

Abstract
Background and Purpose
NADPH oxidases (NOXs) contribute to platelet activation by a largely unknown mechanism. Here, we studied the effect of the novel NOX inhibitor 2-acetylphenothiazine (2-APT) on human platelet functional responses and intracellular signaling pathways.
Experimental Approach
The generation of superoxide ions was assessed by single cell imaging on adhering platelets using dihydroethidium (DHE), while other reactive oxygen species (ROS) were detected with 5-(and-6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2-DCFDA). Whole blood thrombus formation, washed platelet aggregation, integrin αIIbβ3 inside-out signalling, Syk phosphorylation and PKC activation were analysed to understand the functional consequences of NOX inhibition by 2-APT in platelets.
Key Results
Superoxide ion generation stimulated by platelet adhesion on collagen and fibrinogen was significantly inhibited by 2-APT in concentration-dependent manner (IC50 = 306 nM and 227 nM, respectively), whereas cumulative ROS accumulation was not affected by this pharmacological agent. 2-APT also abolished collagen-dependent whole blood thrombus formation and washed platelet aggregation in response to collagen but not thrombin. The activation of integrin αIIbβ3 and PKC in response to the GPVI-specific agonist collagen-related peptide (CRP) was significantly reduced, whereas the same responses to thrombin were not significantly affected by 2-APT. Finally, Syk activation in response to collagen but not thrombin was inhibited by 2-APT.
Conclusions and Implications
Taken together, our results suggest that 2-APT attenuates GPVI-specific signalling and is a novel inhibitor of collagen-induced platelet responses. Therefore, NOXs could represent a novel target for the discovery of anti-thrombotic drugs.
Keywords: platelet, NADPH oxidase, reactive oxygen species, GPVI, 2-acetylphenothiazine, dihydroethidium, aggregation, thrombus formation, adhesion
Introduction
Platelets are small anucleate cells that play a critical role in haemostasis. Recent evidence also suggests their involvement in the stimulation of angiogenesis (Pula et al., 2010) and tissue repair (Nurden et al., 2008). In addition to these critical physiological functions, platelets also participate in the development and progression of life-threatening pathologies, such as thrombosis (Nieswandt et al., 2011) and atherosclerosis (May et al., 2008). The understanding of platelet regulation is therefore a primary objective in cardiovascular science and a potential starting point for the development of novel pharmacological tools.
Besides classical agonist-induced protein phosphorylation pathways (Harper and Poole, 2010; Stegner and Nieswandt, 2011), platelet activation is also regulated by reactive oxygen species (ROS). The dependence of platelet activation on ROS generation is thought to be the basis of platelet inhibition and anti-thrombotic effect of antioxidants (Freedman, 2008). Platelets respond to the extracellular redox state (Iuliano et al., 1997) and generate different ROS upon activation (Krotz et al., 2004). In particular, superoxide ions from exogenous sources or endogenously produced by platelets significantly increased collagen-dependent aggregation and thrombus formation (Krotz et al., 2002). Amongst superoxide ion-generating enzymes, NOX2 (or Gp91phox) and different regulatory subunits of the NOX complex (p22phox, p47phox and p67phox) have been detected in human platelets (Seno et al., 2001; Chlopicki et al., 2004; Pignatelli et al., 2004; Dharmarajah et al., 2010). Pharmacological inhibition of NOXs attenuated platelet aggregation in response to different agonists (Krotz et al., 2002; Begonja et al., 2005; Pignatelli et al., 2006), and the genetic ablation of NOX2 resulted in the reduction of platelet recruitment to the vascular wall in hypercholesterolaemic mice (Stokes et al., 2007). Although the molecular mechanism of platelet regulation by superoxide ions is still largely undefined, several possible pathways have been proposed, including increasing the bioavailability of platelet-derived ADP (Krotz et al., 2002), scavenging of endothelial cell- and platelet-derived NO (Clutton et al., 2004), directly potentiating integrin αIIbβ3 activation (Begonja et al., 2006) or collagen receptor GPVI signalling (Arthur et al., 2007; 2008).
In this study, we investigated the expression of different NOX isoforms and discovered that, besides NOX2, NOX1 was also expressed in human platelets. We therefore investigated the effect of the novel NOX1-selective inhibitor 2-acetylphenothiazine (2-APT) on platelet superoxide anion generation, overall ROS production, thrombus formation, platelet aggregation and intracellular signalling (Gianni et al., 2010). 2-APT significantly inhibited superoxide ion formation but had no effect on overall ROS generation. In parallel, 2-APT significantly inhibited aggregation and thrombus formation in response to collagen and appeared to affect the signalling of the collagen receptor GPVI. The effect of the non-selective NOX inhibitor diphenylene iodonium (DPI) on platelet responses was very similar and not additive to 2-APT. By contrast, apocynin, despite displaying a very similar effect on superoxide ions compared with other inhibitors, also inhibited overall ROS generation and adhesion onto fibrinogen, possibly suggesting off-target effects of this inhibitor in human platelets. Taken together, this study shows that 2-APT is a potent inhibitor of GPVI signalling and collagen-dependent platelet activation. Our results suggest that existing and novel chemical entities inhibiting NOX enzymes might represent novel antithrombotic agents.
Methods
Human whole blood and platelet isolation
Human blood was drawn from healthy volunteers by median cubital vein venepuncture under local ethics committee approval. Sodium citrate was used as anti-coagulant (0.5% w/v for platelet isolation – 0.1% w/v for whole blood experiments). Washed platelets were isolated as previously described (Pula and Poole, 2008). Briefly, platelet rich plasma (PRP) was separated from whole blood by centrifugation (200× g, 15 min), and platelets were separated from PRP by a second centrifugation step (400× g, 10 min), in the presence of prostaglandin E1 (40 ng·mL−1) and indomethacin (10 μM). Platelets were then resuspended in a modified Tyrode's-HEPES buffer (145 mM NaCl, 2.9 mM KCl, 10 mM HEPES, 1 mM MgCl2, 5 mM glucose, pH 7.3) at a density of 4 × 108 cells·mL−1.
Intracellular superoxide ion and ROS formation assay
Glass coverslips were coated with either 0.1 mg·mL−1 fibrinogen or 0.1 mg·mL−1 fibrillar collagen I from equine tendons (Horm collagen). Platelets were diluted to a density of 4 × 107·mL−1 and dispensed over the coverslip in a live cell imaging chamber. After 1 min, that we estimated necessary to permit a sufficient number of platelets to reach the bottom of the imaging chamber, platelets were treated with 10 μM dihydroethidium (DHE) for superoxide ion detection (Robinson et al., 2008) or 20 μM 5-(and-6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2-DCFDA) for overall ROS measurement. Where indicated, NOX inhibitors or ROS scavengers were added to the live cell chamber [10 nM-10 μM 2-APT, 0.5 mM apocynin, 100 μM DPI or 1 mM N-acetylcysteine (NAC)]. Oxidation-dependent dye conversion and fluorescence generation were monitored by confocal imaging immediately after fluoroprobe addition to avoid signal saturation by basal levels of ROS and continued for a period of 10 min (λex 405 nm for DHE and 490 nm for DCFDA). Images were collected every 10 s using a Zeiss 510 LSM confocal microscope equipped with a 40× oil immersion lens and single cell fluorescence was quantified using Zeiss LSM Examiner software (Zeiss, Jena, Germany). Single platelet fluorescence values were utilized to construct response curves (mean ± SEM) and fluorescence values at time point 10 min were used for the statistical analysis.
Thrombus formation assay
The Bioflux200 system (Fluxion, South San Francisco, CA) was used to analyse thrombus formation in human whole blood under flow. Microchannels were coated with 0.1 mg·mL−1 fibrinogen or 0.1 mg·mL−1 collagen I (monomeric collagen from calf skin), before blocking with 1% BSA and washing with modified Tyrode's buffer. PRP was isolated from blood by centrifugation (200× g, 15 min) and incubated for 1 h with Calcein™ (5 μg·mL−1) at 37°C to facilitate thrombus visualisation in whole blood. PRP was then added to the red blood cell fraction to reconstitute whole blood with physiological blood cell density (including platelets), and thrombus formation (on collagen) or single platelet adhesion (on fibrinogen) were visualized by fluorescence microscopy. Where indicated, NOX inhibitors were added to the blood at the indicated concentrations 5 min before the start of the flow assay (0.5 μM 2-APT, 0.5 mM apocynin or 100 μM DPI). Platelet adhesion under flow conditions was tested on collagen I (shear rate = 1000·s−1) and human fibrinogen (shear rate = 200·s−1). Representative pictures were taken at time 10 min and surface area coverage was measured using Bioflux200 Software (version 2.4).
Platelet aggregation assay by suspension turbidimetry
Washed human platelets at physiological density (i.e. 4 × 108·mL−1) were stimulated with 10 μg·mL−1 fibrillar collagen I from equine tendons (Horm collagen) or 0.1 unit·mL−1 human thrombin. The aggregation experiments were performed over 10 min, with agonist addition at time 1 min, and recordings continued for further 9 min. Where indicated, platelets where preincubated with 10 nM–10 μM 2-APT, 0.5 mM apocynin or 100 μM DPI. Aggregation was measured by turbidimetry at 37°C using a 490D aggregometer (Chrono-Log Corporation, Havertown, PA), as previously described (Pula et al., 2005).
Analysis of integrin αIIbβ3 activation by flow cytometry
Washed human platelets (4 × 107·mL−1) were incubated for 5 min with 0.5% v/v FITC-conjugated PAC-1 antibody (#340507, BD Biosciences, Franklin Lakes, NJ). Where indicated, NOX inhibitors were co-incubated with the antibodies. Integrin activation was obtained by 5 min incubation with 5 μg·mL−1 collagen-related peptide (CRP) or 0.1 unit·mL−1 thrombin. Following 1 in 10 dilution in PBS (pH 7.4: 140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4), platelets were analysed by flow cytometry for FITC staining using FACSCantoII (Becton and Dickinson, Oxford, UK); and the results were analysed using FACSDiva software (Becton and Dickinson).
NOX expression, Syk phosphorylation and PKC activity analysis by immunoblot
For NOX expression analysis, washed platelets were lysed with RIPA buffer (1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, 5 mM EDTA, 50 mM Tris pH 7.4) containing protease and phosphatase inhibitors. As previously described (Pula et al., 2006), for PKC activity analysis, washed platelets were treated with 1 mM EGTA immediately before stimulation and activated with 5 μg·mL−1 CRP or 0.1 unit·mL−1 thrombin for 5 min under stirring conditions using a 490D aggregometer (Chrono-Log Corporation). For Syk phosphorylation analysis, washed platelets were stimulated by 10 μg·mL−1 fibrillar collagen I from equine tendons (Horm collagen) or 0.1 unit·mL−1 thrombin in the presence of 1 mM EGTA. In both cases, cell lysis was achieved with mild cell lysis (MCL) buffer [20 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton] containing protease and phosphatase inhibitors. For Syk phosphorylation analysis, lysates were pre-cleared with protein A/G Plus-Agarose for 3 h at 4°C, before overnight incubation with 2 μg·mL−1 anti-Syk antibody at 4°C. Immunocomplexes were precipitated by incubation with protein A/G Plus-Agarose for 2 h at 4°C. For both techniques, proteins were then resolved by SDS-PAGE and immunoblotted using selective antibodies for different NOX enzymes (NOX1, #SAB4200097, Sigma-Aldrich Life Science Poole UK; NOX2, #ab80508, Abcam, Cambridge, UK; NOX4, #ab62148, Abcam, Cambridge, UK; NOX5, #110400, Abcam), phosphorylated Syk (#2710, Cell Signalling Technology, Danvers, US), Syk (#sc51703, Santa Cruz Biotechnology, Santa Cruz, CA), phospho-PKC substrate (#2261, Cell Signalling Technology) or actin (#A3853, Sigma-Aldrich Life Science).
Data analysis
Data are expressed throughout as mean ± SEM and presented using Prism 5 (GraphPad Software, La Jolla, CA). Statistical significance was analysed by t-test (for comparisons of two groups) or one-way anova with Bonferroni post test (for multiple comparisons).
Materials
Research grade reagents for buffer and solution preparation, human thrombin, 2-APT, Apocynin, DPI and NAC were obtained from Sigma (Poole, UK). For platelet aggregation and adhesion, fibrinogen, and collagen I (collagen monomers from calf skin) were obtained from Sigma (Poole, UK), fibrillar collagen I from equine tendons (Horm collagen) was purchased from Axis-Shield (Dundee, UK), and CRP was a gift from Prof A Poole (Physiology and Pharmacology, University of Bristol, UK) and Prof R Farndale (Biochemistry, University of Cambridge, UK). For live platelet imaging, DHE and CM-H2-DCFDA were supplied by Molecular Probes (Life Technologies, Paisley, UK). FITC-conjugated anti-active integrin αIIbβ3 (PAC-1) antibodies for flow cytometry were purchased from Becton-Dickinson. For immunoprecipitation and immunoblot, anti-NOX1/2/4/5 antibodies were from Abcam, anti-phospho-Syk (Tyr525/Tyr526) was obtained from Cell Signaling Technology (New England Biolabs, Hitchin, UK); whereas antibodies against Syk were from Santa Cruz Biotechnology Inc. (Santa Cruz, CA) and Becton-Dickinson. Anti-actin antibodies and phosphatase inhibitors (cocktail I and II) were from Sigma, whereas protease inhibitors (mini-Complete) were from Roche Applied Science (Burgess Hill, UK).
Results
Although five enzymes belong to the NOX family (NOX1, NOX2/gp91phox, NOX3, NOX4 and NOX5), only the expression of NOX2 has been so far tested and reported in human platelets. Here, we used commercially available antibodies to test the expression of NOX1, NOX4 and NOX5, besides NOX2. Protein extracts from HUVECs were used as positive control for these experiments because they have been shown to express NOX1, NOX2, NOX4 and NOX5 (Cai et al., 2003). As shown in Figure 1, our analysis revealed the expression of both NOX1 and NOX2, but not NOX4 and NOX5.
Figure 1.
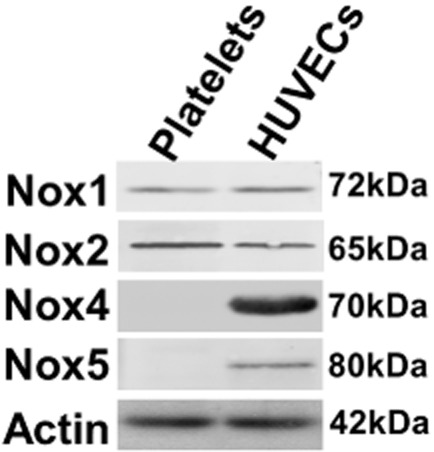
NOX expression in human platelets. Proteins from human platelets were extracted as described in the methods section and separated by SDS/PAGE (20 μg·lane−1). HUVEC extracts were used as positive control for NOX1, NOX2, NOX4 and NOX5. The immunoblotting was performed with anti-NOX1, anti-NOX2, anti-NOX4 or anti-NOX5 antibodies (1 μg·mL−1) and a HRP-conjugated anti-rabbit IgG secondary antibodies. The membranes were also immunoblotted using an anti-actin antibody (1 μg·mL−1) and an HRP-conjugated anti-mouse IgG secondary antibodies to confirm equal lane loading. Enhanced chemiluminescence (ECL) and Hyperfilm™ were used to detect specific immunobands. The results are representative of three independent experiments.
Therefore, besides the non-selective NOX inhibitors apocynin and DPI, we tested the novel inhibitor 2-APT, which has recently been shown to selectively inhibit NOX1 with submicromolar potency (IC50 = 250 nM), while effective on NOX2 only at considerably higher concentrations (IC50 = 5 μM) (Gianni et al., 2010). Initially, intracellular generation of superoxide ion was selectively analysed by single cell fluorescence using DHE at λex 405 nm (Robinson et al., 2008). Platelet adhesion onto fibrillar collagen I or fibrinogen was accompanied by sharp increase in superoxide ion generation with maximal fluorescence reached between 3 and 5 min from platelet adhesion (Figures 2A and 3A respectively). The addition of 0.5 μM 2-APT induced a rapid inhibition of superoxide ion generation occurring in platelets adhering to collagen or fibrinogen (Figures 2B and 3B, respectively). The inhibition of superoxide ion generation by 2-APT was concentration-dependent, with an IC50 of 306 nM and 227 nM on fibrillar collagen I- and fibrinogen-adhering platelets respectively (Figures 2C and 3C). Similarly, apocynin and DPI inhibited superoxide ion generation upon platelet adhesion to collagen and fibrinogen (Figures 2D and 3D respectively). Interestingly the portion of superoxide ion generation that was not inhibited by 2-APT also was resistant to non-selective NOX inhibitors, as shown in experiments in which platelets were co-incubated with 2-APT and either apocynin or DPI (Figures 2D and 3D respectively).
Figure 2.
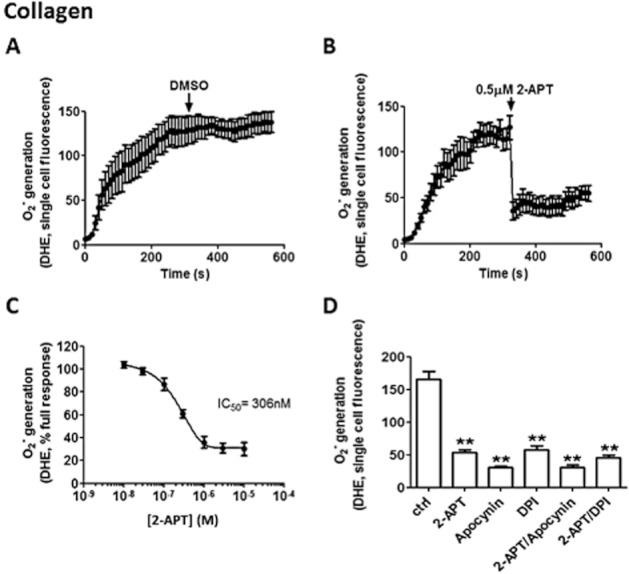
Inhibition of superoxide ion generation by 2-APT in human platelets adhering to collagen. Intracellular superoxide ion generation was tested by DHE-dependent single cell fluorescence imaging of platelets dispensed over collagen-coated glass. DMSO (0.1% v/v) or 2-APT (0.5 μM) was added at the 5 min time point, for which representative time courses are shown in panels A and B respectively. Following treatment with different concentrations of 2-APT (10 nM to 10 μM), the superoxide generation in collagen-adhering platelets was measured and expressed as % of the response in the absence of the inhibitor. The results from three independent experiments were plotted using a variable slope sigmoidal dose–response algorithm, resulting in an IC50 value of 306 nM (C). Non-selective NOX inhibitors apocynin (0.5 mM) and DPI (100 μM) were tested alone or in addition to 2-APT (0.5 μM), and mean (± SEM) single cell fluorescence counts at time point 10 min are shown in panel D. **P < 0.01; one-way anova with Bonferroni post test; n = 6.
Figure 3.

Inhibition of superoxide ion generation by 2-APT in human platelets adhering to fibrinogen. Intracellular superoxide ion generation was tested by DHE-dependent single cell fluorescence imaging of platelets dispensed over fibrinogen-coated glass. DMSO (0.1% v/v) or 2-APT (0.5 μM) were added at the 5 min time point, for which representative time courses are shown in panels A and B respectively. Following treatment with different concentrations of 2-APT (10 nM to 10 μM), the superoxide generation in fibrinogen-adhering platelets was measured and expressed as % of the response in the absence of the inhibitor. The results from three independent experiments were plotted using a variable slope sigmoidal dose–response algorithm, resulting in an IC50 value of 227 nM (C). Non-selective NOX inhibitors apocynin (0.5 mM) and DPI (100 μM) were tested alone or in addition to 2-APT (0.5 μM), and mean (± SEM) single cell fluorescence counts at time point 10 min are shown in panel D. **P < 0.01; one-way anova with Bonferroni post test; n = 6.
Next, we analysed platelet intracellular redox state with CM-H2-DCFDA, which is susceptible to oxidation by and can detect a variety of ROS, including peroxides and hydroxyl radicals. Although slower than the accumulation of intracellular superoxide ions measured with DHE, the increase in intracellular ROS was detected in both fibrillar collagen I- and fibrinogen-adhering platelets (Figure 4, respectively, maximal fluorescence at 5–7 min after adhesion). This response was not inhibited by 2-APT (Figure 4Ai and Bi) or DPI (data not shown), and only apocynin partially reduced ROS accumulation measured by CM-H2-DCFDA (Figure 4Aii and Bii). In contrast, when the ROS scavenger NAC was added to the adhering platelets, the fluorescence increase was completely blocked (Figure 4Aii and Bii). The levels of single cell fluorescence after 10 min in the presence of 2-APT, apocynin, or DPI were compared by one-way anova with Bonferroni post test and, although partial, only the inhibition by apocynin was statistically significant (Figure 4Aiii and 4Biii).
Figure 4.
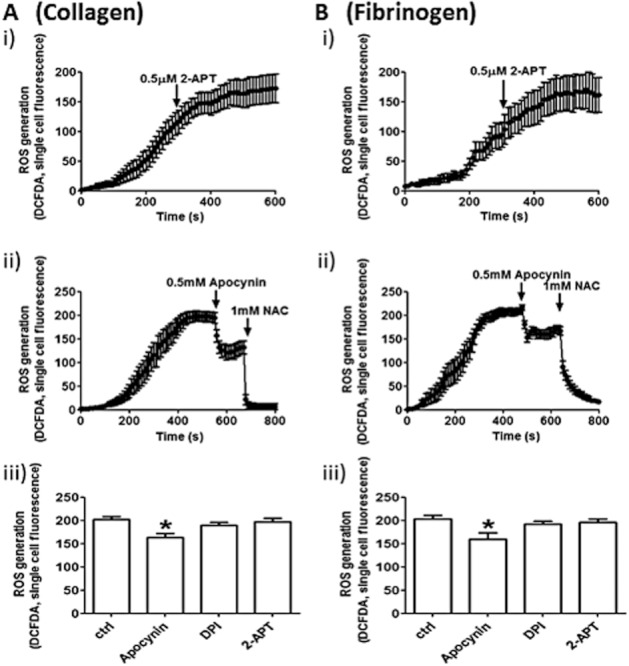
2-APT does not inhibit the overall ROS generation in platelets adhering to collagen or fibrinogen. Human platelets adhering to collagen (A) or fibrinogen (B) were tested for ROS generation using the non-selective redox probe CM-H2-DCFDA. 2-APT (0.5 μM) was added at the 5 min time point, for which representative time courses are shown in panel (i). Representative time courses obtained by adding the non-selective NOX inhibitor apocynin (0.5 mM, t = 500 s) and the ROS scavenger NAC (1 mM, t = 700 s) are shown in panel (ii). The effect of 0.5 μM 2-APT, 0.5 mM apocynin or 100 μM DPI on ROS accumulation was measured as single cell fluorescence counts at 10 min. Data shown in panel (iii) are means (± SEM); n = 5. *P < 0.05; one-way anova with Bonferroni post test.
In order to understand the functional role of superoxide generation and assess the consequence of NOX inhibition by 2-APT on platelet activation, we tested thrombus formation in human whole blood. As previously described (Gutierrez et al., 2008; Konopatskaya et al., 2009), we utilized low shear stress on fibrinogen (200·s−1) and high shear stress on collagen I (1000·s−1), which led to single platelet adhesion on the former and large multicellular complex (thrombus) formation on the latter. Whole blood treatment with 2-APT, apocynin or DPI resulted in the near-complete prevention of thrombus formation on collagen [as shown in Figure 5A, with quantitative analysis in (i) and representative examples in (ii)], whereas platelet adhesion on fibrinogen was partially but significantly inhibited only by apocynin but not 2-APT or DPI [as shown in Figure 5B, with quantitative analysis in (i) and representative examples in (ii)].
Figure 5.
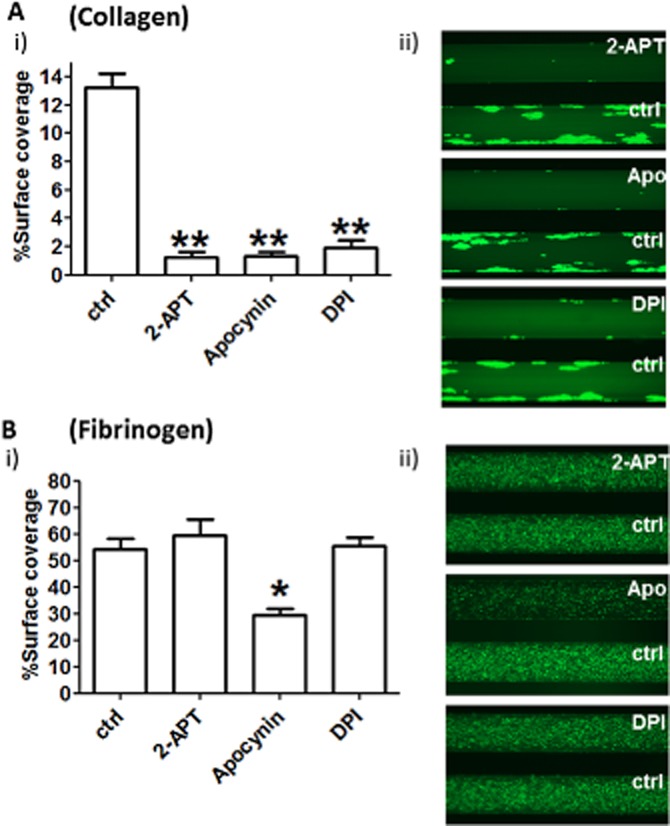
2-APT inhibits thrombus formation on collagen but does not affect platelet adhesion to fibrinogen. Human PRP was isolated from anti-coagulated blood and incubated for 1 h with Calcein™ (5 μg·mL−1). After reconstitution of whole blood by mixing labelled PRP and the red blood cell fraction, thrombus formation on collagen (A) or platelet adhesion on fibrinogen (B) (shear stress 1000·s−1 and 200·s−1, respectively) were tested in the presence of 0.1% v/v DMSO (ctrl), 0.5 μM 2-APT, 0.5 mM apocynin or 100 μM DPI for 10 min. Values of % surface area coverage (mean ± SEM, n = 5) are presented in panel (i), whereas representative pictures of the microchannels after 10 min of flow are shown in panel (ii). Images are representative of five independent experiments. *P < 0.05, **P < 0.01; one-way anova with Bonferroni post test.
Next, platelet aggregation was studied in a turbidimetric assay. Washed platelet aggregation was obtained with either fibrillar collagen I, which activates platelets via receptor GPVI and integrin α2β1 (Stegner and Nieswandt, 2011), or thrombin, a critical physiological agonist, which activates platelet through a signalling pathway distinct from GPVI and initiated by the activation of protease-activated receptors 1 and 4 (Holinstat et al., 2006). As shown by representative aggregation time courses (Figure 6Ai and Bi) and quantitative analysis of five independent experiments (Figure 6Aii and Bii), 2-APT significantly impaired aggregation in response to collagen (Figure 6A), but not thrombin (Figure 6B). Similarly to 2-APT, DPI only inhibited collagen-induced platelet aggregation, while it did not affect thrombin-induced aggregation. On the other hand, apocynin significantly reduced the aggregation in response to both collagen and thrombin.
Figure 6.
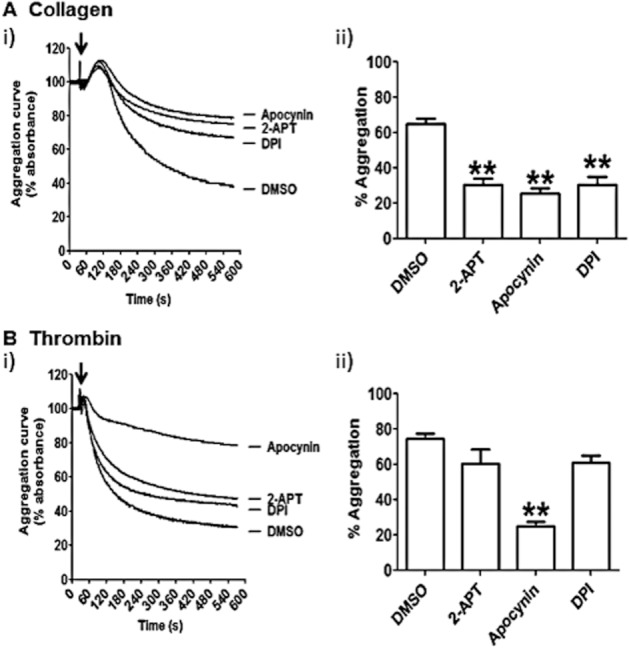
2-APT inhibits collagen-dependent aggregation. Washed platelets were pre-incubated for 5 min with vehicle solution (0.1% v/v DMSO), 0.5 μM 2-APT, 0.5 mM apocynin or 100 μM DPI. Aggregation was measured after stimulation (indicated on the graphs by arrows) by 10 μg·mL−1 fibrillar collagen I (A) or 0.1 unit·mL−1 thrombin (B) and monitored for 9 min by turbidimetry. Representative aggregation traces in the presence of the inhibitors are shown in panel (i). Platelet aggregation was quantified as % decrease in platelet suspension absorbance (means ± SEM) at the final time point as shown in panel (ii). **P < 0.01; one-way anova with Bonferroni post test; n = 5.
In order to understand the mechanism of action of 2-APT and the role of NOX-dependent superoxide ions in platelet activation, we used flow cytometry to test the effect of all three NOX inhibitors on the activation of integrin αIIbβ3, which is the main platelet aggregation receptor. Platelets were stimulated by CRP, which selectively activates GPVI but as distinct from collagen, it does not bind to integrin α2β1 and does not interfere with the flow cytometry assay, or thrombin (Pula and Poole, 2008). All three NOX inhibitors significantly reduced the levels of integrin αIIbβ3 activation in response to CRP (Figure 7Ai), whereas only apocynin inhibited integrin activation in response to thrombin (Figure 7Aii).
Figure 7.
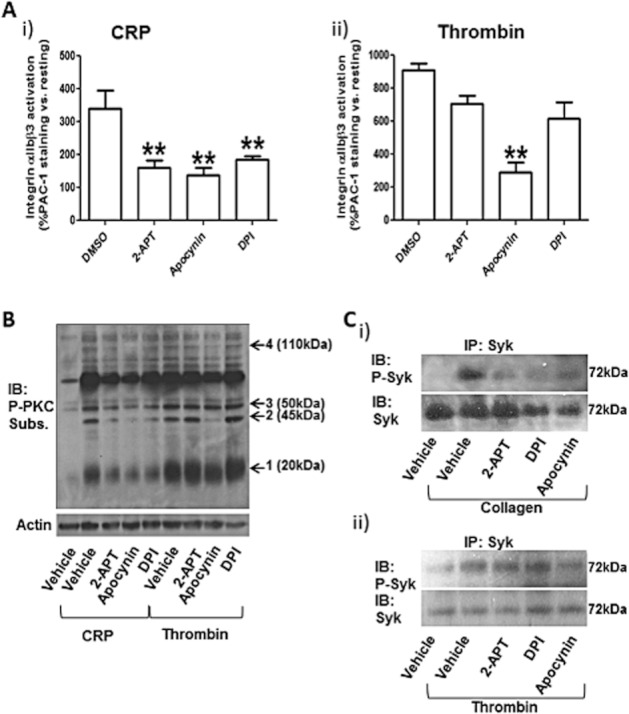
2-APT inhibits GPVI signalling. Integrin αIIbβ3 activation in response to 5 μg·mL−1 CRP (i) or 0.1 unit·mL−1 thrombin (ii) was monitored by flow cytometry using an activation-dependent FITC-conjugated antibody (PAC1) (A). Values are % fold increase over resting platelet (no stimulation) and are means ± SEM (n = 5). **P < 0.01; one-way anova with Bonferroni post test; n = 6 In (B), classical PKC isoform (cPKC) activation was tested by phospho-specific immunoblotting. cPKC-specific phosphorylation profiles showing the number and levels of proteins phosphorylated by these kinases are shown. Platelet activation was obtained by either CRP (5 μg·mL−1) or thrombin (0.1 unit·mL−1) following preincubation for 5 min with vehicle solution (0.1% v/v DMSO), 0.5 μM 2-APT, 0.5 mM apocynin or 100 μM DPI. Arrows numbered 1 to 4 indicate the bands analysed by densitometry in Supporting Information Figure S1. The results are representative of six independent experiments. In (C), Syk activation was also assessed by phospho-specific immunoblotting. Following treatment with NOX inhibitors at the same concentrations indicated above for 5 min, platelet stimulation was obtained with 10 μg·mL−1 collagen (i) or 0.1 unit·mL−1 thrombin (ii) in the presence of 1 mM EGTA for 5 min in continuous stirring (700 r.p.m.). Platelet lysis was performed in MCL buffer, and Syk was immunoprecipitated with 2 μg·mL−1 anti-Syk antibody in the presence of Protein A/G Plus agarose. The immunoprecipitates were resolved by SDS-PAGE and immunostained with anti-phospho-Syk (Tyr525/526) and anti-Syk antibodies. The results are representative of five independent experiments. IB, immunoblotting; IP, immunoprecipitation; Subs., substrate.
Next, we analysed the effect of 2-APT and the non-selective NOX inhibitors on the CRP- and thrombin-dependent activation of PKC, a key player in the signalling of platelet activation (Harper and Poole, 2010). We utilized a phospho-PKC substrate antibody that selectively recognises cellular proteins when phosphorylated at serine residues surrounded by Arg or Lys at the −2 and +2 positions and a hydrophobic residue at the +1 position. This consensus sequences has been shown to be phosphorylated by classical PKC isoforms (cPKCs), amongst which PKCα plays a particularly important role in platelet activation (Konopatskaya et al., 2009). Whole platelet lysates were analysed by immunoblot, resulting in the detection of a cPKC phosphorylation profile for different experimental conditions (i.e. number and intensity of the bands are proportional to the levels of cPKC activation). As shown in Figure 7B (and confirmed by densitometry in Supporting Information Figure S1), stimulation by CRP (lane 2) or thrombin (lane 6) led to a significant activation of cPKCs compared to the resting condition (lane 1). Interestingly, all three NOX inhibitors (lanes 3–5) significantly attenuated cPKC activation in response to CRP, whereas only apocynin (lane 8) showed some inhibitory effect on thrombin-dependent cPKC activation.
In order to confirm the role of NOXs in collagen signalling, we tested collagen- and thrombin-dependent activation of Syk, a key protein kinase for the early stages of GPVI signalling (Getz et al., 2011). As the activation of Syk is accompanied by phosphorylation of Tyr525/526, we utilized phosphorylation-specific antibodies to detect the active state of this kinase after its immunoprecipitation (Pula et al., 2005). Interestingly, collagen- but not thrombin-dependent phosphorylation of Syk was significantly attenuated by all three NOX inhibitors, as shown in Figure 7Ci and Cii, respectively, and in the densitometric analysis shown in Supporting Information Figure S2.
Discussion
In this study, we have demonstrated that 2-APT inhibits collagen-dependent platelet aggregation and thrombus formation at concentrations known to be selectively effective on NOX1 in a recombinant human cell line (Gianni et al., 2010). Although platelet expression of NOX2 (or Gp91phox) and different regulatory subunits of the NOX complex (p22phox, p47phox, and p67phox) has been described (Seno et al., 2001; Chlopicki et al., 2004; Pignatelli et al., 2004; Dharmarajah et al., 2010), the expression of NOX1 had not been reported before this study. In agreement with our pharmacological results, for the first time, we demonstrated that NOX1 is expressed in human platelets at levels comparable with those in endothelial cells (HUVECs), which have previously been shown to express this enzyme (Dworakowski et al., 2008). In our experiments, submicromolar concentrations of 2-APT (as opposed to high micromolar concentrations for apocynin and DPI) impaired collagen-dependent functional responses (aggregation and thrombus formation) and significantly inhibited the formation of superoxide ions, which therefore appear to play a key role in platelet activation. Although we cannot reach any conclusion on which NOX enzyme between NOX1 and NOX2 plays the most significant role in platelet activation at this stage, we provide novel and intriguing data suggesting that NOX activity plays a significant role in the activation-dependent generation of superoxide ions and the collagen-dependent activation of platelets via the GPVI receptor. In view of the recent development of novel and selective NOX inhibitors (Drummond et al., 2011), our observations open new possibilities for the development of novel antithrombotic treatments that target NOX1.
We also tested the effect of the two structurally unrelated non-selective NOX inhibitors apocynin and DPI on human platelet functional responses and signalling. All three tested inhibitors abolished superoxide ion generation, suggesting that they effectively inhibited NOX activity. Moreover, all three inhibitors significantly impaired signalling and platelet functional responses induced by collagen, suggesting a key role for NOXs in collagen-dependent platelet activation. Original observations of a potentiatory effect of superoxide ions on platelet activation are over three decades old (Handin et al., 1977). More recently, superoxide ions have been suggested to stimulate platelet hyperactivity in anoxia/reoxygenation conditions (Leo et al., 1997) and hypercholesterolemia (Stokes et al., 2007). Although the regulation of platelet activation by superoxide ions and other ROS is largely accepted, the molecular mechanisms underlying this phenomenon remain elusive. Initial observations of ROS generation upon platelet activation were conducted by extracellular measurements (Iuliano et al., 1997), whereas only relatively recently it has become possible to collect information on the intracellular generation of ROS in platelets (Begonja et al., 2005). NOXs are associated with intracellular or plasma membranes and depending on their localisation they can generate intracellular or extracellular superoxide ions (Drummond et al., 2011). There are discrepancies in published reports on whether NOX activation in human platelets leads to intracellular or extracellular superoxide ion generation. In some studies, thrombin stimulated intracellular but not extracellular NOX-dependent ROS generation (Krotz et al., 2002; Begonja et al., 2005), whereas in other studies, thrombin stimulated extracellular NOX-dependent ROS generation (Chlopicki et al., 2004). Our experiments with luminol and platelet supernatants appear to be in agreement with the latter position (Supporting Information Figure S3). Similarly, collagen induced extracellular NOX-dependent ROS formation in certain reports (Krotz et al., 2002; Chlopicki et al., 2004), whereas it induced intracellular NOX-dependent ROS formation in other reports (Begonja et al., 2005; Bakdash and Williams, 2008). Although these discrepancies are difficult to explain, some of the problems in interpreting these reports derive from the use of different probes in different studies (e.g. DCFDA, L-012, luminol or lucigenin) and from the fact that the experimental systems in these studies were unable to selectively detect superoxide anions.
In this study, we utilized DHE at an absorbance wavelength (405 nm) that has been shown to selectively detect intracellular superoxide anions (Robinson et al., 2006; 2008). Therefore, we were able to selectively detect a rapid intracellular generation and accumulation of superoxide anions in response to platelet adhesion to either collagen or fibrinogen. This response was significantly impaired by three structurally unrelated NOX inhibitors, which suggested that NOXs are the main source of superoxide anions in activated human platelets. In addition, our experiments with CM-H2-DCFDA demonstrated that platelet adhesion to collagen or fibrinogen also stimulated the intracellular accumulation of ROS, which, in agreement with previous studies, was completely inhibited by the addition of the ROS scavenger NAC (Krotz et al., 2002). Although there are no previous reports of ROS generation in platelets adhering to fibrinogen, the intracellular ROS generation that we observed on collagen has already been reported (Begonja et al., 2005; Bakdash and Williams, 2008). In contrast to previous studies (Begonja et al., 2005), apocynin only partially inhibited collagen-dependent ROS accumulation in our experiments, whereas the other non-selective NOX inhibitor DPI did not inhibit ROS accumulation. This conflict may be due to the differences in the protocols used in the two studies. Our experiments were performed on platelets adhering to adsorbed substrates, whereas Begonja and colleagues performed their experiments in suspension. As a consequence, shear stress did not affect ROS generation in our experiments, whereas the effect of adhesion-dependent cytoskeletal rearrangements and receptor clustering on intracellular signalling and ROS generation were underestimated in Begonja's study. Importantly and similarly to DPI, 2-APT, which effectively inhibited superoxide ion formation in our DHE experiments, did not inhibit intracellular ROS generation in the CM-H2-DCFDA experiments. Taken together, the data presented here suggest that NOX-dependent superoxide anion generation was not the main source of intracellular ROS in platelets (at least in our experimental conditions). In fact, alternative sources of ROS have previously been suggested for human platelets, such as xanthine oxidase, endothelial NOS (eNOS), PLA2 and COXs (Caccese et al., 2000; Freedman et al., 2000; Wachowicz et al., 2002). Another conclusion suggested by our experiments is that the inhibition of ROS accumulation by apocynin in adhering platelets can be due to the off-target antioxidant effects of this inhibitor, which were recently reported (Heumuller et al., 2008; Dharmarajah et al., 2010). In this respect, our data suggest that the use of apocynin in platelet studies can be problematic and should always be accompanied by other NOX inhibitors.
The functional consequences of NOX inhibition were investigated in a whole blood thrombus formation assay under flow conditions. Our results showed that the non-selective NOX inhibitors apocynin and DPI significantly reduced thrombus formation on collagen I. In addition, we also showed that 2-APT had a similar effect and strongly inhibited thrombus formation on collagen-coated surfaces. These observations suggest that collagen-dependent platelet adhesion is positively regulated by NOX-dependent superoxide ion generation, which was suggested in previous studies (Begonja et al., 2005). Platelet adhesion on fibrinogen-coated surfaces was also tested. A lower shear rate (200·s−1) was chosen because of the low resistance to tensile stress of the interaction between fibrinogen and integrin αIIbβ3 on the surface of platelets (Savage et al., 1996). As previously reported, whole blood flow in these conditions led to single platelet adhesion rather than thrombus formation (Gutierrez et al., 2008). DPI and 2-APT did not affect the rate of platelet adhesion, whereas apocynin consistently but only partially inhibited platelet adhesion. It is difficult to interpret these data and there is no previous published study on NOX inhibition of platelet adhesion on fibrinogen under flow. Nonetheless, in view of the efficacy of 2-APT and DPI at inhibiting superoxide ion generation and their complete lack of effect on platelet adhesion on fibrinogen, NOX enzymes do not seem to play a relevant role in platelet adhesion to this substrate. Similarly to our conclusion regarding CM-H2-DCFDA experiments, the apocynin-dependent inhibition of platelet adhesion on fibrinogen is likely to depend on off-target effects of this inhibitor rather than genuine NOX inhibition, because it was not observed with the other NOX inhibitors tested. The limited specificity of apocynin in platelet studies has been reported (Heumuller et al., 2008; Dharmarajah et al., 2010).
Besides adhesion experiments, collagen was also used for the stimulation of platelet aggregation in suspension. In accordance to our results in the thrombus formation assay, NOX-dependent superoxide formation appeared to be critical for collagen-dependent platelet aggregation, as all three NOX inhibitors significantly reduced platelet aggregation in response to this agonist. Similar observations obtained with non-selective NOX inhibitors have been published before this study (Krotz et al., 2002; Chlopicki et al., 2004). Interestingly, also for platelet aggregation apocynin displayed different properties compared to 2-APT and DPI and was the only inhibitor able to inhibit thrombin-induced aggregation. This observation further suggests that apocynin has significant off-target effect on platelets compared with 2-APT and DPI.
Integrin αIIbβ3 and cPKC activation in response to CRP or thrombin and Syk phosphorylation in response to collagen or thrombin were investigated to understand the molecular mechanisms underlying platelet inhibition by 2-APT and to understand the role of NOX activity in platelet regulation. Integrin αIIbβ3 activation was analysed by flow cytometry using an activation-dependent antibody as previously described (Pula et al., 2006), whereas GPVI signalling was assessed by Syk immunoprecipitation followed by phospho-specific immunoblotting (Pula et al., 2005). Integrin αIIbβ3 activation in response to collagen was significantly reduced by all NOX inhibitors (including 2-APT). As GPVI-dependent activation of integrin αIIbβ3 and its consequent binding to fibrinogen are critical steps for platelet aggregation in response to different agonists including collagen (Stegner and Nieswandt, 2011), integrin inhibition by 2APT (and non-selective NOX inhibitors) explain the impairment of collagen-dependent platelet aggregation in our experiments. Interestingly, only apocynin but not 2-APT or DPI inhibited thrombin-induced integrin αIIbβ3 activation, suggesting that NOX activity is not necessary for integrin activation in response to agonists other than collagen. These results are partially in contrast with data previously published by other investigators (Begonja et al., 2005), which showed inhibition of integrin αIIbβ3 activation in response to thrombin by both apocynin and DPI. Despite some minor differences in the experimental conditions used in the two studies (i.e. thrombin concentration, stimulation and buffer composition), this discrepancy remains difficult to explain.
Our results for cPKC activity utilized the GPVI-specific agonist CRP as a stimulus and confirmed that GPVI-dependent signalling depends on NOXs, whereas thrombin signalling is only inhibited by apocynin, possibly as a consequence of off-target effects. Considering the importance of cPKCs in the activation of platelets (Konopatskaya et al., 2009; Harper and Poole, 2010), we believe that these findings are particularly relevant for our understanding of how NOX inhibition by 2-APT affects collagen-dependent platelet activation. Moreover, previous observations of a direct role of cPKCs in the activation of integrin αIIbβ3 (Cifuni et al., 2008) are likely to link our results on integrin αIIbβ3 and cPKC activation (i.e. collagen-dependent cPKC activation is reduced by 2-APT, which results in a decreased integrin αIIbβ3 activation). Although the direct dependence of integrin αIIbβ3 activation on ROS generation has been proposed (Irani et al., 1998; Krotz et al., 2004), our results on platelet adhesion on fibrinogen and thrombin-dependent integrin αIIbβ3 (neither of which were inhibited by 2-APT and DPI) suggest that in human platelets, NOX inhibitors affect integrin αIIbβ3 activation indirectly and only as a consequence of the attenuation of collagen-induced signalling. In view of our results showing attenuation of collagen-dependent Syk activation (a protein kinase critically involved in the early events downstream of GPVI activation) and a previous report that GPVI activation depended on disulphide-dependent dimerization (Arthur et al., 2007), the most likely molecular mechanism underlying the inhibition of collagen-induced responses of platelets by 2-APT is the decrease of superoxide anions available for the catalysis of disulphide bridge formation and consequent dimerization of GPVI receptors (Arthur et al., 2008). Notably, although previous studies reported the dependence of the other major platelet collagen receptor, integrin α2β1, on ROS (Lahav et al., 2003), the use of the GPVI-specific agonist CRP for our integrin αIIbβ3 and cPKC activation experiments confirmed that, in our experimental conditions, NOX inhibition directly affected GPVI signalling.
In summary, this study describes 2-APT as a novel and potent inhibitor of collagen-dependent platelet responses. The direct inhibition of the early events in the signalling cascade of the collagen receptor GPVI is likely to be responsible for the effects of 2-APT on platelets. We also demonstrated for the first time the expression of NOX1 in human platelets. Although we cannot reach any definitive conclusion on whether 2-APT affects platelet activity by inhibiting NOX1 or NOX2, our data suggest that NOXs are important regulatory enzymes in human platelets. In view of the importance of GPVI activation and signalling in the development of cerebral and heart thrombosis (Ollikainen et al., 2004; Kleinschnitz et al., 2007) and the potential of GPVI as a target for the development of antithrombotic drugs with lower risk of haemorrhagic side effects (Mangin et al., 2012), this study might indicate a novel direction for cardiovascular drug discovery.
Acknowledgments
The authors thank the BBSRC and the Royal Society for funding this research. They would also like to thank Dr Y-M Chang (Royal Veterinary College London) for helping with the statistical analysis of the results, and Prof A Poole (Physiology and Pharmacology, University of Bristol, UK) and Prof R Farndale (Biochemistry, University of Cambridge, UK) for donating the CRP used for some of the experiments.
Glossary
- 2-APT
2-acetylphenothiazine
- CRP
collagen-related peptide
- DCFDA
5-(and-6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate
- DHE
dihydroethidium
- DPI
diphenylene iodonium
- eNOS
endothelial NOS
- NAC
N-acetylcysteine
- NOX
NADPH oxidase
- PRP
platelet-rich plasma
- ROS
reactive oxygen species
Conflicts of interest
The authors state no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Densitometric analysis of phospho-PKC substrate immunoblotting of platelets treated with NOX inhibitors and stimulated with either 5 μg·mL−1 CRP or 0.1 unit·mL−1 thrombin. Whole platelet lysates were separated by SDS-PAGE and immunoblotted with an antibody recognizing proteins phosphorylated by classical PKC isoforms within the consensus Arg/Lys-X-Ser-φ-Arg/Lys (with φ = hydrophobic amino acid). Densitometry was performed on five bands indicated in Figure 7B using ImageJ 1.45s (W Rasband, NIH, USA). Data are expressed as arbitrary intensity units and statistical analysis was performed using one-way anova with Bonferroni post test (n = 6).
Figure S2 Densitometric analysis of Syk immunoprecipitation and phospho-specific immunoblotting. Platelets treated with NOX inhibitors or vehicle solution (0.1% v/v DMSO) and stimulated with 10 μg·mL−1 fibrillar collagen I from equine tendons (i) or 0.1 unit·mL−1 thrombin (ii). Following SYK immunoprecipitation the immunoprecipitates were separated by SDS-PAGE and immunoblotted with an antibody recognizing SYK in its phosphorylated at Tyr525/526 residues. Densitometry was performed using ImageJ 1.45s (W Rasband, NIH, USA). Data are expressed as arbitrary intensity units and statistical analysis was performed using one-way anova with Bonferroni post test (n = 5).
Figure S3 Extracellular ROS generation measured by luminol-based luminometry. Platelets were dispensed over collagen- or fibrinogen-coated coverslips and allowed to adhere for 15 min. Supernatants were then collected and 25 μM luminol was added. Following incubation for 15 min at 37°C, samples were analysed by visible light luminometry using a plate reader (Fluostar Optima, BMG Labtech, Ortenberg/Germany). Data are representative of four independent experiments and were analysed by one-way anova with Bonferroni post test (*P < 0.05).
References
- Arthur JF, Shen Y, Kahn ML, Berndt MC, Andrews RK, Gardiner EE. Ligand binding rapidly induces disulfide-dependent dimerization of glycoprotein VI on the platelet plasma membrane. J Biol Chem. 2007;282:30434–30441. doi: 10.1074/jbc.M701330200. [DOI] [PubMed] [Google Scholar]
- Arthur JF, Gardiner EE, Kenny D, Andrews RK, Berndt MC. Platelet receptor redox regulation. Platelets. 2008;19:1–8. doi: 10.1080/09537100701817224. [DOI] [PubMed] [Google Scholar]
- Bakdash N, Williams MS. Spatially distinct production of reactive oxygen species regulates platelet activation. Free Radic Biol Med. 2008;45:158–166. doi: 10.1016/j.freeradbiomed.2008.03.021. [DOI] [PubMed] [Google Scholar]
- Begonja AJ, Gambaryan S, Geiger J, Aktas B, Pozgajova M, Nieswandt B, et al. Platelet NAD(P)H-oxidase-generated ROS production regulates alphaIIbbeta3-integrin activation independent of the NO/cGMP pathway. Blood. 2005;106:2757–2760. doi: 10.1182/blood-2005-03-1047. [DOI] [PubMed] [Google Scholar]
- Begonja AJ, Teichmann L, Geiger J, Gambaryan S, Walter U. Platelet regulation by NO/cGMP signaling and NAD(P)H oxidase-generated ROS. Blood Cells Mol Dis. 2006;36:166–170. doi: 10.1016/j.bcmd.2005.12.028. [DOI] [PubMed] [Google Scholar]
- Caccese D, Pratico D, Ghiselli A, Natoli S, Pignatelli P, Sanguigni V, et al. Superoxide anion and hydroxyl radical release by collagen-induced platelet aggregation – role of arachidonic acid metabolism. Thromb Haemost. 2000;83:485–490. [PubMed] [Google Scholar]
- Cai H, Griendling KK, Harrison DG. The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol Sci. 2003;24:471–478. doi: 10.1016/S0165-6147(03)00233-5. [DOI] [PubMed] [Google Scholar]
- Chlopicki S, Olszanecki R, Janiszewski M, Laurindo FR, Panz T, Miedzobrodzki J. Functional role of NADPH oxidase in activation of platelets. Antioxid Redox Signal. 2004;6:691–698. doi: 10.1089/1523086041361640. [DOI] [PubMed] [Google Scholar]
- Cifuni SM, Wagner DD, Bergmeier W. CalDAG-GEFI and protein kinase C represent alternative pathways leading to activation of integrin alphaIIbbeta3 in platelets. Blood. 2008;112:1696–1703. doi: 10.1182/blood-2008-02-139733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clutton P, Miermont A, Freedman JE. Regulation of endogenous reactive oxygen species in platelets can reverse aggregation. Arterioscler Thromb Vasc Biol. 2004;24:187–192. doi: 10.1161/01.ATV.0000105889.29687.CC. [DOI] [PubMed] [Google Scholar]
- Dharmarajah J, Arthur JF, Sobey CG, Drummond GR. The anti-platelet effects of apocynin in mice are not mediated by inhibition of NADPH oxidase activity. Naunyn Schmiedebergs Arch Pharmacol. 2010;382:377–384. doi: 10.1007/s00210-010-0552-3. [DOI] [PubMed] [Google Scholar]
- Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10:453–471. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dworakowski R, Alom-Ruiz SP, Shah AM. NADPH oxidase-derived reactive oxygen species in the regulation of endothelial phenotype. Pharmacol Rep. 2008;60:21–28. [PubMed] [Google Scholar]
- Freedman JE. Oxidative stress and platelets. Arterioscler Thromb Vasc Biol. 2008;28:s11–s16. doi: 10.1161/ATVBAHA.107.159178. [DOI] [PubMed] [Google Scholar]
- Freedman JE, Li L, Sauter R, Keaney JJ. alpha-Tocopherol and protein kinase C inhibition enhance platelet-derived nitric oxide release. FASEB J. 2000;14:2377–2379. doi: 10.1096/fj.00-0360fje. [DOI] [PubMed] [Google Scholar]
- Getz TM, Mayanglambam A, Daniel JL, Kunapuli SP. Go6976 abrogates GPVI-mediated platelet functional responses in human platelets through inhibition of Syk. J Thromb Haemost. 2011;9:608–610. doi: 10.1111/j.1538-7836.2011.04192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni D, Taulet N, Zhang H, DerMardirossian C, Kister J, Martinez L, et al. A novel and specific NADPH oxidase-1 (Nox1) small-molecule inhibitor blocks the formation of functional invadopodia in human colon cancer cells. ACS Chem Biol. 2010;5:981–993. doi: 10.1021/cb100219n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez E, Petrich BG, Shattil SJ, Ginsberg MH, Groisman A, Kasirer-Friede A. Microfluidic devices for studies of shear-dependent platelet adhesion. Lab Chip. 2008;8:1486–1495. doi: 10.1039/b804795b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handin RI, Karabin R, Boxer GJ. Enhancement of platelet function by superoxide anion. J Clin Invest. 1977;59:959–965. doi: 10.1172/JCI108718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper MT, Poole AW. Diverse functions of protein kinase C isoforms in platelet activation and thrombus formation. J Thromb Haemost. 2010;8:454–462. doi: 10.1111/j.1538-7836.2009.03722.x. [DOI] [PubMed] [Google Scholar]
- Heumuller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schroder K, et al. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- Holinstat M, Voss B, Bilodeau ML, McLaughlin JN, Cleator J, Hamm HE. PAR4, but not PAR1, signals human platelet aggregation via Ca2+ mobilization and synergistic P2Y12 receptor activation. J Biol Chem. 2006;281:26665–26674. doi: 10.1074/jbc.M602174200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irani K, Pham Y, Coleman LD, Roos C, Cooke GE, Miodovnik A, et al. Priming of platelet alphaIIbbeta3 by oxidants is associated with tyrosine phosphorylation of beta3. Arterioscler Thromb Vasc Biol. 1998;18:1698–1706. doi: 10.1161/01.atv.18.11.1698. [DOI] [PubMed] [Google Scholar]
- Iuliano L, Colavita AR, Leo R, Pratico D, Violi F. Oxygen free radicals and platelet activation. Free Radic Biol Med. 1997;22:999–1006. doi: 10.1016/s0891-5849(96)00488-1. [DOI] [PubMed] [Google Scholar]
- Kleinschnitz C, Pozgajova M, Pham M, Bendszus M, Nieswandt B, Stoll G. Targeting platelets in acute experimental stroke: impact of glycoprotein Ib, VI, and IIb/IIIa blockade on infarct size, functional outcome, and intracranial bleeding. Circulation. 2007;115:2323–2330. doi: 10.1161/CIRCULATIONAHA.107.691279. [DOI] [PubMed] [Google Scholar]
- Konopatskaya O, Gilio K, Harper MT, Zhao Y, Cosemans JM, Karim ZA, et al. PKCalpha regulates platelet granule secretion and thrombus formation in mice. J Clin Invest. 2009;119:399–407. doi: 10.1172/JCI34665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krotz F, Sohn HY, Gloe T, Zahler S, Riexinger T, Schiele TM, et al. NAD(P)H oxidase-dependent platelet superoxide anion release increases platelet recruitment. Blood. 2002;100:917–924. doi: 10.1182/blood.v100.3.917. [DOI] [PubMed] [Google Scholar]
- Krotz F, Sohn HY, Pohl U. Reactive oxygen species: players in the platelet game. Arterioscler Thromb Vasc Biol. 2004;24:1988–1996. doi: 10.1161/01.ATV.0000145574.90840.7d. [DOI] [PubMed] [Google Scholar]
- Lahav J, Wijnen EM, Hess O, Hamaia SW, Griffiths D, Makris M, et al. Enzymatically catalyzed disulfide exchange is required for platelet adhesion to collagen via integrin alpha2beta1. Blood. 2003;102:2085–2092. doi: 10.1182/blood-2002-06-1646. [DOI] [PubMed] [Google Scholar]
- Leo R, Pratico D, Iuliano L, Pulcinelli FM, Ghiselli A, Pignatelli P, et al. Platelet activation by superoxide anion and hydroxyl radicals intrinsically generated by platelets that had undergone anoxia and then reoxygenated. Circulation. 1997;95:885–891. doi: 10.1161/01.cir.95.4.885. [DOI] [PubMed] [Google Scholar]
- Mangin PH, Tang C, Bourdon C, Loyau S, Freund M, Hechler B, et al. A humanized GPVI mouse model to assess the antithrombotic efficacy of anti-GPVI agents. J Pharmacol Exp Ther. 2012;341:156–163. doi: 10.1124/jpet.111.189050. [DOI] [PubMed] [Google Scholar]
- May AE, Seizer P, Gawaz M. Platelets: inflammatory firebugs of vascular walls. Arterioscler Thromb Vasc Biol. 2008;28:s5–10. doi: 10.1161/ATVBAHA.107.158915. [DOI] [PubMed] [Google Scholar]
- Nieswandt B, Pleines I, Bender M. Platelet adhesion and activation mechanisms in arterial thrombosis and ischaemic stroke. J Thromb Haemost. 2011;9(Suppl. 1):92–104. doi: 10.1111/j.1538-7836.2011.04361.x. [DOI] [PubMed] [Google Scholar]
- Nurden AT, Nurden P, Sanchez M, Andia I, Anitua E. Platelets and wound healing. Front Biosci. 2008;13:3532–3548. doi: 10.2741/2947. [DOI] [PubMed] [Google Scholar]
- Ollikainen E, Mikkelsson J, Perola M, Penttila A, Karhunen PJ. Platelet membrane collagen receptor glycoprotein VI polymorphism is associated with coronary thrombosis and fatal myocardial infarction in middle-aged men. Atherosclerosis. 2004;176:95–99. doi: 10.1016/j.atherosclerosis.2004.03.021. [DOI] [PubMed] [Google Scholar]
- Pignatelli P, Sanguigni V, Lenti L, Ferro D, Finocchi A, Rossi P, et al. gp91phox-dependent expression of platelet CD40 ligand. Circulation. 2004;110:1326–1329. doi: 10.1161/01.CIR.0000134963.77201.55. [DOI] [PubMed] [Google Scholar]
- Pignatelli P, Di Santo S, Buchetti B, Sanguigni V, Brunelli A, Violi F. Polyphenols enhance platelet nitric oxide by inhibiting protein kinase C-dependent NADPH oxidase activation: effect on platelet recruitment. FASEB J. 2006;20:1082–1089. doi: 10.1096/fj.05-5269com. [DOI] [PubMed] [Google Scholar]
- Pula G, Poole AW. Critical roles for the actin cytoskeleton and cdc42 in regulating platelet integrin alpha2beta1. Platelets. 2008;19:199–210. doi: 10.1080/09537100701777303. [DOI] [PubMed] [Google Scholar]
- Pula G, Crosby D, Baker J, Poole AW. Functional interaction of protein kinase Calpha with the tyrosine kinases Syk and Src in human platelets. J Biol Chem. 2005;280:7194–7205. doi: 10.1074/jbc.M409212200. [DOI] [PubMed] [Google Scholar]
- Pula G, Schuh K, Nakayama K, Nakayama KI, Walter U, Poole AW. PKCdelta regulates collagen-induced platelet aggregation through inhibition of VASP-mediated filopodia formation. Blood. 2006;108:4035–4044. doi: 10.1182/blood-2006-05-023739. [DOI] [PubMed] [Google Scholar]
- Pula G, Garonna E, Dunn WB, Hirano M, Pizzorno G, Campanella M, et al. Paracrine stimulation of endothelial cell motility and angiogenesis by platelet-derived deoxyribose-1-phosphate. Arterioscler Thromb Vasc Biol. 2010;30:2631–2638. doi: 10.1161/ATVBAHA.110.215855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM, et al. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci U S A. 2006;103:15038–15043. doi: 10.1073/pnas.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson KM, Janes MS, Beckman JS. The selective detection of mitochondrial superoxide by live cell imaging. Nat Protoc. 2008;3:941–947. doi: 10.1038/nprot.2008.56. [DOI] [PubMed] [Google Scholar]
- Savage B, Saldivar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell. 1996;84:289–297. doi: 10.1016/s0092-8674(00)80983-6. [DOI] [PubMed] [Google Scholar]
- Seno T, Inoue N, Gao D, Okuda M, Sumi Y, Matsui K, et al. Involvement of NADH/NADPH oxidase in human platelet ROS production. Thromb Res. 2001;103:399–409. doi: 10.1016/s0049-3848(01)00341-3. [DOI] [PubMed] [Google Scholar]
- Stegner D, Nieswandt B. Platelet receptor signaling in thrombus formation. J Mol Med (Berl) 2011;89:109–121. doi: 10.1007/s00109-010-0691-5. [DOI] [PubMed] [Google Scholar]
- Stokes KY, Russell JM, Jennings MH, Alexander JS, Granger DN. Platelet-associated NAD(P)H oxidase contributes to the thrombogenic phenotype induced by hypercholesterolemia. Free Radic Biol Med. 2007;43:22–30. doi: 10.1016/j.freeradbiomed.2007.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachowicz B, Olas B, Zbikowska HM, Buczynski A. Generation of reactive oxygen species in blood platelets. Platelets. 2002;13:175–182. doi: 10.1080/09533710022149395. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.