthesis and pharmacological characterization of novel peptides acting as non-selective NOP/opioid agonists and their in vivo evaluation as spinal analgesics in monkeys. Thus, some [X1] substituted N/OFQ analogues were synthesized and evaluated pharmacologically in calcium mobilization experiments performed in CHO cells expressing the human NOP or classical opioid receptors as well as chimeric G-proteins that force Gi-coupled receptors to signal via the PLC-IP3-Ca2+ pathway (Camarda et al., 2013; Fischetti et al., 2009). From these experiments, [Dmt1]N/OFQ(1–13)-NH2 was selected as the most potent and least selective agonist. The pharmacological profile of this peptide was then evaluated in vitro in (i) membranes of CHO cells expressing the NOP, μ, δ or κ opioid receptor studied with receptor binding and stimulation of [35S]-GTPγS binding experiments for μ opioid and NOP receptors; (ii) membranes of the rat cerebral cortex or spinal cord in the [35S]-GTPγS assay; and (iii) the electrically stimulated guinea pig ileum, a pharmacological preparation expressing both μ opioid and NOP receptors. The in vivo activity of [Dmt1]N/OFQ(1–13)-NH2 was investigated by injecting the peptide i.t. and measuring its effects in the tail withdrawal assay in non-human primates.
Methods
Calcium mobilization
CHO cells stably co-expressing the human NOP, κ or μ opioid receptor and the C-terminally modified Gαqi5 and CHO cells expressing the δ opioid receptor and the GαqG66Di5 protein were maintained in culture medium consisting of Dulbecco's MEM/HAM'S F-12 (50/50) supplemented with 10% fetal calf serum, penicillin (100 IU·mL−1), streptomycin (100 μg·mL−1), fungizone (2.5 μg·mL−1), geneticin (G418; 200 μg·mL−1) and hygromycin B (200 mg·mL−1) as described previously (Camarda et al., 2013; Fischetti et al., 2009; Camarda and Calo', 2013). Cell cultures were kept at 37°C in 5% CO2 humidified air. In all cases, experimental cultures were free from selection agents (hygromycin B, G418). When confluence was reached (3–4 days), cells were subcultured as required using trypsin//EDTA and used for experimentation. Cells were seeded at a density of 50 000 cells per well into 96-well black, clear-bottom plates. After 24 h incubation, the cells were loaded with medium supplemented with 2.5 mM probenecid, 3 μM of the calcium sensitive fluorescent dye Fluo-4 AM and 0.01% pluronic acid, for 30 min at 37°C. Afterwards, the loading solution was aspirated and 100 μL per well of assay buffer: HBSS supplemented with 20 mM HEPES, 2.5 mM probenecid and 500 μM Brilliant Black (Sigma-Aldrich, St. Louis, MO, USA) was added. Serial dilutions of ligands for experimental use were made in HBSS/HEPES (20 mM) buffer (containing 0.02% BSA fraction V). After placing both plates (cell culture and compound plate) into the FlexStation II (Molecular Device, Union City, CA, US), fluorescence changes were measured at room temperature. Online additions were carried out in a volume of 50 μL per well.
Receptor binding
These were performed using freshly prepared membranes from CHO cells expressing human μ, δ, κ opioid or NOP receptors essentially according to McDonald et al. (2003). Briefly, membranes (100 μg per tube) were incubated in 0.5 mL volumes with a fixed concentration of [3H]-diprenorphine ([3H]-DPN, 0.8 nM) for μ, δ and κ opioid receptors or [3H]-UFP-101 (1 nM) and increasing concentrations of [Dmt1]N/OFQ(1–13)-NH2 and a range of reference ligands as described in the Results section. Non-specific binding was assessed using naloxone for μ, δ and κ opioid receptors for [3H]-DPN or J-113397 for [3H]-UFP-101. Bound and free radioligand were separated following 1 h incubation at room temperature by vacuum filtration.
[35S]-GTPγS stimulation binding
This was performed using freshly prepared membranes from CHO cells expressing human μ opioid or NOP receptors (50 μg per tube) or frozen rat cerebral cortex or spinal cord membranes (100 μg per tube) essentially according to McDonald et al. (2003). Briefly, membranes were incubated at 30°C in 0.5 mL of assay buffer supplemented with [35S]-GTPγS (∼160 pM) and increasing concentrations of [Dmt1]N/OFQ(1–13)-NH2 and a range of reference ligands as described in the Results section. Non-specific binding was defined in the presence of unlabelled GTPγS. Bound and free radioligand were separated by vacuum filtration. Rats (Charles River Italia, Lecco, Italy) were handled according to guidelines published in the European Communities Council directives (86/609/EEC) and National regulations (D.L. 116/92). They were housed in 425 × 266 × 155 mm cages (Techniplast, Milsn, Italy), eight per cage, under standard conditions (22°C, 55% humidity, 12 h light/dark cycle, lights on at 07:00 h ) with food (MIL, standard diet, Morini, Reggio Emilia, Italy) and water ad libitum. The total number of rats used was 10. Rats were killed by isofluorane overdose. These experiments and protocols were approved by the University of Ferrara Ethic Committee for Animal Research. All studies involving animals are reported in accordance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath et al., 2010).
Electrically stimulated guinea pig ileum
Segments of ileum were taken from male albino guinea pigs (300–350 g). The total number of guinea pigs used was 4. The bioassay experiments were performed as previously described (Bigoni et al., 1999). The tissues were suspended in 5 mL organ baths containing Krebs solution (composition in mM: NaCl 118.5, KCl 4.7, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25, CaCl2 1.8, glucose 10, hexamethonium bromide 2.2 and benadril 1.37) oxygenated with 95% O2 and 5% CO2. The temperature was set at 37°C. A resting tension 1 g was applied to the tissues. Tissues were stimulated through two platinum ring electrodes with supramaximal rectangular pulses of 1 ms duration and 0.05 Hz frequency. The electrically evoked contractions were measured isotonically by means of Basile strain gauge transducers and recorded with a personal computer-based acquisition system (Power Lab, 4/25, ADInstruments, Sydney, Australia). After an equilibration period of about 60 min, the contractions induced by electrical field stimulation were stable. At this time, cumulative concentration–response curves to agonists were performed (0.5 log unit steps). Guinea pigs (Pampaloni, Siena, Italy) were handled according to guidelines published in the European Communities Council directives (86/609/EEC) and National Regulations (D.L. 116/92). They were housed in 425 × 266 × 155 mm cages (Techniplast), five per cage, under standard conditions (22°C, 55% humidity, 12 h light/dark cycle, light on at 07:00 h) with food (MIL, standard diet, Morini, Reggio Emilia) and water ad libitum. Guinea pigs were killed by isofluorane overdose. These experiments and protocols were approved by the University of Ferrara Ethic Committee for Animal Research.
Monkey tail withdrawal assay
Ten adult intact male and female rhesus monkeys (Macaca mulatta) with body weights ranging between 6.8 and 12.5 kg were used. The monkeys were housed individually with free access to water and were fed approximately 25 biscuits (Purina Monkey Chow; Ralston Purina, St. Louis, MO, USA) and fresh fruit daily. No monkey had exposure to any opioid drug 1 month before the present study. The monkeys were housed in facilities accredited by the American Association for the Accreditation of Laboratory Animal Care. The studies were conducted in accordance with the University Committee on the Use and Care of Animals in the University of Michigan (Ann Arbor, MI, USA) and the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health (Bethesda, MD, USA).
The warm water tail-withdrawal assay was used to evaluate thermal antinociceptive effects of the test compound (Ko et al., 2006). Briefly, monkeys were seated in primate restraint chairs, and the lower part of their shaved tails (approximately 15 cm) were immersed in a thermal flask containing water maintained at either 42, 46 or 50°C. Tail-withdrawal latencies were measured using a computerized timer by an experimenter who did not know dosing conditions. In each test session, monkeys were evaluated once with three temperatures given in a random order. If the monkeys did not remove their tails within 20 s (cutoff), the flask was removed and a maximum time of 20 s was recorded. Test sessions began with determining a control value at each temperature. Subsequent tail-withdrawal latencies were determined every 30 min after i.t. administration. The same group of subjects (n = 4) was tested in a 3 h time course by using a single dosing procedure.
Scratching behaviour, inferred as a response to itch sensation (Ko et al., 2004), was recorded on videotape while the monkeys were in their home cages. A scratch was defined as one short-duration (<1 s) episode of scraping contact of the forepaw or hind paw on the skin surface of other body parts. Scratching responses were scored by individuals who were blinded to experimental conditions. Each recording session was conducted for 15 min per test session that occurred every 30 min after i.t. administration. The same group of subjects (n = 6) was tested in a 3 h time course by using a single dosing procedure.
For i.t. administration in monkeys, [Dmt1]N/OFQ(1–13)-NH2 was administered at a total volume of 1 mL. The detailed description for i.t. drug delivery can be referred to previous studies (Ko et al., 2006). All experiments using i.t. administration in monkeys were conducted with a 10-day inter-injection interval.
Drugs
All peptides were prepared and purified as previously described (Guerrini et al., 1997). All tissues culture media and supplements were from Invitrogen (Paisley, UK). Peptides and naloxone were solubilized in distilled water. J-113397 was solubilized in DMSO at the concentration of 10 mM with successive dilutions made in saline. Stock solutions were kept at −20°C until use. For in vivo experiments peptides were dissolved in sterile saline solution just before injections.
Data analysis and terminology
The drug and receptor nomenclature and terminology adopted in this paper conforms to the guide to receptors and channels (Alexander et al., 2011). All data are expressed as means ± SEM of n experiments. For potency values 95% confidence limits were indicated.
Calcium mobilization data are expressed in fluorescence intensity units (FIU) as % over the baseline. Receptor binding data are expressed as % displacement. [35S]-GTPγS data are expressed as stimulation factor that is the ratio between specific agonist stimulated [35S]-GTPγS binding and basal specific binding. Guinea-pig ileum data are expressed as % of the control twitch induced by electrical field stimulation.
Affinity values are showed as pKi calculated using the Cheng and Prusoff equation:
Agonist potencies are given as pEC50 = the negative logarithm to base 10 of the molar concentration of an agonist that produces 50% of the maximal possible effect. Concentration response curve to agonists were fitted with the following equation:
where X is the agonist concentration.
Antagonist potencies were derived from the Gaddum Schild equation:
Assuming a slope value equal to unity, where CR indicates the ratio between agonist potency in the presence and absence of antagonist.
For in vivo monkey studies, mean values (mean ± SEM) were calculated from individual values for all behavioural end points. Comparisons were made for the same monkeys across all test sessions. Data were analysed by a two-way repeated ANOVA followed by the Newman–Keuls test for multiple comparisons. The criterion for significant differences was set at P < 0.05.
Results
In CHONOP cells stably expressing the Gαqi5 chimeric protein, N/OFQ evoked a concentration-dependent stimulation of calcium release displaying high potency [pEC50 9.30 (CL95% 9.05–9.55) ] and maximal effect (240 ± 14% over the basal values) while dermorphin was inactive up to 10 μM (Figure 1, left panel). Opposite results were obtained in CHOMOP, CHO cells stably expressing the Gαqi5 chimeric protein and human μ opioid receptors, where dermorphin concentration-dependently stimulated calcium mobilization [pEC50 8.17 (CL95% 7.93–8.41) ]; Emax 130 ± 12% over the basal values) while N/OFQ was inactive up to 1 μM (Figure 1, right panel). Under the same experimental conditions, N/OFQ analogues were assayed in both cell lines. Table 1 summarizes the results obtained in this series of experiments. The amide form of N/OFQ displayed similar potency, maximal effects and selectivity of action as the natural peptide. [Tyr1]N/OFQ-NH2 displayed a slight reduction in NOP potency [pEC50 9.14 (CL95% 8.93–9.35) ] while being able to activate the μ opioid receptor, although only in the micromolar range of concentrations [pEC50 6.07 (CL95% 5.90–6.24) ]. The substitution of Phe1 with Dmt produced a reduction of NOP potency by 10-fold [pEC50 8.57 (CL95% 8.28–8.86) ] associated with an important increase in μ opioid potency [pEC50 7.05 (CL95% 6.55–7.55) ]. Similar results were obtained when the [Tyr1] and [Dmt1] modifications were applied to the N/OFQ(1–13)-NH2 template. N/OFQ(1–13)-NH2 behaves as a highly potent [pEC50 9.49 (CL95% 9.42–9.56) ] and selective NOP agonist. [Tyr1]N/OFQ(1–13)-NH2 displayed a slight reduction in NOP potency [pEC50 9.16 (CL95% 8.91–9.41) ] and selectivity. [Dmt1]N/OFQ(1–13)-NH2 similar to [Dmt1]N/OFQ-NH2 behaved as a mixed NOP/μ opioid receptor agonist showing only 26-fold selectivity for NOP over μ opioid receptors (Figure 1 and Table 1). From this series of experiments, the compound [Dmt1]N/OFQ(1–13)-NH2 was selected as the most potent and least selective NOP/μ opioid receptor agonist.
Figure 1.
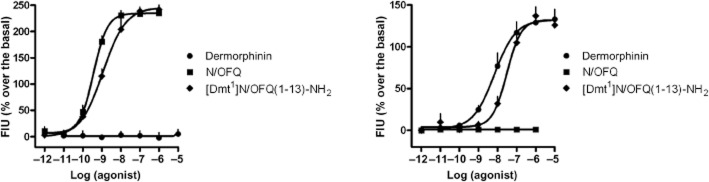
Calcium mobilization experiments. Concentration–response curves to N/OFQ, dermorphin, and [Dmt1]N/OFQ(1–13)-NH2 in CHO cells stably expressing the Gαqi5 chimeric protein and the NOP (left panel) and μ opioid (right panel) human recombinant receptor. Data are the mean ± SEM of four separate experiments performed in duplicate.
Table 1.
Effects of standard and novel agonists in calcium mobilization experiments performed in CHO cells stably expressing the human NOP or μ opioid receptor and the Gαqi5 protein
| NOP | μ | ||||
|---|---|---|---|---|---|
| pEC50(CL95%) | Emax ± SEM | pEC50(CL95%) | Emax ± SEM | μ/NOP | |
| Dermorphin | <5 | – | 8.17 (7.93–8.41) | 133 ± 12% | <0.0007 |
| N/OFQ | 9.30 (9.05–9.55) | 235 ± 14% | <5 | – | >20.000 |
| N/OFQ-NH2 | 9.49 (9.42–9.56) | 255 ± 13% | <5 | – | >30.000 |
| [Tyr1]N/OFQ-NH2 | 9.14 (8.93–9.35) | 289 ± 14% | 6.07 (5.90–6.24) | 121 ± 16% | 1174 |
| [Dmt1]N/OFQ-NH2 | 8.57 (8.28–8.86) | 259 ± 7% | 7.05 (6.55–7.55) | 97 ± 10% | 33 |
| N/OFQ(1–13)-NH2 | 9.49 (9.42–9.56) | 222 ± 10% | <5 | – | >30.000 |
| [Tyr1]N/OFQ(1–13)-NH2 | 9.16 (8.91–9.41) | 235 ± 17% | 6.01 (5.67–6.49) | 105 ± 6% | 1412 |
| [Dmt1]N/OFQ(1–13)-NH2 | 8.94 (8.39–9.49) | 242 ± 12% | 7.52 (7.18–7.86) | 126 ± 18% | 26 |
The values are the means of three to four separate experiments performed in duplicate.
[Dmt1]N/OFQ(1–13)-NH2 affinity for NOP and classical opioid receptors was assessed in displacement binding experiments performed in membranes of CHO cells transfected with human recombinant receptors and compared with affinities of standard ligands. In CHONOP cell membranes, N/OFQ displaced the radioligand with a pKi value of 10.18 (CL95% 10.01–10.35). N/OFQ(1–13)-NH2 bound the receptor with a pKi of 10.60 (CL95% 10.41–10.79). The NOP selective antagonist J-113397 displayed an affinity of 9.44 (CL95% 9.29–9.59). [Dmt1]N/OFQ(1–13)-NH2 displaced the radioligand with a pKi value of 10.59 (CL95% 10.40–10.78) (Figure 2, left top panel). In CHOMOP cell membranes, [Dmt1]N/OFQ(1–13)-NH2 displaced [3H]-DPN with a pKi of 10.48 (CL95% 10.30–10.66). The standard ligands dermorphin and naloxone showed pKi values of 8.90 (CL95% 8.72–9.08) and 8.95 (CL95% 8.68–9.22), respectively (Figure 2, right top panel). In CHODOP cell membranes, [Dmt1]N/OFQ(1–13)-NH2 showed a pKi value of 9.43 (CL95% 9.27–9.59), where the standard δ opioid receptor agonist DPDPE, the selective δ opioid receptor antagonist naltrindole and the non-selective opioid receptor antagonist naloxone displaced [3H]-DPN with pKi values of 7.29 (CL95% 7.09–7.49), 9.74 (CL95% 9.53–9.95) and 7.46 (CL95% 7.33–7.59), respectively (Figure 2, left bottom panel). Finally, [Dmt1]N/OFQ(1–13)-NH2 showed a pKi value of 9.83 (CL95% 9.45–10.21) in CHOKOP cell membranes. The κ opioid receptor agonist dynorphin A displayed a pKi value of 10.71 (CL95% 10.14–11.28), where the κ opioid receptor selective antagonist nor-binaltorphimine and the opioid universal ligand naloxone displaced [3H]-DPN with pKi values of 10.14 (CL95% 9.51–10.79) and 8.44 (CL95% 8.25–8.63), respectively (Figure 2, right bottom panel). Thus, receptor binding experiments not only confirmed that [Dmt1]N/OFQ(1–13)-NH2 binds with high affinity to NOP and μ opioid receptor sites, but also demonstrated that the peptide behaves as a potent κ and δ opioid receptor ligand. On this basis, a separate series of experiments were performed to investigate the pharmacological activity of [Dmt1]N/OFQ(1–13)-NH2 at κ and δ opioid receptors.
Figure 2.
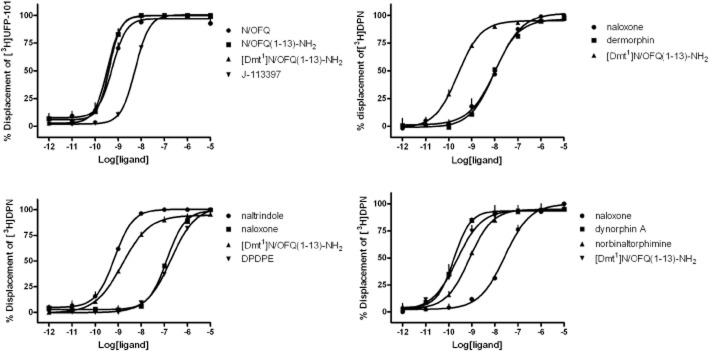
Receptor binding experiments. Competition binding curves to [Dmt1]N/OFQ(1–13)-NH2 and standard ligands in membranes of CHO cells expressing NOP (top left panel), μ (top right panel), δ (bottom left panel) or κ (bottom right panel) opioid receptors. [3H]-UFP-101 was used as radioligand for the NOP and [3H]-DPN for classical opioid receptors. Data are the mean ± SEM of 3 separate experiments performed in duplicate.
In CHOKOP cells stably expressing the Gαqi5 chimeric protein, dynorphin A evoked a concentration-dependent stimulation of calcium release displaying high potency [pEC50 8.92 (CL95% 8.53–9.31) ] and maximal effect (230 ± 15% over the basal values) while N/OFQ was inactive up to 1 μM. [Dmt1]N/OFQ(1–13)-NH2 mimicked the stimulating effects of dynorphin A showing similar maximal effects, but 20-fold lower potency [pEC50 7.61 (CL95% 7.31–7.91) ] (Figure 3, left panel). In CHODOP cells stably expressing the GαqG66Di5 chimeric protein, DPDPE concentration-dependently stimulated calcium mobilization { [pEC50 8.10 (CL95% 7.90–8.30) ]; Emax 121 ± 14% over the basal values} while N/OFQ was inactive up to 1 μM (Figure 3, right panel). [Dmt1]N/OFQ(1–13)-NH2 induced a concentration-dependent stimulation of calcium release showing maximal effects similar to DPDPE, but lower potency (pEC50 7.24 (CL95% 6.60–7.88)) (Figure 3, right panel).
Figure 3.

Calcium mobilization experiments. Concentration–response curves to N/OFQ, [Dmt1]N/OFQ(1–13)-NH2, and standard opioid agonists (dynorphin A and DPDPE for κ and δ opioid receptors, respectively) in CHO cells stably expressing chimeric G proteins and the κ (left panel) and δ (right panel) human recombinant opioid receptor. Data are the mean ± SEM of four separate experiments performed in duplicate.
In CHONOP cell membranes, N/OFQ stimulated [35S]-GTPγS binding in a concentration-dependent manner with a pEC50 value of 8.52 (CL95% 7.92–9.12) and Emax of 4.80 ± 0.37. N/OFQ(1–13)-NH2 and [Dmt1]N/OFQ(1–13)-NH2 mimicked the stimulating effect of the natural peptide showing similar potency and maximal effects. The μ opioid receptor agonist dermorphin produced a weak stimulation only at the highest concentration tested that is 10 μM (Figure 4, left panel). In contrast, in CHOMOP cell membranes, dermorphin, stimulated [35S]-GTPγS binding in a concentration-dependent manner with high potency and maximal effects [pEC50 7.74 (CL95% 7.56–7.91); Emax of 5.47 ± 0.13]. [Dmt1]N/OFQ(1–13)-NH2 mimicked the stimulant effect of the opioid peptide showing similar maximal effects and even higher potency [pEC50 8.19 (CL95% 8.00–8.39) ]. In these cell membranes, N/OFQ was found to be inactive up to μM concentrations (Figure 4, right panel).
Figure 4.
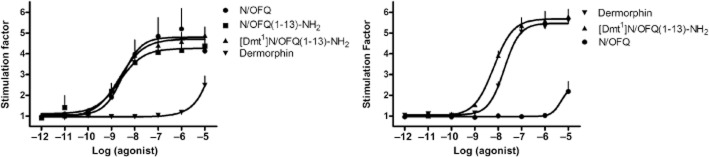
[35S]-GTPγS binding experiments. Concentration–response curves to N/OFQ, N/OFQ(1–13)-NH2, dermorphin, and [Dmt1]N/OFQ(1–13)-NH2 in membranes of CHO cells stably expressing the NOP (left panel) or μ (right panel) human recombinant opioid receptors. Data are the mean ± SEM of five separate experiments performed in duplicate.
In rat cerebral cortex membranes (Figure 5), N/OFQ stimulated [35S]GTPγS binding in a concentration-dependent manner with a pEC50 value of 7.82 (CL95% 7.49–8.15) and Emax of 1.40 ± 0.03. N/OFQ(1–13)-NH2 mimicked the stimulatory effect of N/OFQ with similar maximal effects, but higher potency [pEC50 8.48 (CL95% 8.09–8.87) ]. The MOP agonist dermorphin displayed a relatively low potency in this preparation and this prevented a precise determination of its maximal effects. [Dmt1]N/OFQ(1–13)-NH2 produced a stimulation of [35S]GTPγS binding with a pEC50 value of 8.01 (CL95% 7.969–8.32) and Emax of 1.68 ± 0.04; of note, the maximal effect elicited by [Dmt1]N/OFQ(1–13)-NH2 was significantly higher than those produced by the other ligands (Figure 5).
Figure 5.
[35S]-GTPγS binding experiments. Concentration–response curves to N/OFQ, N/OFQ(1–13)-NH2, dermorphin, and [Dmt1]N/OFQ(1–13)-NH2 in membranes of the rat cerebral cortex. Data are the mean ± SEM of four separate experiments performed in duplicate.
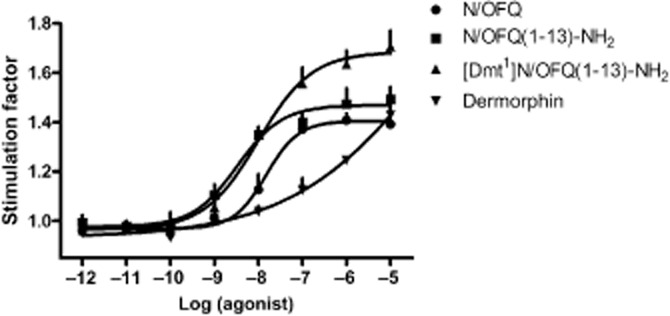
In rat spinal cord membranes N/OFQ and N/OFQ(1–13)-NH2 produced superimposable results (pEC50 ≍ 7.6; Emax ≍ 1.25). Dermorphin displayed a lower potency [pEC50 of 6.41 (CL95% 5.66–7.17) ] but higher maximal effect (1.42 ± 0.10). [Dmt1]N/OFQ(1–13)-NH2 produced a stimulation of [35S]GTPγS binding with similar potency to N/OFQ [pEC50 7.81 (CL95% 7.47–8.16) ] and maximal effects higher than those elicited by dermorphin (Figure 6, top left panel). In this preparation, the stimulatory effects of N/OFQ, dermorphin and [Dmt1]N/OFQ(1–13)-NH2 were challenged with the NOP selective antagonist J-113397 and the universal opioid receptor antagonist naloxone. As shown in Figure 6 top right panel, the effects of N/OFQ were resistant to naloxone while sensitive to J-113397 (pKB 7.95). In contrast, the action of dermorphin was antagonized by naloxone (pKB 8.07), but not by J-113397 (bottom left panel). As shown in Figure 6 bottom right panel, the stimulating effect elicited by [Dmt1]N/OFQ(1–13)-NH2 was sensitive to both naloxone and J-113397. Co-application of the two antagonists did not produce a further shift in the concentration-response curve to [Dmt1]N/OFQ(1–13)-NH2.
Figure 6.

[35S]-GTPγS binding experiments. Concentration–response curves to N/OFQ, N/OFQ(1–13)-NH2, dermorphin, and [Dmt1]N/OFQ(1–13)-NH2 in membranes of the rat spinal cord (top left panel). Effects of naloxone and J-113397 versus N/OFQ (top right panel), dermorphin (bottom left panel), and [Dmt1]N/OFQ(1–13)-NH2 (bottom right panel). Data are the mean ± SEM of five separate experiments performed in duplicate.
In the electrically stimulated guinea pig ileum, N/OFQ inhibited the twitch response in a concentration-dependent manner [pEC50 8.26 (CL95% 8.16–8.36), Emax = 40 ± 2% inhibition of control twitch]. The μ opioid receptor agonist dermorphin mimicked the effect of N/OFQ being, however, more potent (pEC50 8.61 (CL95% 8.50–8.72) and efficacious (Emax = 80 ± 2% inhibition of control twitch). [Dmt1]N/OFQ(1–13)-NH2 inhibited the electrically induced twitch showing similar potency and maximal effects as dermorphin (Figure 7, top left panel). The inhibitory action of N/OFQ was not affected by naloxone, but was antagonized by J-113397 (pKB 7.87) (Figure 7, top right panel). In contrast, the effects of dermorphin were sensitive to naloxone (pKB 8.55) but not J-113397 (Figure 7, bottom left panel). Finally, the effects of [Dmt1]N/OFQ(1–13)-NH2 were challenged with J-113397, naloxone and the cocktail of the two antagonists. As shown in Figure 7 bottom right panel, naloxone antagonized the inhibitory effect of [Dmt1]N/OFQ(1–13)-NH2 producing a rightward shift of the concentration-response curve and no modifications of maximal effects; a pKB value of 7.97 was derived from these experiments. J-113397 1 μM was also able to counteract [Dmt1]N/OFQ(1–13)-NH2 effects by producing a slight displacement to the right of the concentration–response curve associated with a reduction in maximal effect; a pKB value of 5.96 was derived from these experiments. When the two antagonists were assayed together, they displayed an additive effect.
Figure 7.
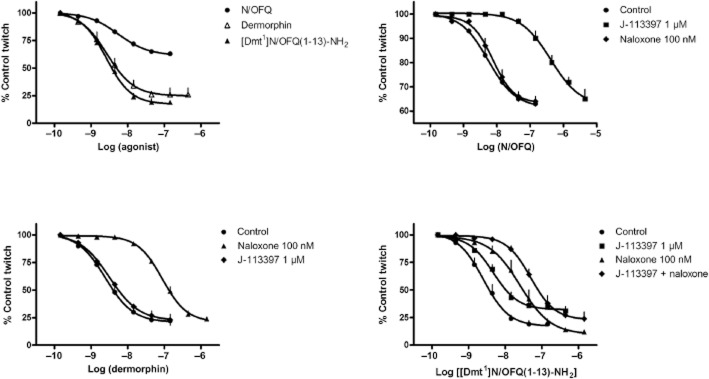
Electrically stimulated guinea-pig ileum. Concentration-response curves to N/OFQ, dermorphin, and [Dmt1]N/OFQ(1–13)-NH2 (top left panel). Effects of naloxone and J-113397 vs N/OFQ (top right panel), dermorphin (bottom left panel), and [Dmt1]N/OFQ(1–13)-NH2 (bottom right panel). Data are the mean ± SEM of four separate experiments performed in duplicate.
Finally, the in vivo effects of [Dmt1]N/OFQ(1–13)-NH2 on pain transmission were evaluated in non-human primates after spinal administration (Figure 8). In the dose range 1 and 10 nmol, the peptide produced dose-dependent antinociceptive effects. Of note at 10 nmol [Dmt1]N/OFQ(1–13)-NH2 elicited maximal antinociceptive effects (top left panel) without causing itch/scratching responses (top right panel). By contrast, supramaximal doses of peptide, 30 and 100 nmol, still produced full antinociceptive effects (bottom left panel) that were, however, associated with robust scratching responses (bottom right panel).
Figure 8.

Monkey tail withdrawal assay. Effect of low (top panels) and high (bottom panels) doses of [Dmt1]N/OFQ(1–13)-NH2 on nociception (left panels) and scratching behaviour (right panels). Data are the mean ± SEM of data (four separate experiments for left panels and six experiments for right panels). The asterisk represents a significant difference from the vehicle condition from the time point 30 min to the corresponding time point for each dose (*P < 0.05).
Discussion
Based on the recent evidence of a synergistic antinociceptive effect in response to the simultaneous activation of spinal NOP and opioid receptors (Hu et al., 2009; Ko and Naughton, 2009), the present study was carried out with the aim of identifying, pharmacologically characterize and evaluate mixed NOP/opioid agonists as innovative spinal analgesics. [Dmt1]N/OFQ(1–13)-NH2 was identified in the calcium mobilization primary screening assay as the most potent and least selective agonist. The pharmacological activity of the peptide was then confirmed in various in vitro assays performed on recombinant human receptors (receptor binding, [35S]-GTPγS binding) as well as at native animal receptors expressed in the rat cerebral cortex and spinal cord and in the guinea pig ileum. In vivo in the tail withdrawal assay performed in monkeys [Dmt1]N/OFQ(1–13)-NH2 produced dose-dependent antinociceptive effects. Of note, while [Dmt1]N/OFQ(1–13)-NH2 displayed similar potency to N/OFQ in vitro, the peptide was approximately 30-fold more potent in vivo and elicited longer-lasting effects. These results corroborate the hypothesis that non-selective NOP/opioid agonists may behave as innovative spinal analgesics and [Dmt1]N/OFQ(1–13)-NH2 is a prototype for this class of drugs.
The calcium mobilization assay used for screening the NOP/opioid receptor ligands has been validated in previous studies. In particular, the pharmacological profile of the human NOP receptor coupled with calcium signalling has been assessed with a rather large panel of ligands encompassing full and partial agonist as well as pure antagonist activity (Camarda et al., 2013). Similar experiments were performed to investigate the pharmacological profile of human classical opioid receptors although, in this case, the panel of ligands investigated was relatively small (Camarda and Calo, 2013). The results obtained with the calcium assay were virtually identical to those described in the literature using classical assays for Gi-coupled receptors (i.e. cAMP levels or stimulation of [35S]-GTPγS binding in cells expressing recombinant receptors, inhibition of electrically induced contractions in isolated tissues). For instance, the correlation analysis of results obtained with NOP ligands in the calcium mobilization and in the mouse vas deferens assays yielded a correlation coefficient r2 of 0.90 (Camarda et al., 2013).
The design of non-selective NOP/opioid agonists was based on the following evidence: (i) N/OFQ(1–13)-NH2 maintains the same potency and efficacy as the natural peptide (Calo et al., 1996; Guerrini et al., 1997); (ii) the substitution of Phe1 with Tyr in N/OFQ as well as N/OFQ(1–13)-NH2 sequences causes a reduction in selectivity for NOP over classical opioid receptors (Calo et al., 1997; Varani et al., 1999); and (iii) the substitution of Tyr1 with Dmt in opioid peptide sequences increases ligand potency (Salvadori et al., 1995; Schiller, 2010). The results obtained in the calcium assay demonstrated that this design strategy was indeed successful. In fact, the substitution of Phe with Tyr in position 1 generated less selective peptides. However, both [Tyr1]N/OFQ-NH2 and [Tyr1]N/OFQ(1–13)-NH2 were more than 1000-fold more potent at NOP than at μ opioid receptors. These results are in line with previous findings. In fact, [Tyr1]N/OFQ-NH2 and [Tyr1]N/OFQ(1–13)-NH2 were able to bind to both NOP and μ opioid receptor sites in guinea pig brain membranes but with higher affinity at the former receptor (Varani et al., 1999). Moreover, in the electrically stimulated guinea pig ileum [Tyr1]N/OFQ(1–13)-NH2, at low concentrations (<30 nM), produced naloxone-resistant inhibitory effects; however, at higher concentrations, the opioid antagonist partially counteracted the action of the peptide (Varani et al., 1999). Finally, when tested in vivo, [Tyr1]N/OFQ mimicked the effect of the natural peptide decreasing systemic arterial pressure in the rat (Champion and Kadowitz, 1997) and eliciting erectile activity in the cat (Champion et al., 1998). The non-natural amino acid Dmt has been widely and successfully used in the past for generating highly potent ligands for opioid receptors (Bryant et al., 2003; Schiller, 2010) and also, opioid/neurotensin hybrid peptides (Kleczkowska et al., 2010). The ability of this residue to increase opioid receptor affinity compared with Tyr has been confirmed in the present study. In fact [Dmt1]N/OFQ-NH2 and [Dmt1]N/OFQ(1–13)-NH2 displayed a slight decrease in NOP potency associated with a substantial increase in potency at μ opioid receptors. As a consequence, the selectivity of these peptides for NOP over μ opioid receptors dropped to only ≍30-fold. Because [Dmt1]N/OFQ(1–13)-NH2 displayed slightly higher potency and lower selectivity compared with [Dmt1]N/OFQ-NH2, it was selected as candidate for further studies.
Very high NOP/ μ opioid receptor affinity of [Dmt1]N/OFQ(1–13)-NH2 was demonstrated in receptor binding experiments performed using membranes prepared from CHO cells expressing NOP or classical opioid receptors. These experiments also demonstrated that the peptide is capable of binding to δ and κ opioid receptors, although with lower affinity. Functional calcium mobilization studies indicated that [Dmt1]N/OFQ(1–13)-NH2 behaves as a full agonist also at δ and κ opioid receptors. [Dmt1]N/OFQ(1–13)-NH2 affinity in receptor binding experiments matched its potency in calcium mobilization studies and the same rank order that is NOP > μ ≥ κ > δ has been determined for [Dmt1]N/OFQ(1–13)-NH2 in the two sets of data. In [35S]-GTPγS assay, the peptide behaved as potent full agonist both at NOP and at μ opioid receptors. In these experiments, [Dmt1]N/OFQ(1–13)-NH2 displayed, in line with calcium mobilization data, higher potency at NOP than at μ opioid receptors, but its ratio of selectivity (2) was substantially lower than that derived from calcium mobilization studies. Collectively, these results clearly demonstrated that [Dmt1]N/OFQ(1–13)-NH2 behaved as a universal NOP/opioid full agonist at recombinant human receptors.
The pharmacological activity of the peptide was then reassessed at native animal receptors by performing [35S]-GTPγS binding experiments with membranes from the rat cerebral cortex and spinal cord and bioassay experiments in the guinea pig ileum. In line with previous findings, N/OFQ and dermorphin stimulated [35S]-GTPγS binding in the rat cerebral cortex and spinal cord membranes (Sim et al., 1995; Albrecht et al., 1998; Narita et al., 1999a,b). In both preparations, [Dmt1]N/OFQ(1–13)-NH2 behaved as a potent agonist producing maximal effects higher than those elicited by the selective agonists. The receptor mechanism involved in the stimulant effects of [Dmt1]N/OFQ(1–13)-NH2 in rat spinal cord membranes has been investigated in receptor antagonist experiments. While the stimulating effects of N/OFQ and dermorphin were sensitive to J-113397 and naloxone, respectively, that elicited by [Dmt1]N/OFQ(1–13)-NH2 was counteracted by both molecules. This result suggests that stimulation of [35S]-GTPγS binding by N/OFQ and dermorphin derives from the selective activation of NOP and opioid receptors, respectively, while that elicited by [Dmt1]N/OFQ(1–13)-NH2 is due to the simultaneous activation of both types of receptors. This view is corroborated by findings obtained in the electrically stimulated guinea pig ileum. In this preparation, J-13397 and naloxone selectively antagonized the inhibitory effects of N/OFQ and dermorphin, respectively, with pKB values similar to those obtained in the rat spinal cord membranes and the literature (Calo et al., 1997; Bigoni et al., 2000). In contrast, the inhibitory action of [Dmt1]N/OFQ(1–13)-NH2 was sensitive to both antagonists and a profound shift to the right of the concentration–response curve to the agonist was obtained when J-113397 and naloxone were co-applied. Collectively, these findings clearly demonstrate that [Dmt1]N/OFQ(1–13)-NH2 acts as a potent and mixed NOP/opioid full agonist at native animal receptors expressed in the periphery and in the CNS.
In the spinal cord, NOP and opioid (particularly the μ opioid receptor) receptor stimulation elicits antinociceptive effects via similar cellular mechanism, that is presynaptic inhibition of neurotransmitter release from primary sensory neurons (Zeilhofer and Calo, 2003). Moreover, recent studies performed in non-human primates suggest that the simultaneous activation of NOP and opioid receptors produces synergistic antinociceptive effects (Hu et al., 2009; Ko and Naughton, 2009). This evidence prompted us to assess the spinal antinociceptive properties of [Dmt1]N/OFQ(1–13)-NH2 in non-human primates. It should be emphasized that in this species, antinociceptive effects in response to spinal administration of NOP agonists are behaviourally selective while those elicited by μ opioid receptor agonists are always associated with scratching (Ko et al., 2004; 2006). In monkeys, [Dmt1]N/OFQ(1–13)-NH2 induced significant antinociceptive effects at the dose of 3 nmol and full antinociception at 10 nmol. Interestingly, over this range of doses, the antinociceptive effect of [Dmt1]N/OFQ(1–13)-NH2 was not associated with scratching. Compared with N/OFQ (Ko et al., 2006; Ko and Naughton, 2009), [Dmt1]N/OFQ(1–13)-NH2 was found to be about ∼10–30-fold more potent and elicited longer-lasting effects. These results contrast to the similar NOP potency displayed by [Dmt1]N/OFQ(1–13)-NH2 and N/OFQ or N/OFQ(1–13)-NH2 in vitro. It has been demonstrated that [desPhe1]N/OFQ is a major metabolite of N/OFQ when the peptide is given i.t. (Ko et al., 2006). Therefore, the presence of the non-natural amino acid Dmt in position 1 may reduce susceptibility to enzymatic degradation. This may cause an increase in peptide potency and duration of action in vivo, where metabolism is likely to be more relevant than in vitro. However, it is unlikely that the huge increase in [Dmt1]N/OFQ(1–13)-NH2 potency is solely due to increased metabolic stability. On the other hand, it is known that μ opioid peptide agonists are very potent analgesics when delivered spinally (see Malmberg and Yaksh, 1992). However, the analgesic effects elicited by selective μ opioid receptor activation are always associated with scratching while those elicited with submaximal doses of [Dmt1]N/OFQ(1–13)-NH2 are not associated with this unwanted effect. It is therefore suggested that the high potency of the synthetic peptide mainly derives from its ability to simultaneously activate spinal NOP and opioid receptors. This simultaneous receptor activation produced behaviourally selective synergistic antinociceptive effects as demonstrated in previous monkey studies performed with subthreshold doses of morphine and N/OFQ (Ko and Naughton, 2009) or the potent and selective NOP agonist UFP-112 (Hu et al., 2009). [Dmt1]N/OFQ(1–13)-NH2 induced scratching behaviour similar to morphine only at higher supramaximal doses (i.e. 30 and 100 nmol) (Ko et al., 2006; Ko and Naughton, 2009). These results further suggest that the high antinociceptive potency of [Dmt1]N/OFQ(1–13)-NH2 may derive from its mixed NOP/opioid agonist activity. However this is just a proposal for interpreting our findings, in fact, receptor antagonist and knockout studies are required to firmly attribute the in vivo actions of [Dmt1]N/OFQ(1–13)-NH2 to NOP and/or opioid receptor activation.
I.t infusions of analgesics have been increasingly utilized during the last two decades for the treatment of persistent cancer pain. With recent technological advances in the field, this therapeutic option has been extended to moderate or severe pain related to cancer and non-cancer origins (Smith et al., 2008). However, only morphine and ziconotide (Kress et al., 2009) have been approved for i.t. administration; thus, there is a strong medical need for novel drugs to be used as spinal analgesics. In this regard non-selective NOP/opioid agonists such as [Dmt1]N/OFQ(1–13)-NH2 may represent an interesting option. Indeed, the synergistic antinociceptive effect generated by the simultaneous activation of NOP and MOP receptors may offer important advantages: (i) during acute administration a complete analgesic effect can be achieved with fewer or even elimination of the side effects associated with the full activation of a single receptor; and (ii) during chronic treatment the desired level of analgesia can maintained for longer (i.e. reduction in tolerance liability) because the analgesic action does not derive from the full and prolonged activation of a single receptor. However, these are intriguing speculations that need rigorous experimental validation.
In conclusion, the present study describes the design, synthesis and in vitro pharmacological characterization of [Dmt1]N/OFQ(1–13)-NH2, a potent mixed NOP/opioid agonist. The spinal administration of this peptide in non-human primates elicits potent antinociceptive effects similar to those produced by combinations of NOP and opioid receptor agonists. These results suggest that [Dmt1]N/OFQ(1–13)-NH2 could be considered the prototype of a novel class of spinal analgesics worthy of consideration for clinical development.
Acknowledgments
We would like to thank Colette Cremeans, Erin Gruley and Yong-Gong Shi for technical assistance, and E. Kostenis for the generous gift of chimeric G-protein plasmids. This study was supported by funds from the University of Ferrara (FAR grants to GC and SS), The Italian Ministry of University (FIRB grant to GC and RG) and the United States National Institutes of Health, NIAMS and NIDA (R01-AR-059193 grant to MCK).
Glossary
- N/OFQ
nociceptin/orphanin FQ
- NOP receptor
N/OFQ peptide receptor
Conflicts of interest
The authors declare that there are no conflicts of interest.
References
- Albrecht E, Samovilova NN, Oswald S, Baeger I, Berger H. Nociceptin (orphanin FQ): high-affinity and high-capacity binding site coupled to low-potency stimulation of guanylyl-5′-O-(gamma-thio)-triphosphate binding in rat brain membranes. J Pharmacol Exp Ther. 1998;286:896–902. [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigoni R, Giuliani S, Calo G, Rizzi A, Guerrini R, Salvadori S, et al. Characterization of nociceptin receptors in the periphery: in vitro and in vivo studies. Naunyn Schmiedebergs Arch Pharmacol. 1999;359:160–167. doi: 10.1007/pl00005338. [DOI] [PubMed] [Google Scholar]
- Bigoni R, Calo G, Rizzi A, Guerrini R, De Risi C, Hashimoto Y, et al. In vitro characterization of J-113397, a non-peptide nociceptin/orphanin FQ receptor antagonist. Naunyn Schmiedebergs Arch Pharmacol. 2000;361:565–568. doi: 10.1007/s002100000220. [DOI] [PubMed] [Google Scholar]
- Bryant SD, Jinsmaa Y, Salvadori S, Okada Y, Lazarus LH. Dmt and opioid peptides: a potent alliance. Biopolymers. 2003;71:86–102. doi: 10.1002/bip.10399. [DOI] [PubMed] [Google Scholar]
- Calo G, Rizzi A, Bogoni G, Neugebauer V, Salvadori S, Guerrini R, et al. The mouse vas deferens: a pharmacological preparation sensitive to nociceptin. Eur J Pharmacol. 1996;311:R3–R5. doi: 10.1016/0014-2999(96)00563-8. [DOI] [PubMed] [Google Scholar]
- Calo G, Rizzi A, Bodin M, Neugebauer W, Salvadori S, Guerrini R, et al. Pharmacological characterization of nociceptin receptor: an in vitro study. Can J Physiol Pharmacol. 1997;75:713–718. [PubMed] [Google Scholar]
- Camarda V, Calo G. Chimeric G-proteins in fluorimetric calcium assays: experience with opioid receptors. Methods Mol Biol. 2013;937:293–306. doi: 10.1007/978-1-62703-086-1_18. [DOI] [PubMed] [Google Scholar]
- Camarda V, Fischetti C, Anzellotti N, Molinari P, Ambrosio C, Kostenis E, et al. Pharmacological profile of NOP receptors coupled with calcium signaling via the chimeric protein Gαqi5. Naunyn Schmiedebergs Arch Pharmacol. 2009;379:599–607. doi: 10.1007/s00210-009-0396-x. [DOI] [PubMed] [Google Scholar]
- Champion HC, Kadowitz PJ. [Tyr1]-nociceptin, a novel nociceptin analog, decreases systemic arterial pressure by a naloxone-insensitive mechanism in the rat. Biochem Biophys Res Commun. 1997;234:309–312. doi: 10.1006/bbrc.1997.6629. [DOI] [PubMed] [Google Scholar]
- Champion HC, Bivalacqua TJ, Wang R, Hellstrom WJ, Kadowitz PJ. [Tyr1]-nociceptin and nociceptin have similar naloxone-insensitive erectile activity in the cat. J Androl. 1998;19:747–753. [PubMed] [Google Scholar]
- Fischetti C, Camarda V, Rizzi A, Pela′ M, Trapella C, Guerrini R, et al. Pharmacological characterization of the nociceptin/orphanin FQ non peptide antagonist Compound 24. Eur J Pharmacol. 2009;614:50–57. doi: 10.1016/j.ejphar.2009.04.054. [DOI] [PubMed] [Google Scholar]
- Guerrini R, Calo G, Rizzi A, Bianchi C, Lazarus LH, Salvadori S, et al. Address and message sequences for the nociceptin receptor: a structure-activity study of nociceptin-(1-13)-peptide amide. J Med Chem. 1997;40:1789–1793. doi: 10.1021/jm970011b. [DOI] [PubMed] [Google Scholar]
- Hu E, Calo G, Guerrini R, Ko M. Long lasting antinociceptive spinal effects in primates of the novel nociceptin/orphanin FQ receptor agonist UFP-112. Pain. 2009;148:107–113. doi: 10.1016/j.pain.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khroyan TV, Zaveri NT, Polgar WE, Orduna J, Olsen C, Jiang F, et al. SR 16435 [1-(1-(bicyclo[3.3.1]nonan-9-yl)piperidin-4-yl)indolin-2-one], a novel mixed nociceptin/orphanin FQ/mu-opioid receptor partial agonist: analgesic and rewarding properties in mice. J Pharmacol Exp Ther. 2007;320:934–943. doi: 10.1124/jpet.106.111997. [DOI] [PubMed] [Google Scholar]
- Khroyan TV, Polgar WE, Cami-Kobeci G, Husbands SM, Zaveri NT, Toll L. The first universal opioid ligand, (2S)-2-[(5R,6R,7R,14S)-N-cyclopropylmethyl-4,5-epoxy-6,14-ethano-3-hydroxy -6-methoxymorphinan-7-yl]-3,3-dimethylpentan-2-ol (BU08028): characterization of the in vitro profile and in vivo behavioral effects in mouse models of acute pain and cocaine-induced reward. J Pharmacol Exp Ther. 2011;336:952–961. doi: 10.1124/jpet.110.175620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleczkowska P, Kosson P, Ballet S, Van den Eynde I, Tsuda Y, Tourwe D, et al. PK20, a new opioid-neurotensin hybrid peptide that exhibits central and peripheral antinociceptive effects. Mol Pain. 2010;6:86. doi: 10.1186/1744-8069-6-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko MC, Naughton NN. Antinociceptive effects of nociceptin/orphanin FQ administered intrathecally in monkeys. J Pain. 2009;10:509–516. doi: 10.1016/j.jpain.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko MC, Song MS, Edwards T, Lee H, Naughton NN. The role of central mu opioid receptors in opioid-induced itch in primates. J Pharmacol Exp Ther. 2004;310:169–176. doi: 10.1124/jpet.103.061101. [DOI] [PubMed] [Google Scholar]
- Ko MC, Wei H, Woods JH, Kennedy RT. Effects of intrathecally administered nociceptin/orphanin FQ in monkeys: behavioral and Mass Spectrometric Studies. J Pharmacol Exp Ther. 2006;318:1257–1264. doi: 10.1124/jpet.106.106120. [DOI] [PubMed] [Google Scholar]
- Ko MC, Woods JH, Fantegrossi WE, Galuska CM, Wichmann J, Prinssen EP. Behavioral effects of a synthetic agonist selective for nociceptin/orphanin FQ peptide receptors in monkeys. Neuropsychopharmacology. 2009;34:2088–2096. doi: 10.1038/npp.2009.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress HG, Simpson KH, Marchettini P, Ver Donck A, Varrassi G. Intrathecal therapy: what has changed with the introduction of ziconotide. Pain Pract. 2009;9:338–347. doi: 10.1111/j.1533-2500.2009.00308.x. [DOI] [PubMed] [Google Scholar]
- Lambert DG. The nociceptin/orphanin FQ receptor: a target with broad therapeutic potential. Nat Rev Drug Discov. 2008;7:694–710. doi: 10.1038/nrd2572. [DOI] [PubMed] [Google Scholar]
- McDonald J, Barnes TA, Okawa H, Williams J, Calo G, Rowbotham DJ, et al. Partial agonist behaviour depends upon the level of nociceptin/orphanin FQ receptor expression: studies using the ecdysone-inducible mammalian expression system. Br J Pharmacol. 2003;140:61–70. doi: 10.1038/sj.bjp.0705401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmberg AB, Yaksh TL. Isobolographic and dose-response analyses of the interaction between intrathecal mu and delta agonists: effects of naltrindole and its benzofuran analog (NTB) J Pharmacol Exp Ther. 1992;263:264–275. [PubMed] [Google Scholar]
- Meunier JC, Mollereau C, Toll L, Suaudeau C, Moisand C, Alvinerie P, et al. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature. 1995;377:532–535. doi: 10.1038/377532a0. [DOI] [PubMed] [Google Scholar]
- Mustazza C, Bastanzio G. Development of nociceptin receptor (NOP) agonists and antagonists. Med Res Rev. 2011;31:605–648. doi: 10.1002/med.20197. [DOI] [PubMed] [Google Scholar]
- Narita M, Mizoguchi H, Oji DE, Dun NJ, Hwang BH, Nagase H, et al. Identification of the G-protein-coupled ORL1 receptor in the mouse spinal cord by [35S]-GTPgammaS binding and immunohistochemistry. Br J Pharmacol. 1999a;128:1300–1306. doi: 10.1038/sj.bjp.0702907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Mizoguchi H, Sora I, Uhl GR, Tseng LF. Absence of G-protein activation by mu-opioid receptor agonists in the spinal cord of mu-opioid receptor knockout mice. Br J Pharmacol. 1999b;126:451–456. doi: 10.1038/sj.bjp.0702330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazzaro C, Rizzi A, Salvadori S, Guerrini R, Regoli D, Zeilhofer HU, et al. UFP-101 antagonizes the spinal antinociceptive effects of nociceptin/orphanin FQ: behavioral and electrophysiological studies in mice. Peptides. 2007;28:663–669. doi: 10.1016/j.peptides.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Reinscheid RK, Nothacker HP, Bourson A, Ardati A, Henningsen RA, Bunzow JR, et al. Orphanin FQ: a neuropeptide that activates an opioidlike G protein-coupled receptor. Science. 1995;270:792–794. doi: 10.1126/science.270.5237.792. [DOI] [PubMed] [Google Scholar]
- Rizzi A, Gavioli EC, Marzola G, Spagnolo B, Zucchini S, Ciccocioppo R, et al. Pharmacological Characterization of the Nociceptin/Orphanin FQ Receptor Antagonist SB-612111 [(-)-cis-1-Methyl-7-[[4-(2,6-dichlorophenyl)piperidin-1-yl]methyl]-6,7,8,9-tetrahydro-5H benzocyclohepten-5-ol]: In vivo Studies. J Pharmacol Exp Ther. 2007a;321:968–974. doi: 10.1124/jpet.106.116780. [DOI] [PubMed] [Google Scholar]
- Rizzi A, Spagnolo B, Wainford RD, Fischetti C, Guerrini R, Marzola G, et al. In vitro and in vivo studies on UFP-112, a novel potent and long lasting agonist selective for the nociceptin/orphanin FQ receptor. Peptides. 2007b;28:1240–1251. doi: 10.1016/j.peptides.2007.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvadori S, Attila M, Balboni G, Bianchi C, Bryant SD, Crescenzi O, et al. Delta opioidmimetic antagonists: prototypes for designing a new generation of ultraselective opioid peptides. Mol Med. 1995;1:678–689. [PMC free article] [PubMed] [Google Scholar]
- Schiller PW. Bi- or multifunctional opioid peptide drugs. Life Sci. 2010;86:598–603. doi: 10.1016/j.lfs.2009.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim LJ, Selley DE, Childers SR. In vitro autoradiography of receptor-activated G proteins in rat brain by agonist-stimulated guanylyl 5′-[gamma-[35S]thio]-triphosphate binding. Proc Natl Acad Sci USA. 1995;92:7242–7246. doi: 10.1073/pnas.92.16.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HS, Deer TR, Staats PS, Singh V, Sehgal N, Cordner H. Intrathecal drug delivery. Pain Physician. 2008;11:S89–S104. [PubMed] [Google Scholar]
- Spagnolo B, Calo G, Polgar WE, Jiang F, Olsen CM, Berzetei-Gurske I, et al. Activities of mixed NOP and mu-opioid receptor ligands. Br J Pharmacol. 2008;153:609–619. doi: 10.1038/sj.bjp.0707598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toll L, Khroyan TV, Polgar WE, Jiang F, Olsen C, Zaveri NT. Comparison of the antinociceptive and antirewarding profiles of novel bifunctional nociceptin receptor/mu-opioid receptor ligands: implications for therapeutic applications. J Pharmacol Exp Ther. 2009;331:954–964. doi: 10.1124/jpet.109.157446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varani K, Rizzi A, Calo G, Bigoni R, Toth G, Guerrini R, et al. Pharmacology of [Tyr1]nociceptin analogs: receptor binding and bioassay studies. Naunyn Schmiedebergs Arch Pharmacol. 1999;360:270–277. doi: 10.1007/s002109900074. [DOI] [PubMed] [Google Scholar]
- Zeilhofer HU, Calo G. Nociceptin/orphanin FQ and its receptor–potential targets for pain therapy? J Pharmacol Exp Ther. 2003;306:423–429. doi: 10.1124/jpet.102.046979. [DOI] [PubMed] [Google Scholar]