

Abstract
BACKGROUND AND PURPOSE
The class III PI3K inhibitor, 3-methyladenine (3-MA), is commonly used to selectively block autophagy. Recent findings suggest a strong relationship between autophagy and lipid turnover. Here, we explore the effect of 3-MA on adipocyte lipolysis.
EXPERIMENTAL APPROACH
Assays were performed in 3T3-L1 cells. Cells were treated with 3-MA and wortmannin, a pan PI3K and autophagy inhibitor. Pharmacological and genetic manipulation of endogenous autophagic and lipolytic pathways was used to ascertain the contribution of 3-MA to the observed effects on lipolysis.
KEY RESULTS
3T3-L1 cells that were exposed to 3-MA showed a consistent increase in lipolysis, approximately 50% over basal levels. The effect of 3-MA was not secondary to autophagic inhibition as treatment of 3T3-L1 cells with wortmannin yielded no such changes. Dosing and time course experiments showed that 3-MA's ability to activate lipolysis was more sensitive than its inhibitory effect on autophagy. Knockdown of adipose triglyceride lipase (ATGL) negated the stimulatory effect of 3-MA by >90%, indicating that 3-MA enhanced ATGL-dependent hydrolysis of triacylglycerols. Additionally, the lipolytic effect of 3-MA was dependent on the activation of PKA and 3-MA induced a rapid and potent elevation of intracellular cAMP levels in adipocytes.
CONCLUSIONS AND IMPLICATIONS
Cumulatively, we show that 3-MA potently modulated a cellular mechanism and its underlying signalling pathways not associated with autophagy. Furthermore, we describe a novel stimulatory effect on a major signalling pathway. Our findings provide valuable information to studies employing 3-MA as a specific inhibitor for PI3K and autophagy.
Keywords: 3-methyladenine, PI3-kinase, autophagy, lipolysis, PKA, cAMP, triglyceride hydrolysis, adipocyte, adipose tissue, 3T3-L1
Introduction
Macro-autophagy (autophagy hereafter) is a self-digestive mechanism essential for regulation and maintenance of cellular homeostasis (Ito et al., 2007). The relationship between autophagy and lipid metabolism is a topic that has garnered increasing interest and relevance in recent years as obesity rates have steadfastly increased. Autophagy and triacylglycerol (TAG) hydrolysis are catabolic cellular processes that are activated during periods of nutrient deprivation (Dong and Czaja, 2011; Kovsan et al., 2011). These mechanisms share markedly similar responses both biochemically and functionally, in response to a limited nutrient supply (Gibbons et al., 2000; Finn and Dice, 2006).
Dysfunction within the autophagic machinery itself, or within the associated signalling pathways, can lead to a variety of pathological conditions (Ravikumar et al., 2010). Autophagy serves as a catabolic mechanism to recycle damaged cellular components to be used as substrates for energy production. In autophagy, sequestered components such as damaged organelles, proteins and lipids become trapped in double-membrane-bound compartments called autophagosomes (Kundu and Thompson, 2008). Once trapped within autophagosomes, the contents are subsequently degraded following fusion with lysosomes.
For autophagy induction to proceed properly, the class III PI3K and beclin 1 (a tumour suppressor protein) are required for a proximal step during autophagy activation. Beclin 1 and class III PI3K form a highly conserved core complex that is instrumental for the localization of essential autophagic proteins (such as Atg5 and Atg7) to the pre-autophagosome (Kang et al., 2011). Furthermore, the core complex, in particular the class III PI3K, Vps34, is responsible for the production of the pro-autophagic class III PI3K product, phosphatidylinositol 3-phosphate (PI-3-P; Ravikumar et al., 2010). PI-3-P is believed to act as a substrate or scaffold for autophagosome assembly and aids in recruitment of additional key autophagy mediators, and is therefore required for autophagy.
As PI3K is essential for the activation of functional autophagy, it has become a well-characterized target for autophagy inhibition. The pan PI3K inhibitor wortmannin and the class III PI3K inhibitor 3-methyladenine (3-MA) are widely used to block autophagic activation (Huang and Sinicrope, 2010; Petiot et al., 2000; Seglen and Gordon, 1982). Inhibition of PI3K at the core complex inhibits autophagosome formation and the autophagic machinery ultimately fails to engage properly (Seglen and Gordon, 1982). In addition to pharmacological inhibition of autophagy, ablation of either Atg 5 or Atg7, two key autophagy proteins required for autophagosome recruitment and assembly (Codogno and Meijer, 2006), is enough to inhibit autophagy. Both pharmacological and genetic inhibitions of autophagy are applicable under both nutrient-deprived and basal conditions.
Fatty acids are essential substrates for energy production and membrane lipid biogenesis. They are stored as triacylglycerols (TAGs) in lipid droplets of adipocytes and non-adipocyte cells. During instances of increased metabolic demand such as extended fasting and exercise, free fatty acids (FFAs) and glycerol are released by the process of lipolysis that is catalysed by hydrolytic enzymes called lipases. Adipose triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL) are the two prominent cytosolic lipases involved in mediating adipose lipolysis with ATGL mediating the rate-limiting first step in the hydrolysis of TAGs via lipolysis (Ahmadian et al., 2010; Greenberg et al., 2011; Lafontan and Langin, 2009; Lass et al., 2010). Recent evidence, such as that shown by Singh et al. (Singh et al., 2009a; Singh et al., 2009b), clearly shows the autophagic mechanism also mediates fasting-induced lipolysis in both mouse liver and cultured hepatocytes. Specifically, lipid droplets are delivered to lysosomes by autophagosomes, where lysosomal acid lipase acts to hydrolyse TAGs and cholesteryl esters.
In this present study, we initially attempted to uncover the relationship between lipolysis catalysed by cytosolic lipases and lipid turnover mediated by autophagy under basal nutrient conditions. By using differentiated 3T3-L1 adipocytes as a model for lipolysis as previously described (Green et al., 2004; Yang et al., 2008; 2010), we studied glycerol output from cells where basal autophagy was suppressed by PI3K inhibitors and genetic manipulation of autophagy-related genes. Our data presented in this study demonstrate a novel stimulatory effect of 3-MA on lipolysis that is dependent on PKA activation but independent of autophagic inhibition.
Methods
Cell culture
3T3-L1 fibroblasts were maintained in high glucose Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Grand Island, NY, USA) containing 10% newborn calf serum (Lonza, Basal, Switzerland), 1% penicillin/streptomycin (Invitrogen) and 1% glutamine (Invitrogen). For differentiation into mature adipocytes, 3T3-L1 fibroblasts were switched to media as described earlier, containing 10% fetal bovine serum (Sigma, St. Louis, MO, USA) in lieu of newborn calf serum. In addition, this media also contained 1 µg·mL−1 insulin, 0.25 µM dexamethasone and 0.5 mM IBMX. After 3 days of treatment, dexamethasone and IBMX were removed from the culture media. After 2 days of insulin only treatment, cells were then maintained in DMEM media containing 10% fetal bovine serum, 1% pen/strep and 1% glutamine. For drug treatment, all compounds were dissolved in phenol red-free DMEM (Invitrogen) containing 0.2% fatty acid-free BSA (Sigma). All cells were kept at 37°C in a 10% CO2 atmosphere.
Cell lysis and immunoblotting
All standard immunoblotting procedures were followed. After specified treatments, cells were washed twice with ice-cold PBS (pH 7.5) and then lysed in buffer containing 50 mM Tris, 1% Triton-X 100, 131 mM sodium chloride, 1 mM sodium vanadate, 1 mM sodium diphosphate, 1 mM sodium fluoride, 1 mM EDTA and protease inhibitors (1 mini tablet per 7 mL of volume). Following centrifugation at 10 000×g for 10 min, the supernatant was collected and normalized. Quantification of protein concentration was accomplished via the DC protein assay kit (Bio-Rad, Hercules, CA, USA). 2× Laemmli sample buffer was added to the lysates in a 1:1 ratio. Equal amounts of samples were then resolved on SDS-PAGE and transferred onto nitrocellulose membranes (Bio-Rad). Following transfer, membranes were blocked in 5% non-fat milk and then probed with 1° and 2° antibodies before chemiluminescent detection using Amersham ECL substrate (GE Healthcare, Waukesha, WI, USA) and a Fuji LAS-4000 Imaging System.
Small interfering ribonucleic acid gene knockdown
Gene knockdown in 3T3-L1 adipocytes was achieved by an electroporation method as described previously (Inoue et al., 2006). Stealth small interfering ribonucleic acid (siRNA) oligonucleotides were purchased from Invitrogen. For ATGL knockdown, two siRNAs directed against murine isoform were combined for maximum knockdown efficiency. The sequences for the first set of oligonucleotides are as follows: sense 5′-UCA GAC GGA GAG AAC GUC AUC AUA U-3′ and anti-sense 3′-AUA UGA UGA CGU UCU CUC CGU CUG A-5′. The second set of oligonucleotides comprised sense 5′-CCA GGC CAA UGU CUG CAG CAC AUU U-3′ and anti-sense 3′-AAA UGU GCU GCA GAC AUU GGC CUG G-5′. Atg 5 knockdown was achieved using one siRNA directed against the murine isoform. This set of oligonucleotides comprised sense 5′-AUU CCA UGA GUU UCC GGU UGA UGG C-3′ and anti-sense 3′-GCC AUC AAC CGG AAA CCU ACG C-5′. Stealth RNAi negative control MED GC (Invitrogen #10620312) was used for all control knockdowns.
Lipolysis measurement
Lipolysis was measured as glycerol released per amount of protein present in the samples (µM·mg−1). Media was collected from the cells at various time points throughout the experiments. The amount of glycerol released was determined using the AdipoSIGHT Lipolysis Assay Kit (ZenBio, Research Triangle Park, NC, USA) according to the manufacturer's instructions. Following glycerol release measurements, the data were then normalized to the protein concentration of each sample, via methods described earlier.
Measurement of cAMP
cAMP levels were quantified using the Direct cAMP Assay Kit (Sigma) according to the manufacturer's instructions. cAMP was measured as (pmol·mL−1) following normalization of samples by protein concentration. Compounds used for treatment were dissolved in phenol red-free DMEM containing 0.2% fatty acid-free BSA as described previously.
Data analysis
Data are shown as mean ± SD. A minimum number of six (n= 6) independent experiments were used for statistical analysis. Paired Student's t-test, one-way ANOVA including post-tests, or two-way ANOVA including Bonferroni post-tests were used for data comparison as indicated per experiment, and differences were considered significant when P < 0.05.
Materials
Anti-LC3 antibody (#PM036) was purchased from MBL International (Woburn, MA, USA) and used at 1:1000 dilution. Anti-Perilipin A (#ab61682) was used at 1:3000, anti-ATGL (#2439B) and anti-P62 (#5114) were used at 1:1000, and purchased from Abcam (Cambridge, MA, USA) and Cell Signaling (Danvers, MA, USA), respectively. Anti-β actin antibody (#A1978) was used for all loading controls at 1:5000 and was purchased from Sigma. Secondary HRP antibodies were used at 1:5000 and purchased from Pierce Biotechnology (Rockford, IL, USA). All dilutions mentioned earlier are for immunoblotting. Wortmannin was purchased from Invitrogen. 3-MA, isoproterenol, 3-isobutyl-1-methylxanthine (IBMX), dexamethasone and H-89 dihydrochloride hydrate (H89) were purchased from Sigma.
Results
Treatment with 3-MA affects both autophagy and lipid turnover
Activation of PI3-kinases is a prerequisite for maintaining basal autophagic activity. To evaluate the potential involvement of autophagy in TAG breakdown, we treated 3T3-L1 adipocytes with PI3K inhibitors such as wortmannin and 3-MA, and then measured glycerol release as a function of complete lipolysis. The lipid-laden adipocytes were derived from 3T3-L1 fibroblasts that were induced into adipogenic differentiation via a well-established method (Figure 1A). Accumulation of the LC3-II isoform was used as a determinant for autophagic activity as previously described (Mizushima et al., 2010). p62/SQSTM1 is a protein that forms aggregates which are predominantly degraded by autophagy (Bjorkoy et al., 2005). p62 turnover was used as a secondary assay for measuring autophagy activation as previously described (Barth et al., 2010). In comparison to that with vehicle alone, treatment with 3-MA and wortmannin both significantly reduced intracellular levels of LC3-II and as revealed by immunoblotting (Figure 1B). Additionally, cells treated with the PI3K inhibitors showed increases in p62 expression by immunoblot, indicating effective inhibition of autophagy (Figure 1B). Interestingly, 3-MA treated cells showed a near twofold increase in lipolysis. The level of stimulation was comparable to that incurred by the β-adrenoceptor agonist isoproterenol, a well-known activator of lipolysis (Figure 1C). By contrast, TAG hydrolysis was not altered by treatment with wortmannin. Taken together, these findings suggest that in adipocytes, 3-MA stimulated TAG hydrolysis that was distinct from autophagic inhibition.
Figure 1.
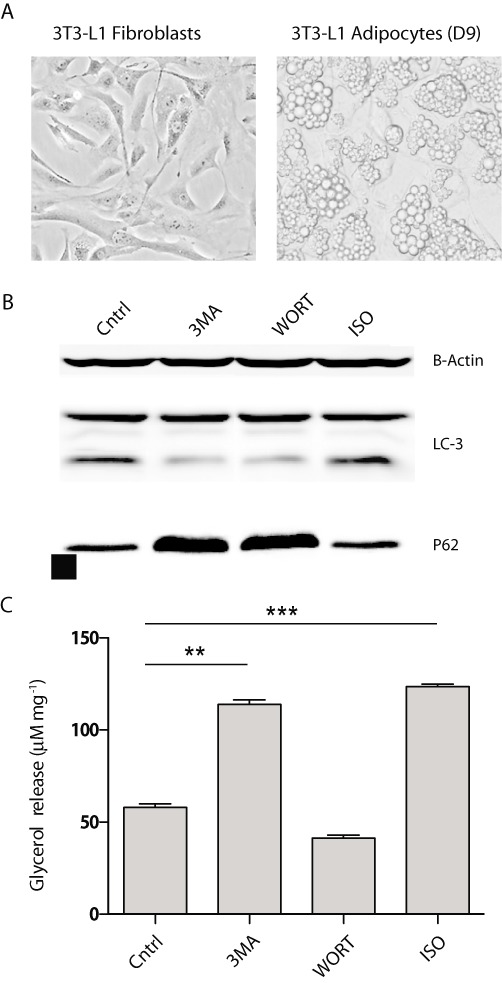
3-MA and wortmannin treatment of 3T3-L1 adipocytes. 3T3-L1 adipocytes were treated for 4 h in the absence of serum with 5 mM of 3-MA, 100 nM of wortmannin (WORT) and 10 µM of isoproterenol (ISO). (A) Phase-contrast micrograph of untreated 3T3-L1 fibroblasts and 3T3-L1 adipocytes. (B) Immunoblotting analysis of LC3 expression and β-actin loading control post-drug treatment. (C) Lipolysis assay measuring glycerol release at 4 h post-treatment. Data are shown as mean ± SD and represent six (n= 6) independent experiments. Statistical significance was determined using paired Student's t-tests (**P < 0.002; ***P < 0.001).
Ablation of the key autophagy mediator Atg5 has no effect on TAG hydrolysis
To further assess if the stimulation of TAG hydrolysis following 3-MA treatment was distinct from autophagic inhibition, we performed loss-of-function experiments by silencing the expression of a key autophagy protein Atg 5. Compared to cells transfected with a non-silencing control siRNA, cells exposed to siRNA directed specifically against mouse Atg 5 retained <5% of the endogenous protein following transfection (Figure 2A). p62 immunoblot demonstrated significant inhibition of autophagy following Atg 5 ablation (Figure 2B). Under basal, 3-MA, and isoproterenol-stimulated conditions, no changes in lipolysis were observed between the control and Atg 5 knockdown groups (Figure 2C). Two-way ANOVA showed no statistically significant difference in the relationship between atg5 ablation and observable changes in lipolysis [F(1,6) = 0.7641, P= 0.4157]. These findings suggested that inhibition of autophagy did not significantly affect lipolysis in adipocytes. Therefore, it is unlikely that the lipolytic stimulation witnessed following 3-MA treatment was secondary to autophagic inhibition.
Figure 2.
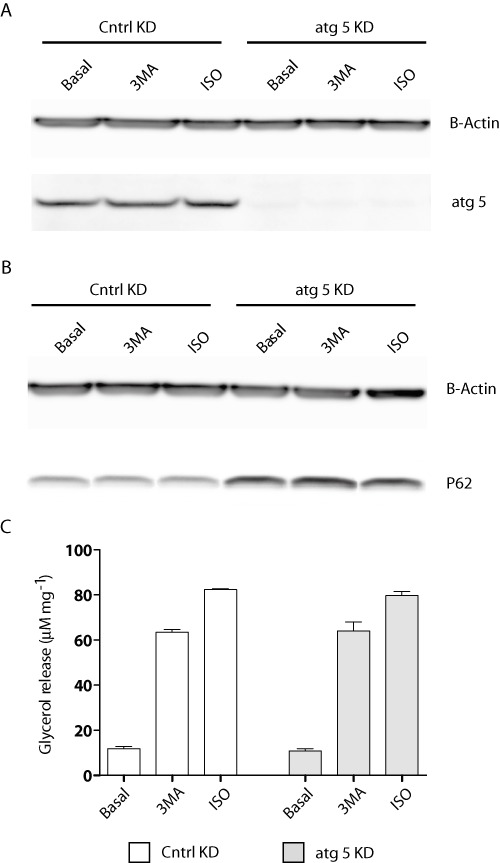
Inhibition of autophagy does not affect TAG hydrolysis. 3T3-L1 adipocytes electroporated with control or Atg 5 siRNA oligos were treated with vehicle alone, 10 µM of isoproterenol (ISO) or 5 mM of 3-MA for a period of 4 h. (A) Immunoblot analysis of Atg 5 protein levels and actin loading control following knockdown and 4 h treatment. (B) Immunoblot analysis of the autophagy marker p62 and actin loading control in both control and Atg 5 knockdown cells in the presence or absence of 4 h isoproterenol or 3-MA treatment. (C) Lipolysis assay measuring glycerol release following Atg 5 knockdown. Data are shown as mean ± SD and represent six (n= 6) independent experiments. Statistical significance was determined using two-way ANOVA including Bonferroni post-tests; no significant changes were observed.
3-MA induced TAG hydrolysis is stable at various dosing and temporal regimes
In order to determine whether 3-MA acutely or chronically stimulated lipolysis, we treated 3T3-L1 adipocytes with 3-MA for various periods of time. When compared with vehicle alone, 3-MA increased lipolysis throughout the entire 6 h span. As shown in (Figure 3A), an approximate 50% increase in glycerol release was observed in cells following 1 hr of 3-MA treatment. Longer treatments resulted in further increase in the rates of glycerol release. However, the fold change induced by 3-MA over the basal level stayed at ∼50% at the subsequent time points. Two-way ANOVA showed statistical significance between treated and non-treated groups [F(1,12) = 1893 P < 0.0001] and in treatment time [F(5,12) = 1563 P < 0.0001]. Therefore, the increase in lipolysis occurred early in the course of the 3-MA treatment and the effect remained stable when the treatment was prolonged. Moreover, a dose–response experiment showed that treatment with 3-MA at concentrations greater than 0.625 mM was sufficient to induce a near-maximal increase in the lipolytic rates (Figure 3B).
Figure 3.
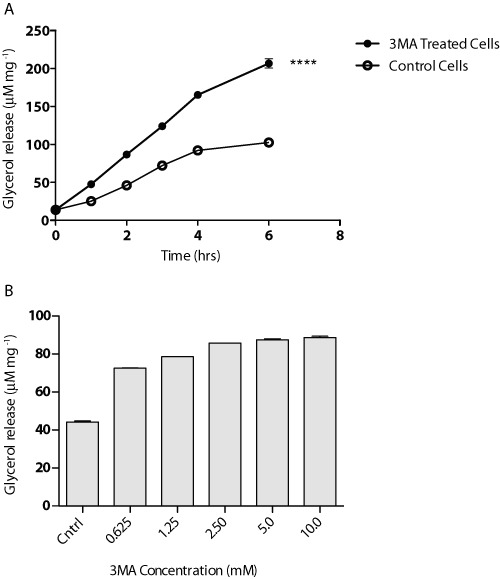
Concentration-response and time course for 3-MA treatment of 3T3-L1 adipocytes. (A) Cells were exposed to 5 mM of 3-MA for 6 h. Media was collected at 1 h intervals and glycerol release was measured. (B) Cells were treated under varying concentrations of 3-MA for a period of 4 h. Following treatment, media was collected and glycerol release was measured. Data are shown as mean ± SD and represent seven (n= 7) independent experiments. Statistical significance was determined using two-way ANOVA including Bonferroni post-tests (****P < 0.0001); all time points were found to be significant as shown.
3-MA stimulated lipolysis is dependent on ATGL
ATGL is the rate-limiting lipase mediating TAG turnover in adipocytes (Haemmerle et al., 2006). To assay the nature of the increase in lipolysis in response to 3-MA treatment, we introduced siRNA via electroporation to knockdown ATGL in mature 3T3-L1 adipocytes. Compared with the protein levels in cells transfected with a non-silencing control siRNA, <5% of ATGL protein was present in cells transfected with a pool of siRNA specifically directed against ATGL (Figure 4A). Cells were then subjected to treatment with 3-MA, using isoproterenol as a positive control. The glycerol release assay revealed that both 3-MA and isoproterenol drastically stimulated glycerol release in the control knockdown cells. In the ATGL knockdown cells, however, the effects of both agents were effectively negated (Figure 4B). Two-way ANOVA showed a statistical significance in relationship to both gene ablation [F(1,6) = 4600, P < 0.0001] and drug treatment [F(2,6) = 2515, P < 0.0001]. These results demonstrate that 3-MA promotes TAG turnover through ATGL-dependent lipolysis.
Figure 4.
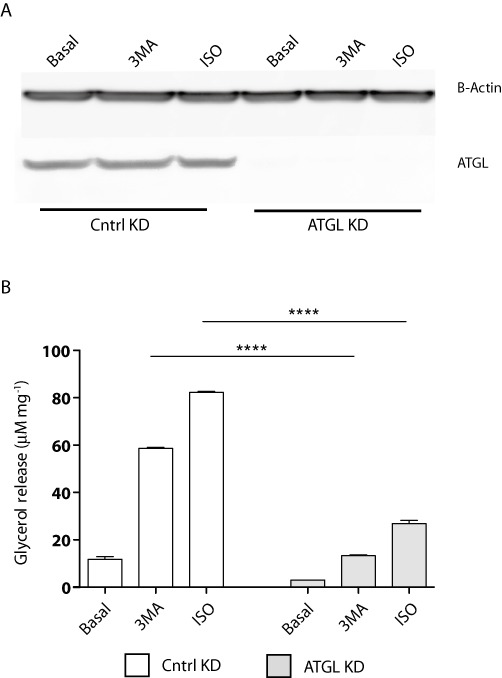
3-MA activates ATGL-mediated lipolysis. 3T3-L1 adipocytes electroporated with control or ATGL-specific siRNA oligonucleotides were treated with vehicle alone, 5 mM of 3-MA or 10 µM of isoproterenol (ISO) for 4 h. (A) Immunoblotting analysis of ATGL expression and actin loading following siRNA knockdown of ATGL. (B) Lipolysis assay measuring glycerol release following ATGL knockdown. Data are shown as mean ± SD and represent six (n= 6) independent experiments. Statistical significance was determined using two-way ANOVA including Bonferroni post-tests (****P < 0.0001).
3-MA stimulates activation of cAMP-dependent protein kinase (PKA)
As 3-MA and isoproterenol share similar patterns of lipolytic stimulation, we next directly evaluated the effect of 3-MA on PKA activation. We treated 3T3-L1 adipocytes with 3-MA, isoproterenol or another cAMP-elevating agent IBMX and then analysed hyperphosphorylation of a well-known PKA substrate perilipin (Zhang et al., 2002). As revealed by immunoblotting in Figure 5A, stimulation of cells with isoproterenol or IBMX predictably promoted an upward shift of the lipid droplet coat protein perilipin. This shift of perilipin was due to phosphorylation by PKA, since it could be abolished by co-treatment of cells with a selective PKA inhibitor H89. Of interest, 3-MA was able to induce the same shift of perilipin that was sensitive to H89, indicating an efficient activation of PKA by 3-MA. In conjunction with hyperphosphorylation of perilipin, phosphorylation of HSL in response to 3-MA treatment was also detected by immunoblotting with an antibody specific for PKA-phosphorylated HSL (Figure 5A). As with perilipin, HSL phosphorylation induced by 3-MA was abolished upon inhibition of PKA by co-treatment of cells with H89. Additionally, phosphorylation of perilipin and HSL did not appear to be secondary to the inhibition of PI3Ks as wortmannin did not confer similar effects. Finally, glycerol release assays demonstrated that the effect produced by 3-MA on lipolysis was reduced to basal levels when PKA was inhibited by H89 (Figure 5B). One-way ANOVA showed a statistically significant difference in regard to drug treatment [F(8,9) = 325, P < 0.0001]. Therefore, like β-adrenoceptor agonists, 3-MA stimulated lipolysis via activation of PKA in adipocytes.
Figure 5.
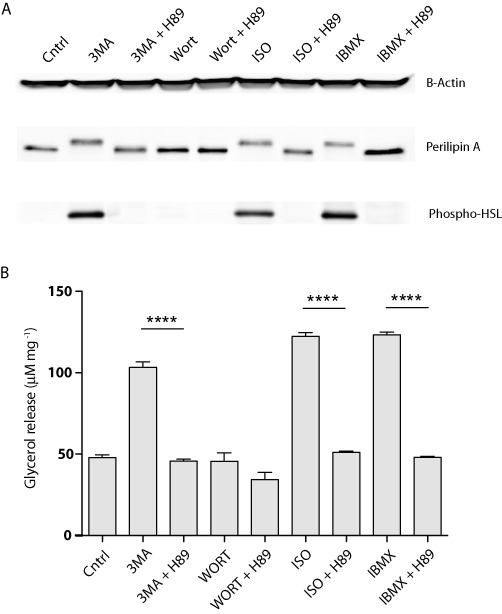
3-MA stimulates lipolysis in a PKA-dependent manner. 3T3-L1 adipocytes were treated for 4 h with 5 mM of 3-MA, 100 nM of wortmannin (Wort), 10 µM of isoproterenol (ISO) or 0.5 mM of IBMX in the presence or absence of 10 µM of H89. (A) Immunoblotting analysis of perilipin A, phospho-HSL and actin loading control 4 h post-treatment. (B) Lipolysis assay measuring glycerol release following 4 h drug treatment. Data are shown as mean ± SD and represent six (n= 6) independent experiments. Statistical significance was determined using one-way ANOVA including Bonferroni post-tests (****P < 0.0001).
cAMP levels increase dramatically in response to 3-MA treatment
After seeing potent activation of PKA as a result of 3-MA treatment, we next assayed intracellular cAMP levels in response to 3-MA treatment. First, 3T3-L1 adipocytes were treated with 5 mM of 3-MA for varying periods of time. cAMP levels increased by threefold over basal for both isoproterenol-treated positive control cells and 3-MA-treated cells (Figure 6A). Two-way ANOVA showed statistical significance in respect to both drug treatment [F(2,15) = 2868, P < 0.0001] and treatment time [F(4,15) = 1899, P < 0.0001]. The observed increases peaked at 15 min following treatment and remained constant throughout the 2 h collection period. Second, cells were treated with 3-MA in combination with IBMX, a non-selective phosphodiesterase inhibitor commonly used to prevent cAMP turnover (Figure 6B). Again, two-way ANOVA revealed statistical significance in respect to drug treatment [F(3,20) = 829.2, P < 0.0001] and treatment time [F(4,20) = 577.5, P < 0.0001]. As expected, IBMX treatment alone markedly increased intracellular cAMP levels. However, the effects of 3-MA and IBMX were not additive as the combination treatment with 3-MA and IBMX failed to further increase the peak concentration of cAMP induced by IBMX alone at the 15-min time point. Moreover, there was a concentration-related effect of 3-MA on the elevation of cAMP levels. As shown in Figure 6C, 3-MA at concentrations as low as 0.625 mM was effective in promoting the accumulation of cAMP (Figure 6C) and subsequent downstream activation of PKA as reported earlier (data not shown). Furthermore, cAMP accumulation was also stimulated in undifferentiated 3T3-L1 fibroblasts treated with 3-MA (Figure 6D). Despite the cell type-specific differences in the dose responses, these results indicate that the stimulatory effect of 3-MA on cAMP/PKA is not restricted to adipocytes.
Figure 6.
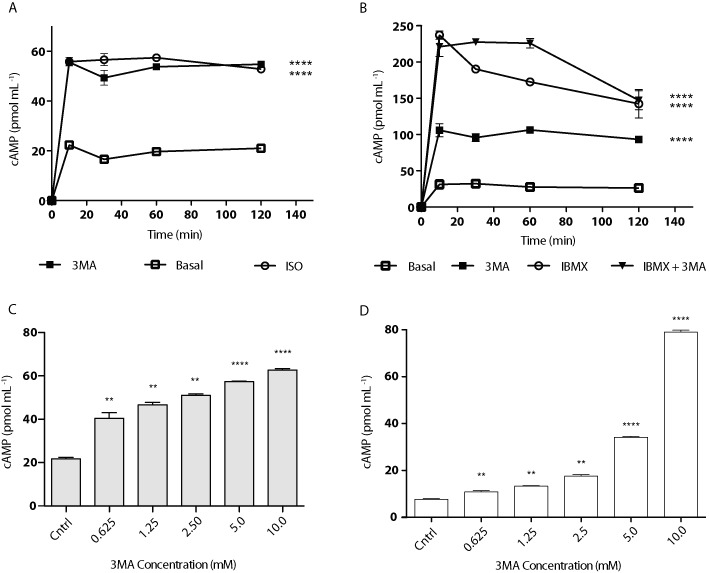
3-MA potently increases intracellular cyclic-AMP levels. 3T3-L1 adipocytes were treated with vehicle alone, 3-MA, isoproterenol (ISO) or IBMX as shown. (A) Time course assay showing direct measurement of intracellular cAMP following treatment with 5 mM of 3-MA or 10 µM of isoproterenol. (B) Time course assay showing direct measurement of intracellular cAMP following treatment with 5 mM of 3-MA, 0.5 mM of IBMX or combination. (C) Concentration-response assay showing direct measurement of intracellular cAMP following 30 min treatment of 3T3-L1 adipocytes with 3-MA at varying concentrations. (D) Concentration response assay showing direct measurement of intracellular cAMP following 30 min treatment of 3T3-L1 fibroblasts with 3-MA at varying concentrations. Data are shown as mean ± SD and represent six (n= 6) independent experiments. Statistical significance for Figure 6A and B was determined using two-way ANOVA including Bonferroni post-tests (****P < 0.0001); all time points except for 0 min were significant for each treatment condition. For Figure 6C and D, significance was determined using paired Student's t-tests (**P < 0.003; ****P < 0.0001).
Discussion and conclusions
Traditionally, 3-MA has been described as a specific autophagy inhibitor and thus has been and continues to be used extensively in autophagy research (Ge et al., 2008). Increasing evidence, however, suggests that 3-MA is not as specific as once believed (Wu et al., 2010). Our findings demonstrate that 3-MA produces a metabolic effect that is not related to its inhibition of autophagy, thus providing a novel piece to this new evidence. While there is no doubt that 3-MA is a potent inhibitor of autophagy as we have shown, we have also demonstrated that it can act as a potent stimulatory agent for adipocyte lipolysis.
Lipolysis in adipocytes is critically mediated by the cytosolic lipases ATGL and HSL. In response to β-adrenergic stimulation and ultimate PKA activation, both ATGL and HSL undergo translocation onto the surface of LDs (Egan et al., 1992; Brasaemle et al., 2000; Sztalryd et al., 2003; Bezaire et al., 2009; Yang et al., 2010; Wang et al., 2011). Phosphorylation of perilipin by PKA releases perilipin-bound co-lipase CGI-58 (Granneman et al., 2009), thereby stimulating the lipase activity of translocated ATGL. Results from our siRNA-mediated knockdown analyses demonstrate that 3-MA is capable of inducing ATGL-dependent lipolysis in adipocytes. The finding that 3-MA was able to achieve the same level of lipolytic stimulation as seen with isoproterenol is surprising, considering that this β-adrenoceptor agonist is one of the most potent agents used to activate lipolysis in adipocytes (Collins and Surwit, 2001). Moreover, experiments combining PKA inhibition with phospho-specific immunoblotting of perilipin and HSL showed that treatment with 3-MA profoundly up-regulated the PKA activity in adipocytes. Additionally, we found that intracellular cAMP levels increased rapidly and consistently after cells were exposed to 3-MA. The concentration response and time course match precisely those for lipolytic stimulation. During a combination treatment, 3-MA was unable to further increase the peak concentration of cAMP induced by the phosphodiesterase inhibitor IBMX. This latter evidence supports a possibility that 3-MA signals to prevent degradation of intracellular cAMP rather than to promote its production.
Although 3-MA is known to be more selective towards class III PI3Ks, both 3-MA and wortmannin are able to inhibit all classes of PI3K (Wu et al., 2010). The observed effects of 3-MA appear to be independent of PI3K inhibition as wortmannin proved ineffective in activating PKA as well as stimulating adipocyte lipolysis. Moreover, experimental results obtained from 3T3-L1 fibroblasts show that increase in intracellular cAMP and the activation of PKA associated with 3-MA treatment can be present in cell types other than adipocytes, although kinetic differences in inhibition of various isoforms of PI3K could result in varying observations in different cells. If it holds true, it would be important to determine whether the activating effect on PKA would in any way contribute to the autophagic inhibition by 3-MA.
It was recently shown that the autophagic mechanism mediates fasting-induced lipolysis in mouse liver (Singh et al., 2009a). Genetic ablation of a required autophagy protein Atg5 leads to enhanced TG accumulation and decreased fatty acid oxidation in hepatocytes. On the other hand, adipose-specific knockout of autophagy-related gene 7 (Atg7) hinders adipogenic differentiation of pre-adipocyte cells (Zhang et al., 2009; Singh et al., 2009b). In differentiation mature adipocytes, we found that neither PI3K inhibition by wortmannin nor silencing of Atg5 had any effect on lipolytic rates at the basal level and following isoproterenol stimulation. Thus, it is unlikely that the basal autophagy process is actively involved in TAG turnover in differentiated adipocytes, although our data do not exclude a lipolytic role for elevated autophagy as seen under conditions of prolonged nutrient deprivation.
In summary, the present study highlights the existence of a novel metabolic and cellular signalling effect of 3-MA, which raises the question about the specificity of 3-MA as an autophagy inhibitor. Based upon these observations and others, we recommend a cautious approach to future studies utilizing 3-MA to inhibit autophagy, as there are potentially more undescribed effects associated with this compound. For example, 3-MA treatment was previously shown to reduce TG accumulation in differentiating 3T3-L1 cells (Singh et al., 2009b). It would be interesting to determine whether the loss of TG content is due to hindered differentiation caused by autophagic inhibition, or it is due to enhanced lipolysis caused by 3-MA-induced PKA activation.
Acknowledgments
This work was supported by National Institutes of Health DK089178 and a Junior Faculty Award from the American Diabetes Association to J. L. We would like to thank Mr. Xin Lu for his technical expertise and Dr. Traci Czyzyk for her assistance with data analysis.
Glossary
- 3-MA
3-methyladenine
- ATGL
adipose triglyceride lipase
- HSL
hormone-sensitive lipase
- IBMX
3-isobutyl-1-methylxanthine
- siRNA
small interfering ribonucleic acid
- TAG
triacylglycerol
Conflict of interest
We have no conflicts of interest.
Note added in proof
While studying the effects of 3-MA in primary rat hepatocytes, Caro et al. (1988) previously observed slight increase in both glycogen breakdown and intracellular cAMP concentration in response to a 20-min incubation with 5 mM of 3-MA.
References
- Ahmadian M, Wang Y, Sul HS. Lipolysis in adipocytes. Int J Biochem Cell Biol. 2010;42:555–559. doi: 10.1016/j.biocel.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth S, Glick D, Macleod KF. Autophagy: assays and artifacts. J Pathol. 2010;221:117–124. doi: 10.1002/path.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezaire V, Mairal A, Ribet C, Lefort C, Girousse A, Jocken J, et al. Contribution of adipose triglyceride lipase and hormone-sensitive lipase to lipolysis in hMADS adipocytes. J Biol Chem. 2009;284:18282–18291. doi: 10.1074/jbc.M109.008631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasaemle DL, Levin DM, Adler-Wailes DC, Londos C. The lipolytic stimulation of 3T3-L1 adipocytes promotes the translocation of hormone-sensitive lipase to the surfaces of lipid storage droplets. Biochim Biophys Acta. 2000;1483:251–262. doi: 10.1016/s1388-1981(99)00179-1. [DOI] [PubMed] [Google Scholar]
- Caro LHP, Plomp PJAM, Wolvetang EJ, Kerkhof C, Meijer AJ. 3-methyladenine, an inhibitor of autophagy, has multiple effects on metabolism. Eur J Biochem. 1988;175:325–329. doi: 10.1111/j.1432-1033.1988.tb14200.x. [DOI] [PubMed] [Google Scholar]
- Codogno P, Meijer AJ. Atg5: more than an autophagy factor. Nat Cell Biol. 2006;8:1045–1047. doi: 10.1038/ncb1006-1045. [DOI] [PubMed] [Google Scholar]
- Collins S, Surwit RS. The beta-adrenergic receptors and the control of adipose tissue metabolism and thermogenesis. Recent Prog Horm Res. 2001;56:309–328. doi: 10.1210/rp.56.1.309. [DOI] [PubMed] [Google Scholar]
- Dong H, Czaja MJ. Regulation of lipid droplets by autophagy. Trends Endocrinol Metab. 2011;22:234–240. doi: 10.1016/j.tem.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan JJ, Greenberg AS, Chang MK, Wek SA, Moos MC, Jr, Londos C. Mechanism of hormone-stimulated lipolysis in adipocytes: translocation of hormone-sensitive lipase to the lipid storage droplet. Proc Natl Acad Sci U S A. 1992;89:8537–8541. doi: 10.1073/pnas.89.18.8537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn PF, Dice JF. Proteolytic and lipolytic responses to starvation. Nutrition. 2006;22:830–844. doi: 10.1016/j.nut.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Ge JN, Huang D, Xiao T, Wang Z, Li XL, Xiao H, et al. [Effect of starvation-induced autophagy on cell cycle of tumor cells] Ai Zheng. 2008;27:788–794. [PubMed] [Google Scholar]
- Gibbons GF, Islam K, Pease RJ. Mobilisation of triacylglycerol stores. Biochim Biophys Acta. 2000;1483:37–57. doi: 10.1016/s1388-1981(99)00182-1. [DOI] [PubMed] [Google Scholar]
- Granneman JG, Moore HP, Krishnamoorthy R, Rathod M. Perilipin controls lipolysis by regulating the interactions of AB-hydrolase containing 5 (Abhd5) and adipose triglyceride lipase (Atgl) J Biol Chem. 2009;284:34538–34544. doi: 10.1074/jbc.M109.068478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green A, Rumberger JM, Stuart CA, Ruhoff MS. Stimulation of lipolysis by tumor necrosis factor-alpha in 3T3-L1 adipocytes is glucose dependent: implications for long-term regulation of lipolysis. Diabetes. 2004;53:74–81. doi: 10.2337/diabetes.53.1.74. [DOI] [PubMed] [Google Scholar]
- Greenberg AS, Coleman RA, Kraemer FB, McManaman JL, Obin MS, Puri V, et al. The role of lipid droplets in metabolic disease in rodents and humans. J Clin Invest. 2011;121:2102–2110. doi: 10.1172/JCI46069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haemmerle G, Lass A, Zimmermann R, Gorkiewicz G, Meyer C, Rozman J, et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. 2006;312:734–737. doi: 10.1126/science.1123965. [DOI] [PubMed] [Google Scholar]
- Huang S, Sinicrope FA. Celecoxib-induced apoptosis is enhanced by ABT-737 and by inhibition of autophagy in human colorectal cancer cells. Autophagy. 2010;6:256–269. doi: 10.4161/auto.6.2.11124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Chiang SH, Chang L, Chen XW, Saltiel AR. Compartmentalization of the exocyst complex in lipid rafts controls Glut4 vesicle tethering. Mol Biol Cell. 2006;17:2303–2311. doi: 10.1091/mbc.E06-01-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Koshikawa N, Mochizuki S, Takenaga K. 3-Methyladenine suppresses cell migration and invasion of HT1080 fibrosarcoma cells through inhibiting phosphoinositide 3-kinases independently of autophagy inhibition. Int J Oncol. 2007;31:261–268. [PubMed] [Google Scholar]
- Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovsan J, Bluher M, Tarnovscki T, Kloting N, Kirshtein B, Madar L, et al. Altered autophagy in human adipose tissues in obesity. J Clin Endocrinol Metab. 2011;96:E268–E277. doi: 10.1210/jc.2010-1681. [DOI] [PubMed] [Google Scholar]
- Kundu M, Thompson CB. Autophagy: basic principles and relevance to disease. Annu Rev Pathol. 2008;3:427–455. doi: 10.1146/annurev.pathmechdis.2.010506.091842. [DOI] [PubMed] [Google Scholar]
- Lafontan M, Langin D. Lipolysis and lipid mobilization in human adipose tissue. Prog Lipid Res. 2009;48:275–297. doi: 10.1016/j.plipres.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Lass A, Zimmermann R, Oberer M, Zechner R. Lipolysis – a highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog Lipid Res. 2010;50:14–27. doi: 10.1016/j.plipres.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275:992–998. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–1435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A. 1982;79:1889–1892. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature. 2009a;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, et al. Autophagy regulates adipose mass and differentiation in mice. J Clin Invest. 2009b;119:3329–3339. doi: 10.1172/JCI39228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztalryd C, Xu G, Dorward H, Tansey JT, Contreras JA, Kimmel AR, et al. Perilipin A is essential for the translocation of hormone-sensitive lipase during lipolytic activation. J Cell Biol. 2003;161:1093–1103. doi: 10.1083/jcb.200210169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Bell M, Sreenevasan U, Hu H, Liu J, Dalen K, et al. Unique regulation of adipose triglyceride lipase (ATGL) by perilipin 5, a lipid droplet-associated protein. J Biol Chem. 2011;286:15707–15715. doi: 10.1074/jbc.M110.207779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, et al. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem. 2010;285:10850–10861. doi: 10.1074/jbc.M109.080796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JY, Della-Fera MA, Baile CA. Guggulsterone inhibits adipocyte differentiation and induces apoptosis in 3T3-L1 cells. Obesity (Silver Spring) 2008;16:16–22. doi: 10.1038/oby.2007.24. [DOI] [PubMed] [Google Scholar]
- Yang X, Lu X, Lombes M, Rha GB, Chi YI, Guerin TM, et al. The G(0)/G(1) switch gene 2 regulates adipose lipolysis through association with adipose triglyceride lipase. Cell Metab. 2010;11:194–205. doi: 10.1016/j.cmet.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HH, Halbleib M, Ahmad F, Manganiello VC, Greenberg AS. Tumor necrosis factor-alpha stimulates lipolysis in differentiated human adipocytes through activation of extracellular signal-related kinase and elevation of intracellular cAMP. Diabetes. 2002;51:2929–2935. doi: 10.2337/diabetes.51.10.2929. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci U S A. 2009;106:19860–19865. doi: 10.1073/pnas.0906048106. [DOI] [PMC free article] [PubMed] [Google Scholar]