

Abstract
Background and Purpose
For antibody therapies against receptor targets, in vivo outcomes can be difficult to predict because of target-mediated clearance or antigen ‘sink’ effects. The purpose of this work was to engineer an antibody to the GM-CSF receptor α (GM-CSFRα) with pharmacological properties optimized for chronic, s.c. treatment of rheumatoid arthritis (RA) patients.
Experimental Approach
We used an in silico model of receptor occupancy to guide the target affinity and a combinatorial phage display approach for affinity maturation. Mechanism of action and internalization assays were performed on the optimized antibody in vitro before refining the modelling predictions of the eventual dosing in man. Finally, in vivo pharmacology studies in cynomolgus monkeys were carried out to inform the predictions and support future clinical development.
Key Results
Antibody potency was improved 8600-fold, and the target affinity was reached. The refined model predicted pharmacodynamic effects at doses as low as 1 mg kg−1 and a study in cynomolgus monkeys confirmed in vivo efficacy at 1 mg kg−1 dosing.
Conclusions and Implications
This rational approach to antibody drug discovery enabled the isolation of a potent molecule compatible with chronic, s.c. self-administration by RA patients. We believe this general approach enables the development of optimal biopharmaceuticals.
Keywords: rheumatoid arthritis, GM-CSFR, affinity maturation, phage display, scFv
Introduction
Rheumatoid arthritis (RA), which affects 0.5–1% of the population in the industrialized world (Symmons, 2002), is a systemic autoimmune disease characterized by chronic and progressive inflammation in multiple joints, leading to irreversible tissue damage and deformity that culminates in disability, a lower quality of life and in many cases premature death (Mikuls et al., 2011). It is initially characterized by inflammatory synovitis involving a broad range of cells such as macrophages, neutrophils, B cells, T cells and synovial fibroblasts. Pioneering work by Feldman and Maini (Feldmann et al., 1996) highlighted the central role of TNF-α in this disease, ultimately leading to the approval of four anti-TNF-α therapies (Taylor and Feldmann, 2009) for the successful treatment of RA.
Although targeting TNF-α in RA has provided significant clinical benefits approximately 30–40% of severe RA patients remain refractory to treatment (Singh et al., 2009) and therefore new therapies that can address this area of unmet medical need are still required. Recently, a number of groups have focused their efforts on the GM-CSF pathway. GM-CSF has been shown to be elevated in synovial fluid (Bell et al., 1995; Ottonello et al., 2002) and membrane biopsies (Farahat et al., 1993) from patients with RA. Furthermore, treatment of patients with recombinant GM-CSF for chemotherapy-induced neutropenia or Felty's syndrome was shown to result in a flare in their arthritis, suggesting that GM-CSF may modulate the immune response towards disease. These observations are supported by mechanistic studies in in vivo models of arthritis (Cook et al., 2001; Plater-Zyberk et al., 2007).
Although the immunological rationale for targeting either GM-CSF or its receptor for the treatment of rheumatoid arthritis is compelling, recent changes in treatment paradigms in this disease mean that successful new therapies need not only be efficacious with a good or improved safety profile but must also be formulated in such a way as to allow self administration by patients avoiding the need for inconvenient and costly i.v. infusions. Moreover, new therapies also have to be dosed infrequently (e.g. monthly for patient convenience). Current autoinjector devices for patient self-administration have an upper volume limit of around 1 mL, which, combined with the challenge of formulating any protein at very high concentrations, emphasizes the importance of engineering high potency in proteins used therapeutically.
A key requirement during antibody development is therefore to engineer a molecule with sufficiently high affinity to guarantee effective target occupancy over prolonged periods. Antibodies against cell membrane-associated antigens (receptors) are usually subject to target mediated clearance, or the antigen-sink effect (Roskos et al., 2004). There are two major implications of the potential antigen-sink liability for a receptor antigen. First, the simple Langmuir equation (Langmuir, 1916) can no longer be used to estimate target antigen occupancy (neutralization) in vivo. Second, the expression and turnover rate of the target receptor may have a profound impact on the pharmacokinetics (PK) of antibodies against the target. As a result, the assessment of affinity goals for antibodies against membrane receptors is often challenging.
Here, we describe an integrated approach to engineer an optimal antibody against the GM-CSF receptor α (GM-CSFRα) suitable to meet the needs of the RA market at launch. Using mathematical modelling of receptor occupancy, we define a theoretical affinity target that would achieve high levels of GM-CSFR occupancy over prolonged periods of time. In order to meet this affinity goal, we employ a highly efficient approach of randomly recombining pools of affinity matured variable heavy (VH) and variable light (VL) domains to identify optimal VH/VL domain pairings that work in synergy to achieve large gains in affinity and potency. Using a range of in vitro biological assays, the antibody was then characterized, and the data were used to refine the model. Finally, the antibody was evaluated in vivo in cynomolgus monkeys to determine its PK and pharmacodynamic (PD) profile, both reinforcing our approach and demonstrating the suitability of the molecule for clinical evaluation.
Methods
In silico translational simulations
An in silico mechanistic biomathematical model was constructed to describe the PK of a typical human IgG, binding of the antibody to GM-CSFRα and the internalization of GM-CSFRα and the antibody–receptor complex. The model assumed 50% absolute s.c. bioavailability, 2.5 mL kg−1 day−1 IgG clearance by the reticuloendothelial system, a distribution volume of 64 mL kg−1, and 20 pM GM-CSFRα with a 1 h internalization half-life for the receptor and antibody–receptor complex (Roskos et al., 2004). The following differential equations describe the PK of the antibody 574D04, its binding to GM-CSFR and the internalization of the complex following s.c. administration (CLRES: intrinsic clearance of endogenous IgG by reticuloendothelial system; S0: synthesis/recycling rate of target receptor, GM-CSFRα; kE: receptor internalization rate; kon, koff: association and dissociation rate constants). To avoid model overparameterization, it is assumed that the internalization rates of unoccupied receptor and 574D04-bound receptor are the same. Although no data have been reported to directly support this assumption for GM-CSF receptors, published data for EGF receptors are consistent with this assumption (Wiley et al., 1991).
In the equations, AbSC represents 574D04 at the s.c. dosing site. Bolus 574D04 is administered to this compartment with an amount of F·dose, where F is the absolute s.c. bioavailability. Ab represents 574D04 in the serum compartment. R is the target receptor, GM-CSFRα, and AbR is the antibody–receptor complex. Following antibody optimization, the model parameters were adjusted to reflect the in vitro binding affinity of 574D04 and the internalization half-life of 574D04/GM-CSFRα complex. Simulations were performed to predict GM-CSFRα blockade following single 0.01–10 mg kg−1 i.v. or s.c. administration of 574D04 in humans. The differential equations describing the disposition of 574D04 and interaction with GM-CSFR following i.v. administration are similar to those shown above, except that the dose is directly given to the Ab compartment.
Expression of recombinant GM-CSFRα and phage display antibody isolation
The sequence encoding the human GM-CSFRα extracellular domain with a murine IL-3 signal sequence and an N-terminal FLAG tag was cloned into the mammalian expression plasmid pEF-BOS (Mizushima and Nagata, 1990). Following transient transfection of the plasmid into CHO cells using standard procedures, the cells were cultured and the encoded protein was expressed. The soluble extracellular domain (ECD) of GM-CSFRα was then purified from the CHO culture supernatants on an M2 affinity chromatography column and eluted with free FLAG peptide. Phage display selections were performed essentially as described previously (Vaughan et al., 1996). In brief, a large single chain variable fragment (scFv) human antibody library was panned in a series of selection cycles on the purified GM-CSFRα ECD in order to enrich for scFv antibodies able to bind to the antigen. Following selections, individual scFv were confirmed as binding to GM-CSFRα by phage elisa (Vaughan et al., 1996) and then tested for their ability to inhibit the binding of recombinant GM-CSF to purified GM-CSFRα ECD in a biochemical receptor : ligand binding assay.
Construction of targeted and recombination libraries
Two VHCDR3 and two VLCDR3 libraries, comprising, respectively, randomization of VH95–100, VH100–102, and VL89–94 and VL94–97 (numbered according to Kabat, 1991), were prepared by oligonucleotide-directed mutagenesis, as described (Baker et al., 2003) in the pCantab6 phage display vector (McCafferty et al., 1994). For recombination, the VH library variants from round 2 of selection on recombinant GM-CSFRα ECD were PCR-amplified, digested with SfiI and XhoI restriction enzymes and cloned into plasmids harbouring the VL variant population, also from round 2 of selection, using previously described methods (Vaughan et al., 1996). Phage display selections for improved affinity were performed in principle according to the published method (Hawkins et al., 1992). Briefly, rounds of phage display selection were performed on purified, biotinylated GM-CSFRα, using decreasing concentrations of antigen over successive rounds in order to stringently select for antibodies of improved Kd.
Expression of scFv and IgG4 molecules
For preparation of scFvs, plasmids containing scFv genes were cultured in Escherichia coli, expression induced by isopropyl-d-thiogalactoside and scFv proteins were isolated from the periplasm by osmotic shock followed by capture of the C-terminal His-tag by Ni2-nitrilotriacetic acid chromatography (Vaughan et al., 1996). For IgG expression, the VH and VL chains of selected scFvs were cloned into human IgG expression vectors, expressed and purified as described (Persic et al., 1997).
In vitro functional assays for GMCSFR antagonism
The TF-1 cell proliferation, granulocyte shape change, granulocyte survival and monocyte TNF-α release assays are all described in the Appendix S1.
Schild analysis
The change in forward scatter of human granulocytes was induced by increasing concentrations of GM-CSF using the described method for neutrophil shape change. This dose–response was carried out in the presence of increasing concentrations of 574D04 to produce a rightward shift of the GM-CSF dose–response curve. EC50 values for GM-CSF in the absence and presence of 574D04 were calculated using GraphPad PRISM software (La Jolla, CA, USA), and the dose ratio (DR) was calculated. Linear regression analysis was performed on log [574D04] M (x-axis) versus log [DR-1] (y-axis), and the pA2 value was determined by extrapolating the line on this Schild plot to zero on the y-axis. If the slope of this line is unity, then the pA2 equals the pKb, an estimate of the drugs affinity for the target receptor. A full account of pA2 analysis has been described previously by Kenakin (1982).
BIAcore measurements
The methods for performing BIAcore analysis are described in the Appendix S1.
Internalization measurements
Detailed methods for antibody internalization measurements are provided in the Appendix S1. Briefly, a GM-CSF-dependent mouse fibroblast cell line expressing human GM-CSF receptors (FD-hGM-CSFR), was labelled with carboxyfluorescein succinimidyl ester and stained with Alexa647-574D04, which bound to cell surface GM-CSFR. The cells were transferred to 37°C to start the reaction. The images of cells were captured over time using an OPERA LX high content confocal imaging system (Perkin Elmer, Waltham, MA, USA) and analysed using Acapella software (Perkin Elmer) and a proprietary algorithm specifically designed to quantify intracellular and membrane fluorescence. Rate of fluorescence accumulated inside the cells was calculated by model fitting of the data using SAAMII software (The Epsilon Group, Charlotteville, VA, USA).
Cynomolgus PK and PD assay
All animal care and experimental studies were approved by the SNBL Institutional Animal care and Use Committees (IACUC) and complied with the principles of the the 3Rs (replace, reduce and refine). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., #b1001; McGrath et al., ). The in vivo studies were conducted at SNBL USA LTD. All test substances were well tolerated and the animals were returned to the colony upon study completion. Two male and two female adult cynomolgus monkeys (Macaca fascicularis) were dosed i.v. with a single 10 mg kg−1 infusion of 574D04. Samples for PK analysis were taken before dosing and then at 1 h and on days 1, 2, 4, 8, 14, 21, 28 and 35 after dosing. 574D04 levels in serum samples were measured by antigen capture elisa. Samples for PD analysis were collected before dosing, 1 h and days 2, 8, 14, 21 and 35 after dosing. Shape change response of eosinophils and neutrophils was assessed using an assay based on that described by Sabroe et al. (1999).
In vivo blockade of GM-CSFR with 574D04
Four treatment groups of five male cynomolgus monkeys received PBS or 574D04 (1, 10 or 30 mg kg−1) as a 30 min i.v. infusion 48 h and 1 h before GM-CSF administration. The first dose of GM-CSF was given 30 min following the end of antibody dosing and animals were dosed s.c. twice daily (approximately 8 h apart) for three consecutive days with 5 μg kg−1 recombinant human GM-CSF. Blood for haematology (complete blood count with differential counts) and serum for determination of antibody concentration was collected prior to each antibody infusion, at 30 min and 4 h after GM-CSF dosing on day 1, 4 h after GM-CSF dosing on days 2 and 3, and on days 4, 6 and 8. Statistical analyses were performed with GraphPad Prism 5 software, using a two-way anova with repeated measures. Bonferroni's post tests were performed to compare treatments with the control PBS group.
Results
Theoretical prediction of affinity goal for anti-GM-CSFRα candidate drug
To guide us in determining the affinity required for an effective anti-GM-CSFRα antibody, we developed a mathematical model to predict the theoretical affinity required to occupy greater than 99% (>99%) of the GM-CSF receptors and therefore block the activity of GM-CSF. By testing different theoretical affinities in the model, a prediction was made that an antibody affinity of 100 pM would be sufficient to achieve and maintain >99% occupancy for 28 days at a s.c. dose of 1 mg of drug per kg of patient body weight (mg kg−1) (Figure 1).
Figure 1.
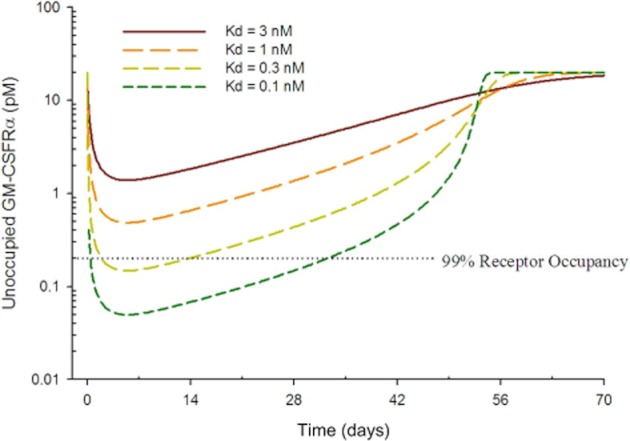
Antibody affinity goal predictions using in silico translational simulations. The biomathematical model assumed 50% s.c. absolute bioavailability, 2.5 mL kg−1 day−1 IgG clearance by the reticuloendothelial system, a distribution volume of 64 mL kg−1 and 20 pM GM-CSFRα with a 1 h internalization rate for the receptor and antibody–receptor complex. Simulations were performed to predict the unoccupied receptor level in humans following a single 1 mg kg−1 s.c. dose.
Isolation of anti-GM-CSFRα scFv and affinity maturation strategy
Having identified a target affinity of 100 pM for an anti-GM-CSFRα antibody, the recombinant human extracellular domain of GM-CSFRα was presented to three different human naïve phage display antibody libraries to isolate a scFv that was specific for GM-CSFRα, i.e., that did not bind to either IL-3Rα, IL-5Rα or the common β chain (Figure S1A). This scFv could dose-dependently inhibit GM-CSF binding to GM-CSFRα (Figure S1B) and could inhibit GM-CSF induced proliferation of TF-1 erythroleukaemic cells (Figure S2). However, the potency of this scFv was low (IC50 > 300 nM) and even upon conversion to a human IgG molecule only achieved a mean IC50 of 43 nM (n = 5) in the TF-1 cell proliferation assay, 237 nM (n = 3) in a granulocyte shape change assay and was unable to inhibit GM-CSF-driven granulocyte survival at concentrations up to 3300 nM. To increase the affinity and potency of this scFv, phage display libraries were first generated to randomise amino acids in the VH and VL CDR3 loops of the scFv. Following affinity selection by phage display, pools of VH and VL genes containing mutations in the CDR3 regions were recombined into a large, combinatorial library that we hypothesized would contain a greater proportion of high-affinity scFv. Pre-recombination variants, with mutations in either VH CDR3 or VL CDR3, were compared with post-recombination variants, with mutations in both VH CDR3 and VL CDR3, in the TF-1 proliferation assay to determine IC50 values for each scFv molecule. ScFv isolated from the post-recombination library were orders of magnitude more potent than scFv from pre-recombination libraries underlining the power of this combinatorial approach (Figure 2A).
Figure 2.

Potency improvements from the combinatorial affinity maturation strategy. (A) IC50 values of scFv antibodies in the TF-1 cell proliferation assay are plotted, showing the improvements in potency of variants compared with the parent antibody. The range of potencies observed following mutation of VHCDR3 alone or VLCDR3 alone are compared with the potencies observed when VHCDR3 and VLCDR3 diversity are combined. (B–E) Comparison of affinity matured 574D04 IgG and parent antibody 28G5 IgG for the neutralization of GM-CSF-induced TF-1 cell proliferation, IC50 = 15 ± 8pM (B); granulocyte shape change, IC50 = 41 ± 30pM (C); monocyte TNF-α release, IC50 = 99 ± 33pM (D); granulocyte survival, IC50 = 911 ± 467pM (E). (B) TF-1 cell proliferation was induced with human recombinant GM-CSF corresponding to EC80 concentration. Cells were incubated with ligand and a dilution series of antibody for 16 h, after which time cell proliferation was quantified by culture for 4–5 h with tritiated thymidine. (C) Human granulocytes were stimulated with human recombinant GM-CSF corresponding to EC80 concentration. Cells were incubated with ligand and a dilution series of antibody for 4 h, after which time cell activation was quantified by an increase in forward scatter (FSC) by flow cytometry. (D) Human monocytes were stimulated with 1 nM human recombinant GM-CSF for 18 h and TNF-α released into the supernatant was quantified by elisa. (E) Human granulocytes were stimulated with 7 pM human recombinant GM-CSF for 68 h, and cell viability was assessed using Alamar Blue. Results are representative of at least three separate experiments. IC50 values were determined using Graphpad PRISM.
Characterization of affinity matured leads as IgG
Lead scFvs were converted to human IgG molecules and re-tested in a panel of in vitro assays that reflected the proposed mechanism of action. We confirmed that these antibodies were significantly improved over the parent antibody (Figure 2). In TF-1 proliferation, granulocyte shape change and monocyte TNF-α release assays, the lead antibody 574D04 had a sub-100 pM potency (15 ± 8, 41 ± 30 and 99 ± 33 pM respectively). In addition, 574D04 was able to inhibit granulocyte survival with sub-nanomolar (911 ± 467 pM) potency, whereas the parent antibody was unable to affect this response at concentrations of up to 3300 nM. The CDR3 recombination approach was able to increase the TF-1 cell proliferation potency of the IgG molecules by 8600-fold (Table 1). Although we obtained significant gains in potency, the 574D04 monovalent Kd value of 139 pM, as determined by surface plasmon resonance, was not within the requirements defined by the initial modelling. Given that measurements of monovalent antibody affinity for recombinant protein may not fully reflect the physiological antibody interaction with cell surface presented GM-CSFR, Schild analysis (Arunlakshana and Schild, 1959) was therefore used to quantify the affinity based on the functional activity of 574D04 (Figure 3A). The Schild analysis of 574D04, using GM-CSF-induced human granulocyte shape change as a suitable assay system, was calculated to be 10.6 (slope = 0.96), equating to an apparent affinity of 27 pM (Figure 3B).
Table 1.
Binding kinetics and TF-1 proliferation IC50 values for wild-type anti-GMCSFR antibody 28G5 and affinity matured antibody variant 574D04
| Assay parameter | Wild type | 574D04 |
|---|---|---|
| kon, M−1 s−1 | 2.45 × 105 | 1.21 × 106 |
| koff, s−1 | 1.75 × 10−2 | 1.70 × 10−4 |
| Kd, nM | 71.4 | 0.14 |
| Kd wild-type/Kd variant | – | 514 |
| TF-1 IC50, nM | 43 | 0.005 |
| TF-1 IC50 wild-type/TF-1 IC50 variant | – | 8600 |
| pH 7.4 Kd, nM | – | 0.145 |
| pH 5.7 Kd, nM | – | 13.4 |
| pH 5.7 Kd/pH 7.4 Kd | – | 92 |
kon = association rate constant; koff = dissociation rate constant; Kd = equilibrium dissociation constant; IC50 = half maximal inhibitory concentration.
Figure 3.

Characterization of 574D04 binding and internalization kinetics. (A) Schild analysis of 574D04 in an assay measuring GM-CSF-induced shape change of human granulocytes. Human peripheral blood granulocytes were stimulated with increasing concentrations of recombinant human GM-CSF (R&D Systems) for 4 h in the presence or absence of a single concentration of 574D04 (0.001, 0.01, 0.1, 1, 10 μg mL−1) to produce a rightward shift in the dose–response curve. Cells were then analysed using forward scatter on a flow cytometer to monitor the change in shape. (B) Schild analysis to determine the pA2 value and thus functional affinity (KD) of 574D04 for the receptor. (C) 574D04 internalization. 574D04 and an isotype control IgG antibody were directly conjugated with Alexa Fluor 647. Internalization studies were performed with AF647-labelled antibodies with mouse FDCP-1 cells overexpressing human GM-CSFR (FD-hGMR). Internalization over time was monitored by translocation of fluorescence from cell membrane to cytosolic compartment, visible as punctate staining of vesicles within cells. Intracellular versus membrane fluorescence was quantified using an OPERA imaging system.
Antibody internalization
As 574D04 binds to a cell surface receptor we also investigated the rate of the antibody internalization following binding. Using fluorescently labelled 574D04, we demonstrated specific binding of the antibody to the surface of FD-hGM-CSFR cells, a GM-CSF-dependent mouse fibroblast cell line that expresses human GM-CSFRα (Figure S3). Following addition to FD-hGM-CSFR, labelled 574D04 accumulated at the cell surface and was internalized within the cell in a time-dependent manner. Internalized antibody was characterized by a punctate staining of vesicles within cells (Figure 3C). The images of cells were captured and analysed using an algorithm designed to quantify intracellular and membrane fluorescence. The rate of fluorescence accumulated inside the cells was calculated to determine the half-life (t½) of 35 ± 6.5 min (n = 7) for internalization, four- to fivefold slower than that described for GM-CSF-mediated receptor internalization with a Ke = 0.12 min−1, t½ of approximately 8 min (Metcalf et al., 1999) or t½ of approximately 7 min (Walker and Burgess, 1987). This slower internalization rate coupled with the observation that direct binding of 574D04 to cells did not evoke any downstream cytokine release (Figure S4A,B) suggested that the antibody-evoked internalization was due to receptor turnover rather than ligand dependent internalization. In addition, we investigated the potential effects of pH changes in the endosomal compartment by measuring the pH dependence of 574D04 binding to GM-CSFRα. As the pH was lowered from pH 7.4 to pH 5.7, we saw a 92-fold reduction in the affinity of the antibody to the receptor (Figure S4C, Table 1), suggesting that antibody dissociation would be strongly favoured in endosomal vesicles. However, further work is required to fully understand the eventual fate of the antibody and receptor.
Theoretical prediction of 574D04 receptor occupancy in humans
The mathematical model was updated to incorporate the new information about affinity and internalization rate of the optimized human antibody 574D04. Simulations using the refined model predicted that a single s.c. dose of 1 mg kg−1 574D04 should maintain 99% GM-CSFRα occupancy for up to 5 weeks (Figure 4).
Figure 4.
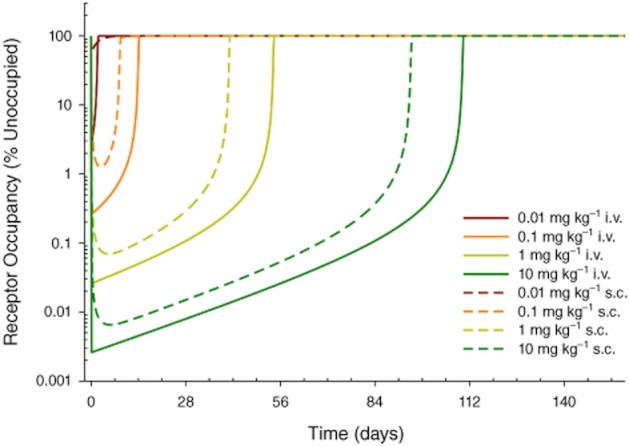
Predicted GM-CSFRα occupancy profiles for 574D04 first-time-in-human dose selections using the revised biomathematical model. Model parameters were adjusted to reflect the in vitro binding affinity of 574D04 and the internalization rate of 574D04/GM-CSFRα complex. Simulations were performed to predict GM-CSFRα blockade following single 0.01–10 mg kg−1 i.v. or s.c. administration of 574D04 in humans.
In vivo activity of 574D04
In order to confirm the in vivo activity of 574D04, species cross-reactivity was investigated and 574D04 shown to antagonize cynomolgus monkey, but not rodent, GM-CSFR. Schild analysis of 574D04 was performed in a cynomolgus granulocyte shape change assay and was found to be 10.8 (slope = 1.1), equating to an apparent affinity of 16 pM (Figure S5), in line with the affinity against human GM-CSFRα of 27 pM. Thus, cynomolgus monkey was determined as the most pharmacologically relevant species in which to assess in vivo efficacy of 574D04.
PK of 574D04 in cynomolgus monkey was assessed following a single 10 mg kg−1 i.v. infusion (Figure 5A). The terminal half-life of 574D04 was estimated as approximately 5.6 days, within the typical range for a human monoclonal antibody in this species. Samples were also taken during the PK study to investigate the PD of 574D04. Using a whole blood assay, a significant shape change (67%) in response to GM-CSF stimulation ex vivo was observed in the eosinophil population before treatment (Figure 5A). Following i.v. infusion of 574D04, responsiveness to GM-CSF was lost and this lack of response to GM-CSF was maintained until day 21. The functional blockade correlated with serum concentrations of 574D04 and suggested that a loss of effective blockade occurred between serum concentrations of 12–71 μg mL−1.
Figure 5.

(A) Relationship between levels of free 574D04 and functional blockade of GM-CSFR in cynomolgus monkeys. Four adult cynomolgus monkeys were dosed i.v. with 10 mg kg−1 574D04. 574D04 levels in serum samples were measured by antigen capture elisa. 574D04 receptor blockade on eosinophils was determined by changes in forward scatter by flow cytometry (shape change) after 1 h ex vivo GM-CSF stimulation of erythrocyte-depleted blood samples. (B,C) In vivo blockade of GM-CSF-induced responses. Recombinant human GM-CSF (5 μg kg−1) was administered twice daily for three consecutive days to cynomolgus monkeys that had previously received PBS, 1 mg kg−1 or 10 mg kg−1 of 574D04. (B) 574D04 levels in serum samples were measured by antigen capture elisa. (C) Complete blood count with differential was monitored at regular intervals. Statistical analyses were performed using a two-way anova with repeated measures and Bonferroni post tests. **P < 0.01, ***P < 0.001, significantly different from control group).
Following this initial demonstration of PK/PD activity, the ability of pre-treatment with 574D04 to suppress an in vivo stimulation with GM-CSF was assessed. Based on the PK/PD analysis, 10 mg kg−1 was selected as an efficacious dose and 1 mg kg−1 as a predicted minimally effective dose. Recombinant human GM-CSF (5μg kg−1) was administered twice daily for three consecutive days to cynomolgus monkeys that had previously received PBS, 1 or 10 mg kg−1 574D04 (Figure 5B). The first dose of GM-CSF stimulated a rapid and transient drop (margination) in circulating leukocytes (69% decrease from baseline, P < 0.001) in the control PBS group (Figure 5C). This margination response was significantly inhibited in the presence of 574D04 after dosing at 1 and 10 mg kg−1 (P < 0.001). A similar trend was noted for neutrophils, basophils, monocytes and eosinophils (84%, 62%, 78% and 98% reductions from baseline, respectively, that were reversed by 574D04) (Figure S6). Continued administration of GM-CSF over 3 days stimulated a leukocytosis (Figure 5C), with significant increases in white blood cells at 52 h (P < 0.01) and 76 h (P < 0.01). This response was inhibited at both 10 mg kg−1 (P < 0.001) and 1 mg kg−1 (P < 0.01 at 76 h) of 574D04.
Discussion and conclusions
Over the last decade, the use of antibodies as a treatment option for chronic diseases has been widely accepted as a viable therapeutic approach by patients, clinicians, regulatory agencies and the pharmaceutical industry. However, due to the high cost of generating these molecules, increased competition and greater expectations of physicians and payers, more consideration has to be given to the initial engineering, design and pharmacology of the antibody to meet the needs of the patient and physicians at launch.
This work describes an integrated approach for engineering a specific, neutralising antibody compatible with s.c. delivery and infrequent dosing intervals. Self-administration by s.c. delivery using spring-driven syringes or autoinjectors is increasingly becoming a key feature for biopharmaceutical drug candidates in RA. Autoinjectors typically have a maximal holding volume of 1 mL and as monoclonal antibodies similar to other proteins tend to reach a solubility limit at around 100 mg mL−1 dosing at approximately 1 mg kg−1 is crucial to ensure device compatibility.
GM-CSF and its receptor are emerging targets of interest for the treatment of RA. Published reports suggest that only 10% of available GM-CSF receptors need to be bound by GM-CSF to elicit a maximal response (Nicola et al., 1988; Hercus et al., 1994). Therefore, this parameter was used in the first model to guide the target antibody affinity required to prevent GM-CSF signalling in vivo. To ensure complete block of GM-CSF function, we chose to aim for >99% occupancy of receptors, at which level the model predicted a target affinity of at least 100 pM for the antibody therapeutic candidate. Given the low starting affinity of our lead antibody (71 nM), which had been isolated by phage display from a naïve antibody library, and the challenging affinity requirements to achieve high receptor occupancy, we took a novel approach to improving affinity.
Previously, saturation mutagenesis has been performed on just one CDR loop (Thompson et al., 1996; Baker et al., 2003; Thom et al., 2006) or sequentially on different loops, using the ‘CDR-walking’ approach (Barbas et al., 1994; Yang et al., 1995; Schier et al., 1996). Alternatively, different CDR loops or regions of a CDR have been optimized in parallel and a small number of optimized loop region sequences recombined to achieve additive gains in potency (Yang et al., 1995; Schier et al., 1996). The novelty of the approach used here was to acknowledge the unpredictable nature of CDR loop synergy and include as many functional VHCDR3 and VLCDR3 loop sequences as possible in a large (>109) combinatorial library. In so doing, we allowed for the emergence of rare, synergistic combinations of CDR loop sequences that could not have been predicted and would not have been isolated by the previous approaches. The overall optimization strategy was highly successful in improving the potency of the final IgG version of the antibody by a factor of 8600-fold. The affinity of the final lead (574D04) for human GM-CSFRα was calculated using Schild analysis (Arunlakshana and Schild, 1959) to be 27 pM, meeting the previously defined affinity requirements of 100 pM.
Having identified an antibody of potentially suitable characteristics for human dosing, it was important to confirm that no further optimization of the molecule was required prior to expensive further investment in human clinical trials. To increase the accuracy of predictions of the dose to man, the PK/PD model required refinement to reflect the complexity of targeting a cell surface receptor. Internalization of receptor-bound antibody is likely to dominate the drug clearance at lower concentration levels when the target receptor is not fully saturated and could constitute a significant sink (Roskos et al., 2004). Indeed, receptor binding and internalization of 574D04 was demonstrated and a quantitative imaging method used to determine the internalization half-life. Although these data were collected using a mouse fibroblast cell line overexpressing human GM-CSFR, a comparable internalization rate has been measured in the human TF-1 cell line, which is not transfected and therefore expresses GM-CSFR at endogenous levels. The method of using confocal microscopy to calculate antibody-receptor-mediated internalization has been previously described for an anti-EGF receptor antibody, Sym004 (Pedersen et al., 2010). Like the antibody 574D04, Sym004 did not induce signalling or phosphorylation but did induce internalization. As the rate of internalization with 574D04 was four- to fivefold slower than ligand-mediated internalization (Walker and Burgess, 1987; Metcalf et al., 1999), we hypothesized that the antibody-bound internalization rate was determined by receptor turnover. This was not formally characterized in this study; however, it has been shown that unoccupied GM-CSFR on mouse peritoneal macrophages internalises with a rate constant Kt of 0.021 min−1 and on BALB/c bone marrow cells with a Kt of 0.009 min−1 equivalent of a t½ of 47 and 111 min, respectively (Nicola et al., 1988), broadly similar to the internalization of the human receptor plus the antibody 574D04. To gain an even greater understanding of this mechanism, it would be of interest to compare these data with a radioimmunoassay that would allow quantification of internalization with and without ligand. In addition, it would also be of interest to determine the fate of the antibody receptor complex once internalized. However, for the purposes of the current study, we have made the assumption that all internalized antibody was degraded.
The revised model, based on the internalization data and the other characteristics of the optimized antibody 574D04, was able to deliver predictions about the proposed clinical dosing. For s.c. dosing, which is strongly preferred as the eventual route for patient self-administration, the model suggests that receptor occupancies >99% could potentially be achieved for the potency-optimized 574D04 given at a dose of 1 mg kg−1 every 5 weeks.
We demonstrated equivalent in vitro activity against cynomolgus monkey GM-CSFR granulocytes confirming the suitability of this species to demonstrate in vivo activity of 574D04. Rodent studies could not be undertaken due to 574D04's lack of cross-reactivity with rodent GM-CSFRα. Therefore, we evaluated 574D04 in cynomolgus monkeys to determine the PK/PD relationship. Although it would have been informative to support our original affinity prediction by conducting a head-to-head comparison of low- and high-affinity GM-CSFR antibodies in cynomolgus monkeys, ethical considerations precluded us from doing so. However, in a single dose PK study, we confirmed that a 10 mg kg−1 dose of the affinity-matured antibody promoted long-term suppression (3–4 half-lives) of GM-CSF activity, and that the PD activity of 574D04 was lost when serum antibody levels dropped below 12–71 μg mL−1. More critically, in a second study, where supra-physiological levels of recombinant GM-CSF were used, we fully inhibited leukocytosis at doses of 574D04 as low as 1 mg kg−1. The increased levels of GM-CSF were above those observed in RA patients. These data therefore demonstrate the potential of 574D04 to fully suppress GM-CSF activity in patients with RA at doses compatible with s.c. delivery. In addition, these findings confirm the theoretical model that we built and the resulting design criteria that we used in the engineering of 574D04.
Recently, the PK and PD results profile of mavrilimumab, the germ-lined derivative of 574D04, from a single ascending dose phase I study in subjects with RA was reported (Burmester et al., 2011). In this human study, it was demonstrated that mavrilimumab at 1 mg kg−1 i.v. significantly inhibited the ex vivo induction of suppressor of cytokine signalling 3 (SOCS3) mRNA by GM-CSF in peripheral blood for up to 2 weeks, suggesting maximal receptor occupancy in man and thus supporting the theoretical model.
In conclusion, we have demonstrated that close interaction between theoretical PK/PD modelling and antibody potency engineering, early in the drug dvelpopment process, resulted in the successful generation of a high-affinity candidate drug, mavrilimumab, suitable for clinical development. The validity of this approach has been borne out in a subsequent clinical study in humans that demonstrated significant inhibition of GM-CSF-mediated effects for up to 2 weeks upon dosing with 1 mg kg−1 mavrilimumab, a result which is well suited to the ultimate goal of patient self-administration. Whilst acknowledging that in this case the theoretical model did not alter the course of action and that further work can be done to find out how broadly this general strategy can be applied, we believe that this integrated approach will be critical for the discovery and development of new biological therapeutic agents.
Acknowledgments
The authors would like to thank Anthony J Coyle for suggestions and encouragement throughout the preparation of this manuscript. Dr P Carter and Prof D Neri for their helpful review of the manuscript prior to submission. We would also like to thank the DNA Chemistry, Tissue Culture and High-Throughput Expression and protein Sciences teams at MedImmune and CSL for their invaluable support of this work.
Glossary
- CDR
complementarity determining region
- ECD
extracellular domain
- IC50
half maximal inhibitory concentration
- Kd
equilibrium dissociation constant
- Kt
internalization rate constant
- PD
pharmacodynamic
- PK
pharmacokinetic
- RA
rheumatoid arthritis
- scFv
single chain variable fragment
- t1/2
half-life
- VH,
variable heavy
- VL,
variable light
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
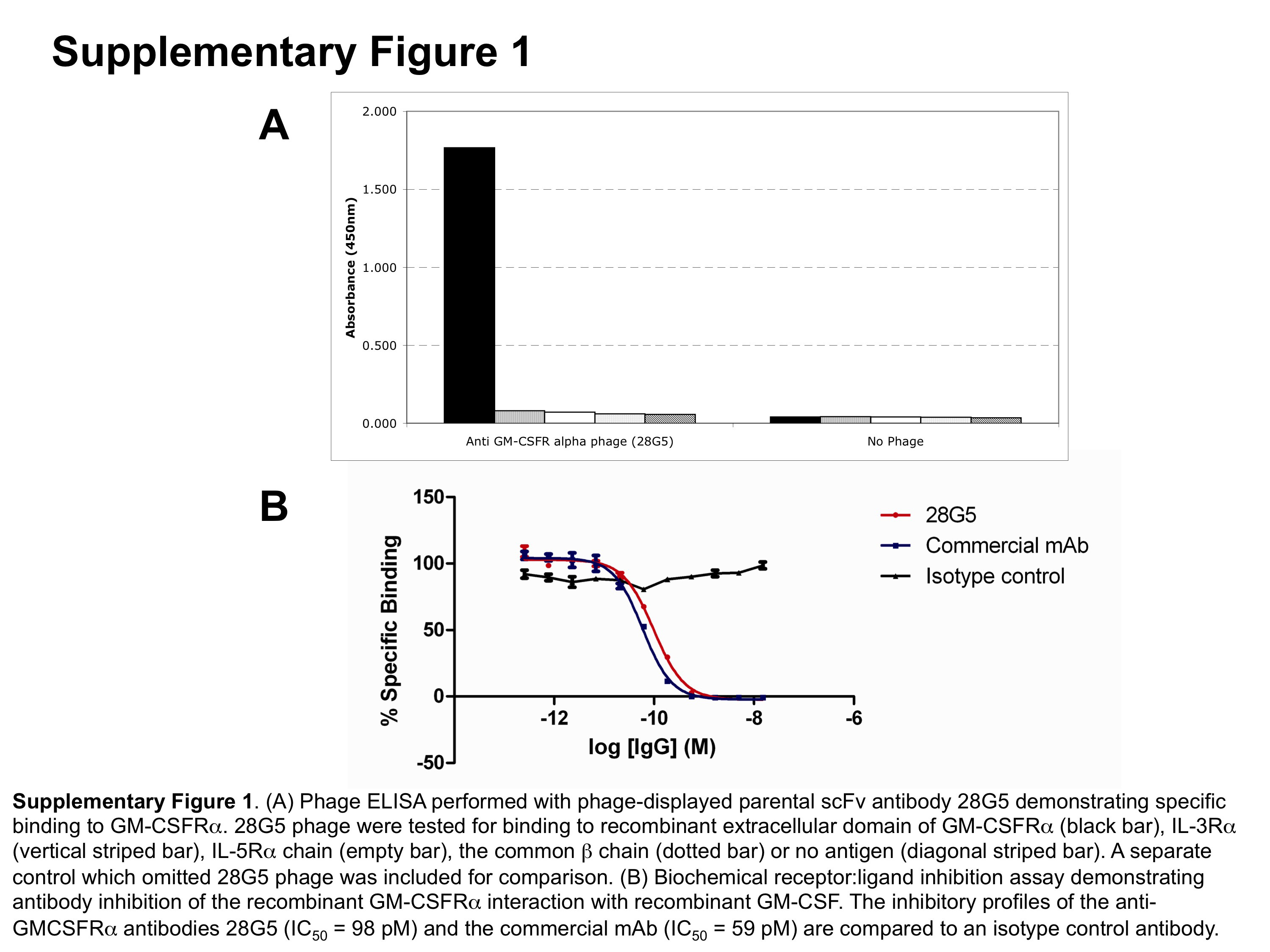
Figure S1 (A) Phage elisa performed with phage-displayed parental scFv antibody 28G5, demonstrating specific binding to GM-CSFRα. 28G5 phage were tested for binding to recombinant extracellular domain of GM-CSFRα, IL-3Rα, IL-5Rα chain, the common β chain or no antigen. A separate control which omitted 28G5 phage was included for comparison. (B) Biochemical receptor : ligand inhibition assay demonstrating antibody inhibition of the recombinant GM-CSFRα interaction with recombinant GM-CSF. The inhibitory profiles of the anti-GMCSFRα antibodies 28G5 (IC50 = 98 pM) and the commercial mAb (IC50 = 59 pM) are compared with an isotype control antibody.
{kind=link}
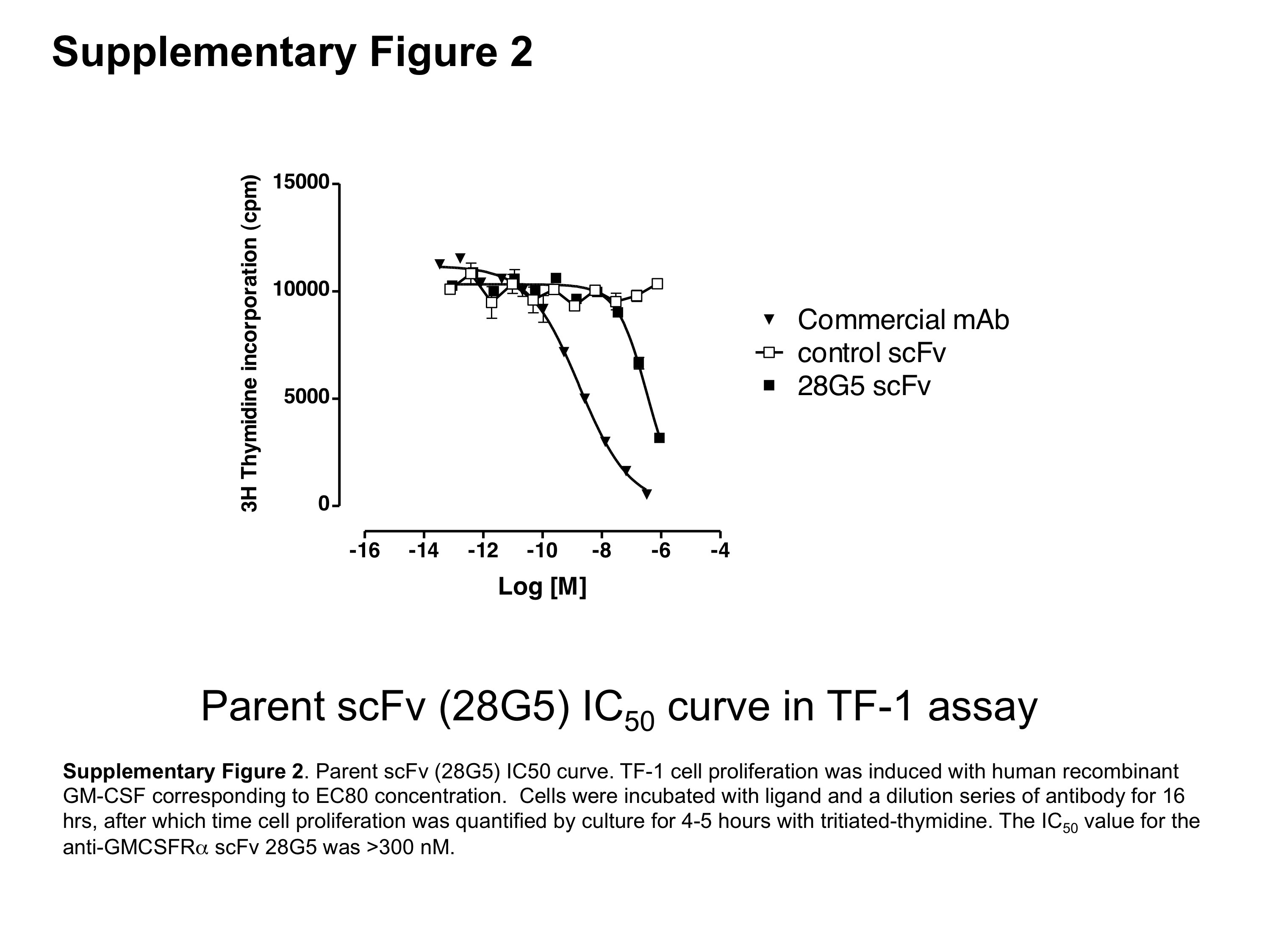
Figure S2 Parent scFv (28G5) IC50 curve. TF-1 cell proliferation was induced with human recombinant GM-CSF corresponding to EC80 concentration. Cells were incubated with ligand and a dilution series of antibody for 16 h, after which time cell proliferation was quantified by culture for 4–5 h with tritiated thymidine. The IC50 value for the anti-GMCSFRα scFv 28G5 was >300 nM.
{kind=link}
Figure S3 Flow cytometry measurement of binding of Alexa647-labelled 574D04 to FD cells transfected with human GM-CSFRα. Flow cytometry traces and geometric mean calculations are shown for Alexa647-labelled 574D04, an Alexa647-labelled lgG4 isotype control and unstained cells. Alexa647-labelled 574D04 shows fluorescence shift as compared with Alexa647-labelled isotype and unstained cells.
{kind=link}
Figure S4 Cytokine production by human PBMCs (mean ± SEM of four donors) in response to 574D04, 10 ng mL–1 human GM-CSF or 5 μg mL–1 PHA. (A) IL-6 production. (B) TNF-α production. (C) The pH dependence of 574D04 binding to GMCSFRα. Binding kinetics was measured by surface plasmon resonance at pH 7.4 and pH 5.7, and the kd was recorded as 0.15 and 13.4 nM, respectively, indicating a 92-fold change in binding affinity over this pH range. Note that the higher binding at pH 5.7 reflects the higher amount of 574D04 IgG captured by the protein-G surface at pH 5.7. Additional controls are 0 nM GMCSFRα at pH 7.4 and 0 nM GMCSFRα at pH 5.7.
{kind=link}
Figure S5 Characterization of 574D04 for cross-reactivity with cynomolgus monkey GM-CSFR. (A) Schild analysis of 574D04 in an assay measuring GM-CSF induced shape change of cynomolgus monkey granulocytes. Cynomolgus peripheral blood granulocytes were stimulated with increasing concentrations of recombinant human GM-CSF (R&D Systems) for 4 h in the presence or absence of a single concentration of 574D04 (0.00025, 0.0025, 0.025, 0.25, 2.5 μg mL–1) to produce a rightward shift in the dose–response curve. Cells were then analysed using forward scatter on a flow cytometer to monitor the change in shape. (B) Schild analysis to determine the pA2 value and thus functional affinity (KD) of 574D04 for the receptor.
{kind=link}
Figure S6 In vivo blockade of GM-CSF-induced responses. Recombinant human GM-CSF (5 μg kg–1) was administered twice daily for three consecutive days to cynomolgus monkeys that had previously received PBS, 1 mg kg–1 or 10 mg kg–1 of 574D04. Complete blood count with differential was monitored at regular intervals. **P < 0.01, ***P < 0.001 versus control group.
{kind=link}
Appendix S1 Supporting methods.
References
- Arunlakshana O, Schild HO. Some quantitative uses of drug antagonists. Br J Pharmacol Chemother. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker KP, Edwards BM, Main SH, Chol GH, Wager RE, Halpern WG, et al. Generation and characterization of LymphoStat-B, a human monoclonal antibody that antagonizes the bioactivities of B lymphocyte stimulator. Arthritis Rheum. 2003;48:3253–3265. doi: 10.1002/art.11299. [DOI] [PubMed] [Google Scholar]
- Barbas CF, Hu D, Dunlop N, Sawyer L, Cababa D, Hendry RM, et al. In vitro evolution of a neutralizing human antibody to human immunodeficiency virus type 1 to enhance affinity and broaden strain cross-reactivity. Proc Natl Acad Sci U S A. 1994;91:3809–3813. doi: 10.1073/pnas.91.9.3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell AL, Magill MK, McKane WR, Kirk F, Irvine AE. Measurement of colony-stimulating factors in synovial fluid: potential clinical value. Rheumatol Int. 1995;14:177–182. doi: 10.1007/BF00262295. [DOI] [PubMed] [Google Scholar]
- Burmester GR, Feist E, Sleeman MA, Wang B, White B, Magrini F. Mavrilimumab, a human monoclonal antibody targeting GM-CSF receptor-alpha, in subjects with rheumatoid arthritis: a randomised, double-blind, placebo-controlled, phase I, first-in-human study. Ann Rheum Dis. 2011;70:1542–1549. doi: 10.1136/ard.2010.146225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook AD, Braine EL, Campbell IK, Rich MJ, Hamilton JA. Blockade of collagen-induced arthritis post-onset by antibody to granulocyte-macrophage colony-stimulating factor (GM-CSF): requirement for GM-CSF in the effector phase of disease. Arthritis Res. 2001;3:293–298. doi: 10.1186/ar318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farahat MN, Yanni G, Poston R, Panayi GS. Cytokine expression in synovial membranes of patients with rheumatoid arthritis and osteoarthritis. Ann Rheum Dis. 1993;52:870–875. doi: 10.1136/ard.52.12.870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annu Rev Immunol. 1996;14:397–440. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- Hawkins RE, Russell SJ, Winter G. Selection of phage antibodies by binding affinity. Mimicking affinity maturation. J Mol Biol. 1992;226:889–896. doi: 10.1016/0022-2836(92)90639-2. [DOI] [PubMed] [Google Scholar]
- Hercus TR, Bagley CJ, Cambareri B, Dottore M, Woodcok JM, Vadas MA, et al. Specific human granulocyte-macrophage colony-stimulating factor antagonists. Proc Natl Acad Sci U S A. 1994;91:5838–5842. doi: 10.1073/pnas.91.13.5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabat EA. Sequences of Proteins of Immunological Interest. 5th edn. Bethesda, MD: U.S. Dept. of Health and Human Services, Public Health Service, National Institutes of Health; 1991. NIH publication; no. 91-3242. [Google Scholar]
- Kenakin TP. The Schild regression in the process of receptor classification. Can J Physiol Pharmacol. 1982;60:249–265. doi: 10.1139/y82-036. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmuir I. The constitution and fundamental properties of solids and liquids. Part I Solids. J Am Chem Soc. 1916;38:2221–2295. [Google Scholar]
- McCafferty J, Fitzgerald KJ, Earnshaw J, Chiswell DJ, Link J, Smith R, et al. Selection and rapid purification of murine antibody fragments that bind a transition-state analog by phage display. Appl Biochem Biotechnol. 1994;47:157–171. doi: 10.1007/BF02787932. discussion 171–173. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C. Guidelines for reportingexperiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf D, Nicola NA, Mifsud S, Di Rago L. Receptor clearance obscures the magnitude of granulocyte-macrophage colony-stimulating factor responses in mice to endotoxin or local infections. Blood. 1999;93:1579–1585. [PubMed] [Google Scholar]
- Mikuls TR, Fay BT, Michaud K, Sayles H, Thiele GM, Caplain L, et al. Associations of disease activity and treatments with mortality in men with rheumatoid arthritis: results from the VARA registry. Rheumatology (Oxford) 2011;50:101–109. doi: 10.1093/rheumatology/keq232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima S, Nagata S. pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res. 1990;18:5322. doi: 10.1093/nar/18.17.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola NA, Peterson L, Hilton DJ, Metcalf D. Cellular processing of murine colony-stimulating factor (Multi-CSF, GM-CSF, G-CSF) receptors by normal hemopoietic cells and cell lines. Growth Factors. 1988;1:41–49. doi: 10.3109/08977198809000245. [DOI] [PubMed] [Google Scholar]
- Ottonello L, Cutolo M, Frumento G, Arduino N, Bertolotto M, Manchini M, et al. Synovial fluid from patients with rheumatoid arthritis inhibits neutrophil apoptosis: role of adenosine and proinflammatory cytokines. Rheumatology (Oxford) 2002;41:1249–1260. doi: 10.1093/rheumatology/41.11.1249. [DOI] [PubMed] [Google Scholar]
- Pedersen MW, Jacobsen HJ, Koefoed K, Hey A, Pyke C, Haurum JS, et al. Sym004: a novel synergistic anti-epidermal growth factor receptor antibody mixture with superior anticancer efficacy. Cancer Res. 2010;70:588–597. doi: 10.1158/0008-5472.CAN-09-1417. [DOI] [PubMed] [Google Scholar]
- Persic L, Roberts A, Wilton J, Cattaneo A, Bradbury A, Hoogenboom HR. An integrated vector system for the eukaryotic expression of antibodies or their fragments after selection from phage display libraries. Gene. 1997;187:9–18. doi: 10.1016/s0378-1119(96)00628-2. [DOI] [PubMed] [Google Scholar]
- Plater-Zyberk C, Joosten LA, Helsen MM, Hepp J, Baeuerle PA, Van Den Berg WB. GM-CSF neutralisation suppresses inflammation and protects cartilage in acute streptococcal cell wall arthritis of mice. Ann Rheum Dis. 2007;66:452–457. doi: 10.1136/ard.2006.057182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskos L, Davis C, Schwab G. The clinical pharmacology of therapeutic monoclonal antibodies. Drug Dev Res. 2004;61:108–120. [Google Scholar]
- Sabroe I, Hartnell A, Jopling LA, Bel S, Ponath PD, Pease JE, et al. Differential regulation of eosinophil chemokine signaling via CCR3 and non-CCR3 pathways. J Immunol. 1999;162:2946–2955. [PubMed] [Google Scholar]
- Schier R, McCall A, Adams GP, Marshall KW, Merritt H, Yim M, et al. Isolation of picomolar affinity anti-c-erbB-2 single-chain Fv by molecular evolution of the complementarity determining regions in the center of the antibody binding site. J Mol Biol. 1996;263:551–567. doi: 10.1006/jmbi.1996.0598. [DOI] [PubMed] [Google Scholar]
- Singh JA, Christensen R, Wells GA, Suarez-Almazor ME, Buchbinder R, Lopez-Olivo MA, et al. Biologics for rheumatoid arthritis: an overview of Cochrane reviews. Cochrane Database Syst Rev. 2009;(4) doi: 10.1002/14651858.CD007848.pub2. CD007848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symmons DP. Epidemiology of rheumatoid arthritis: determinants of onset, persistence and outcome. Best Pract Res Clin Rheumatol. 2002;16:707–722. doi: 10.1053/berh.2002.0257. [DOI] [PubMed] [Google Scholar]
- Taylor PC, Feldmann M. Anti-TNF biologic agents: still the therapy of choice for rheumatoid arthritis. Nat Rev Rheumatol. 2009;5:578–582. doi: 10.1038/nrrheum.2009.181. [DOI] [PubMed] [Google Scholar]
- Thom G, Cockroft AC, Buchanan AG, Candotti CJ, Cohen ES, Lowne D, et al. Probing a protein-protein interaction by in vitro evolution. Proc Natl Acad Sci U S A. 2006;103:7619–7624. doi: 10.1073/pnas.0602341103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J, Pope T, Tung JS, Chan C, Hollis G, Mark G, et al. Affinity maturation of a high-affinity human monoclonal antibody against the third hypervariable loop of human immunodeficiency virus: use of phage display to improve affinity and broaden strain reactivity. J Mol Biol. 1996;256:77–88. doi: 10.1006/jmbi.1996.0069. [DOI] [PubMed] [Google Scholar]
- Vaughan TJ, Williams AJ, Prichard K, Osbourn JK, Pope AR, Earnshaw JC, et al. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat Biotechnol. 1996;14:309–314. doi: 10.1038/nbt0396-309. [DOI] [PubMed] [Google Scholar]
- Walker F, Burgess AW. Internalisation and recycling of the granulocyte-macrophage colony-stimulating factor (GM-CSF) receptor on a murine myelomonocytic leukemia. J Cell Physiol. 1987;130:255–261. doi: 10.1002/jcp.1041300211. [DOI] [PubMed] [Google Scholar]
- Wiley HS, Herbst JJ, Walsh BJ, Lauffenburger DA, Rosenfeld MG, Gill GN, et al. The role of tyrosine kinase activity in endocytosis, compartmentation, and down-regulation of the epidermal growth factor receptor. J Biol Chem. 1991;266:11083–11094. [PubMed] [Google Scholar]
- Yang WP, Green K, Pinz-Sweeney S, Briones AT, Burton DR, Barbas CF., 3rd CDR walking mutagenesis for the affinity maturation of a potent human anti-HIV-1 antibody into the picomolar range. J Mol Biol. 1995;254:392–403. doi: 10.1006/jmbi.1995.0626. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.