

Abstract
BACKGROUND AND PURPOSE
Annexin-A1 (ANX-A1) is an endogenous, glucocorticoid-regulated anti-inflammatory protein. The N-terminal-derived peptide Ac-ANX-A12–26 preserves cardiomyocyte viability, but the impact of ANX-A1-peptides on cardiac contractility is unknown. We now test the hypothesis that ANX-A1 preserves post-ischaemic recovery of left ventricular (LV) function.
EXPERIMENTAL APPROACH
Ac-ANX-A12–26 was administered on reperfusion, to adult rat cardiomyocytes as well as hearts isolated from rats, wild-type mice and mice deficient in endogenous ANX-A1 (ANX-A1–/–). Myocardial viability and recovery of LV function were determined.
KEY RESULTS
Ischaemia–reperfusion markedly impaired both cardiomyocyte viability and recovery of LV function by 60%. Treatment with exogenous Ac-ANX-A12–26 at the onset of reperfusion prevented cardiomyocyte injury and significantly improved recovery of LV function, in both intact rat and wild-type mouse hearts. Ac-ANX-A12–26 cardioprotection was abolished by either formyl peptide receptor (FPR)-nonselective or FPR1-selective antagonists, Boc2 and cyclosporin H, but was relatively insensitive to the FPR2-selective antagonist QuinC7. ANX-A1-induced cardioprotection was associated with increased phosphorylation of the cell survival kinase Akt. ANX-A1−/− exaggerated impairment of post-ischaemic recovery of LV function, in addition to selective LV FPR1 down-regulation.
CONCLUSIONS AND IMPLICATIONS
These data represent the first evidence that ANX-A1 affects myocardial function. Our findings suggest ANX-A1 is an endogenous regulator of post-ischaemic recovery of LV function. Furthermore, the ANX-A1-derived peptide Ac-ANX-A12–26 on reperfusion rescues LV function, probably via activation of FPR1. ANX-A1-based therapies may thus represent a novel clinical approach for the prevention and treatment of myocardial reperfusion injury.
Keywords: annexin-A1, cardioprotection, ischaemia–reperfusion, lipocortin-1, myocardium, post-conditioning, ventricular function
Introduction
Over three decades ago, Flower and Blackwell discovered the 348-amino-acid glucocorticoid-regulated protein, annexin-A1 (ANX-A1), a second messenger in glucocorticoid actions that was originally termed lipocortin-1 (Flower and Blackwell, 1979). ANX-A1 is a 37 kDa protein expressed constitutively in respiratory, renal, brain, vascular and cardiac tissues, as well as circulating inflammatory cells such as neutrophils and monocytes (Dreier et al., 1998; Flower and Blackwell, 1979; Perretti et al., 1996). The unique biological action of annexins is attributed to their distinct N-terminal region (Flower and Blackwell, 1979). Since their original discovery, clarification of the considerable anti-inflammatory properties of both full-length ANX-A1 and its N-terminal-derived peptide mimetic Ac-ANX-A12–26 has emerged largely from the laboratories of Perretti and colleagues (Morand et al., 2006; Perretti et al., 1996; Perretti and Dalli, 2009; Yang et al., 2004). These studies have demonstrated that these anti-inflammatory actions are mediated by the formyl peptide receptor (FPR) family and are largely attributed to FPR2 receptors (previously known as FPRL1 or aspirin-triggered lipoxin A4 receptors) (Perretti et al., 2002; Gavins et al., 2003). Our own studies in contrast have focussed on the protective actions of ANX-A1 peptides specifically at the level of the myocardium in vitro, in settings devoid of inflammatory cells. We demonstrated that Ac-ANX-A12–26 directly protects myocardial preparations against endotoxic shock and metabolic injury (Ritchie et al., 2003; 2005), and the FPR family was again implicated in these actions. Ac-ANX-A12–26 cardioprotection in vitro is comparable to that activated by adenosine or ischaemic preconditioning (Gordon et al., 2003).
Despite clinical advances to accelerate reperfusion post myocardial infarction (MI), via thrombolysis or percutaneous revascularisation interventions, reperfusion injury is still evident post myocardial ischaemia (Moens et al., 2005). An inflammatory response is a contributing component to reperfusion injury, which is independent at least in part, of circulating neutrophils (Goh et al., 2007; Li et al., 2009; Murphy and Steenbergen, 2008; Vinten-Johansen et al., 2005). The myocardium can be further damaged by ischaemic contracture, resulting in incomplete recovery of left ventricular (LV) function as reperfusion ensues (Moens et al., 2005; Goh et al., 2007; Li et al., 2009; Venardos et al., 2009; Tawa et al., 2010). Although the previous studies of Perretti, Gavins and colleagues have shown that ANX-A1 peptides powerfully limit neutrophil-dependent myocardial necrosis (D'Amico et al., 2000; La et al., 2001; Gavins et al., 2005; 2006), their effect specifically on post-ischaemic recovery of LV contractile function is unknown. Interventions that preserve LV contractile function following an ischaemic insult may be particularly relevant clinically, and affect prognosis. The aim of the present study was thus to test the hypothesis that endogenous ANX-A1 and its N-terminal derived peptide Ac-ANX-A12–26 preserve myocardial function post-ischaemic injury. Our ex vivo findings represent the first evidence that both endogenous and exogenous ANX-A1 peptides directly improve recovery of myocardial function during reperfusion, in the intact heart. ANX-A1–based therapies may represent a novel clinical approach for the treatment of myocardial reperfusion injury.
Methods
All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). This investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health (NIH Publications no. 85-23, revised 1996) and the National Health and Medical Research Council of Australia guidelines, and was approved by the Animal Ethics Committee of the Baker IDI Heart & Diabetes Institute. All reagents were purchased from Sigma Aldrich (St. Louis, MO) except where indicated.
Simulated ischaemia–reperfusion (I–R) in adult rat cardiomyocytes
Cultured ventricular cardiomyocytes isolated from adult male Sprague–Dawley rats (∼280 g, weight best suited for myocyte yield) (Ritchie et al., 2005; Garreffa et al., 2006; Goh et al., 2007) were subjected to 90 min hypoxia (95% N2–5% CO2) and 2.5 h subsequent reoxygenation at 37°C as previously described (Goh et al., 2007). The ANX-A1 N-terminal -derived peptide Ac-ANX-A12–26 (Ac-Ala-Met-Val-Ser-Glu-Phe-Leu-Lys-Gln-Ala-Trp-Phe-Ile-Glu-Asn-Glu-Glu-Gln-Glu-Tyr-Val-Gln-Thr-Val-Lys-OH, synthesized by Mimotopes, Clayton, Australia) was dissolved in Krebs buffer, at 0.3 μM, and added to cardiomyocytes at one of two different time points, either for the full duration of hypoxia–reoxygenation (H–R), or at the time of reoxygenation. Cardiomyocyte injury was determined at the end of reoxygenation, on LDH (IU·L−1) activity in the culture medium (Goh et al., 2007), expressed as % of paired control normoxic cardiomyocytes, as well as on cell viability using trypan blue exclusion (Ritchie et al., 2005).
I–R in intact adult rat hearts ex vivo
Hearts isolated from adult male Sprague–Dawley rats (11 weeks old, 374 ± 8 g) were buffer-perfused at constant flow (10 mL·min−1) at 37°C as previously described (Goh et al., 2007). Following equilibration (∼20 min), hearts were subjected to 30 min global, no-flow ischaemia with 30 min reperfusion, conditions previously shown to limit recovery of LV function to approximately 60% of pre-ischaemic levels. Both isovolumic LV function (via intraventricular fluid-filled balloon catheter) and markers of myocardial injury were continuously assessed, including LDH (IU·L−1, expressed fold of pre-ischaemic baseline) and creatine kinase (CK, IU·L−1) (Ritchie et al., 2005; Goh et al., 2007). Ac-ANX-A12–26 (0.3 μM, dissolved in Krebs buffer), in the absence or presence of either the non-selective FPR1/FPR2 antagonist Boc2 (10 μM, synthesized by GL Biochem Shanghai Limited, China), the FPR1-selective antagonist cyclosporin-H (CsH, 1 μM, synthesized by Chemieliva Pharmaceutical Co Ltd, Hong Kong, China) or the FPR2-selective antagonist QuinC7 (10 μM, synthesized by Anthem Biosciences Private Limited, Bangalore, India). Antagonist concentrations were chosen based on previous studies (Dufton and Perretti, 2010; Ritchie et al., 2005; Stenfeldt et al., 2007; Zhou et al., 2007), and were added to the perfusion buffer at the onset of reperfusion. Additional experimental groups included time-control normoxic (sham) hearts, studied in the absence or presence of Ac-ANX-A12–26 (added after 1 h drug-free perfusion, analogous to the time point in which reperfusion was commenced in the hearts subjected to ischaemia). Hearts were then snap-frozen and stored at −80°C until biochemical analysis. Post-ischaemic recovery of LV function was assessed on LV developed pressure (LVDP) and its derivatives LV ± dP/dt and rate-pressure product (RPP), all expressed as % of the within-heart pre-ischaemic baseline. The area under the curve (AUC) for each of these parameters during reperfusion was calculated using GraphPad Prism v5 (GraphPad Software Inc, La Jolla, CA, USA).
LV phosphorylation of the cell survival kinase Akt, as well as phosphorylation of phospholamban (which inhibits sarco(endo)plasmic Ca2+-ATPase, SERCA) were determined following I–R, using phospho-specific antibodies (Cell Signaling Technology, Beverly, MA), expressed as the ratio of phosphorylated to total protein (Goh et al., 2007; Venardos et al., 2009). Results were compared to drug-free hearts subjected to I–R and to sham (time control) normoxic hearts.
I–R in adult mouse hearts ex vivo
Male and female ANX-A1 wild-type (ANX-A1+/+) and ANX-A1−/− mice were generated on a mixed 129/SvJ × C57BL/6 background (kindly provided by Prof RJ Flower, William Harvey Research Institute) as described previously (Hannon et al., 2003; Yang et al., 2009) and studied at ∼21 weeks of age. Hearts were removed from anaesthetized mice following thoracotomy and perfused with Kreb's buffer on a Langendorff perfusion apparatus under constant perfusion pressure (80 mmHg) and temperature (37°C). This setting of constant perfusion pressure is more physiologically relevant than the setting of constant flow used above. Isovolumic function was again determined using a fluid-filled latex balloon positioned in the LV, from which LVDP, LV ± dP/dt, RPP and LVEDP were derived. Following 20 min equilibration under normoxic perfusion, hearts were subjected to global no-flow ischaemia for 22.5 min, followed by restoration of flow for a further 40 min, conditions previously shown to limit recovery of LV function to approximately 60% of pre-ischaemic levels (Venardos et al., 2009). ANX-A12–26 (0.3 μM, dissolved in Krebs buffer) was added to the perfusion buffer of subsets of either wild-type or ANX-A1−/− mouse hearts, at the onset of reperfusion. At the end of the experiment, hearts were then snap-frozen and stored at −80°C until biochemical analysis. Expression of FPR1 and FPR2 in mouse myocardium was determined by real-time PCR. Results were expressed relative to within-heart pre-ischaemic baseline and were compared with drug-free wild-type hearts, subjected either to I–R or to shams.
Real-time PCR
Total RNA isolated from cells was prepared using the RNeasy Mini Kit (QIAGEN, Hilden, Germany). Total RNA was reverse transcribed into cDNA using Superscript III reverse transcriptase and Oligo dT20 (Invitrogen, Carlsbad, CA, USA). Quantitative PCR was performed on a Rotor-Gene 3000 (Corbett Research, Mortlake, NSW, Australia) using power SYBR Green PCR master mix (Applied Biosystems, Scoresby, VIC, Australia) according to the manufacturer's protocol. Mouse primers for the determination of FPR1, FPR2 and β-actin mRNA levels were used (Yang et al., 2004; Rondepierre et al., 2009). Analysis of relative change in gene expression was calculated according to the 2–ΔΔCt method using the housekeeping gene (β-actin) as the control.
Statistical analysis
Results were expressed as mean ± SEM, with n representing the number of different cardiomyocyte preparations studied, or the number of animals per treatment group in the heart respectively. Statistical evaluation of the cardiomyocyte studies used one-way repeated-measures anova with the Student–Newman–Keuls (SNK) method for multiple comparisons (cardiomyocyte studies). For the whole heart studies, two-way anova was used to compare baseline characteristics across genotype and sex in mice, experimental groups over multiple time points, or two-way repeated-measures anova with SNK post hoc analysis for multiple comparisons was used to compare experimental groups over multiple time points, or Mann–Whitney rank sum test for single, non-parametric time point comparisons. P < 0.05 was accepted as significant.
Results
Ac-ANX-A12–26 protects adult rat cardiomyocytes from H–R injury
H–R significantly increased LDH release from cardiomyocytes (P < 0.001) and significantly reduced cardiomyocyte viability (P < 0.05, Figure 1). Ac-ANX-A12–26 completely prevented cardiomyocyte LDH release, whether present for the duration of H–R, or when only added at the start of reoxygenation (Figure 1A, both P < 0.001). Similar Ac-ANX-A12–26 cardioprotective effects were evident on cardiomyocyte viability; the beneficial actions of Ac-ANX-A12–26 were comparable whether the peptide was administered from the start of H–R, or when administered only on reoxygenation (Figure 1B, P < 0.05). Ac-ANX-A12–26 had no effect when present for the analogous time points under normoxia. These results confirm that Ac-ANX-A12–26 elicits direct protective actions on cardiomyocytes subjected to metabolic injury comparable to those in H–R injury, and which are comparable whether treatment is commenced concomitant with the insult or at the onset of post-insult recovery.
Figure 1.
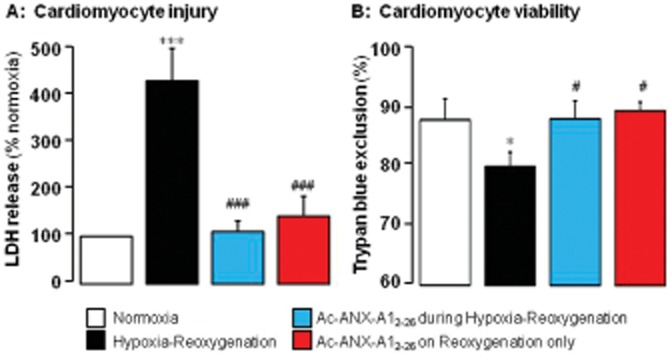
(A) Ac-ANX-A12–26 (0.3 μM) prevents adult rat cardiomyocyte hypoxia–reoxygenation (H–R) injury in vitro (assessed by measuring LDH activity) whether present for the full duration of H–R (n = 11) or only post-H–R (n = 15 cardiomyocyte preparations). (B) H–R-induced loss of cardiomyocyte viability (assessed by measuring trypan blue exclusion) was also prevented by Ac-ANX-A12–26 at both time points (n = 8 cardiomyocyte preparations). *P < 0.05 and ***P < 0.001 versus paired untreated control, and #P < 0.05 and ###P < 0.001 Ac-ANX-A12–26-treated versus untreated H–R cardiomyocytes from the same cardiomyocyte preparation on SNK.
Ac-ANX-A12–26 protects adult rat hearts from I–R injury via FPRs
Sixty-five adult male rats, aged 10.8 ± 0.3 weeks old with bodyweight 374 ± 8 g, were used to study I–R ex vivo. At baseline, hearts exhibited LVDP of 68 ± 3 mmHg and LVEDP of 3.5 ± 0.5 mmHg. As shown in Figure 2, release of LDH from the myocardium was significantly elevated over the time course of reperfusion (P < 0.005). Similarly, the recovery of both LVDP and LV + dP/dt (both P < 0.001) were significantly impaired. At the onset of reperfusion, these markers of LV function were significantly reduced compared with their pre-ischaemic baseline, and remained depressed for the duration of reperfusion. Ac-ANX-A12–26 (0.3 μM), added to the reperfusion buffer, attenuated myocardial release of LDH during reperfusion (Figure 2A). Ac-ANX-A12–26 did not affect recovery of LVDP in the first 2 min of reperfusion. However, a significant improvement in LVDP was evident from 5 min of reperfusion, and LVDP was completely recovered by 10 min reperfusion (P < 0.001, Figure 2B). The beneficial effects of Ac-ANX-A12–26 on both LDH release and the recovery of LVDP were also evident on area under curve (AUC) analysis. Similar Ac-ANX-A12–26 cardioprotection was evident on LV + dP/dt (Figure 2C), RPP (P < 0.0001, Figure 3A), LV-dP/dt (P < 0.0001, Figure 3B) and cardiac CK release (Table 1). Ac-ANX-A12–26 cardioprotection was accompanied by increased phosphorylation of the cell survival kinase Akt relative to total Akt content (P < 0.05, Figure 3C). Preliminary evidence of enhanced phosphorylation of the SERCA regulator, phospholamban was also shown (P < 0.05, Figure 3C). As shown in Table 2, Ac-ANX-A12–26 did not significantly affect LVDP or RPP in normoxic rat hearts, when added to the perfusion buffer after one hour of drug-free perfusion (the analogous time point in which ischaemic hearts underwent reperfusion), although LV ± dP/dt were modestly elevated. Furthermore compared with shams, Ac-ANX-A12–26 did not enhance Akt phosphorylation in normoxic hearts (n = 3; result not shown).
Figure 2.
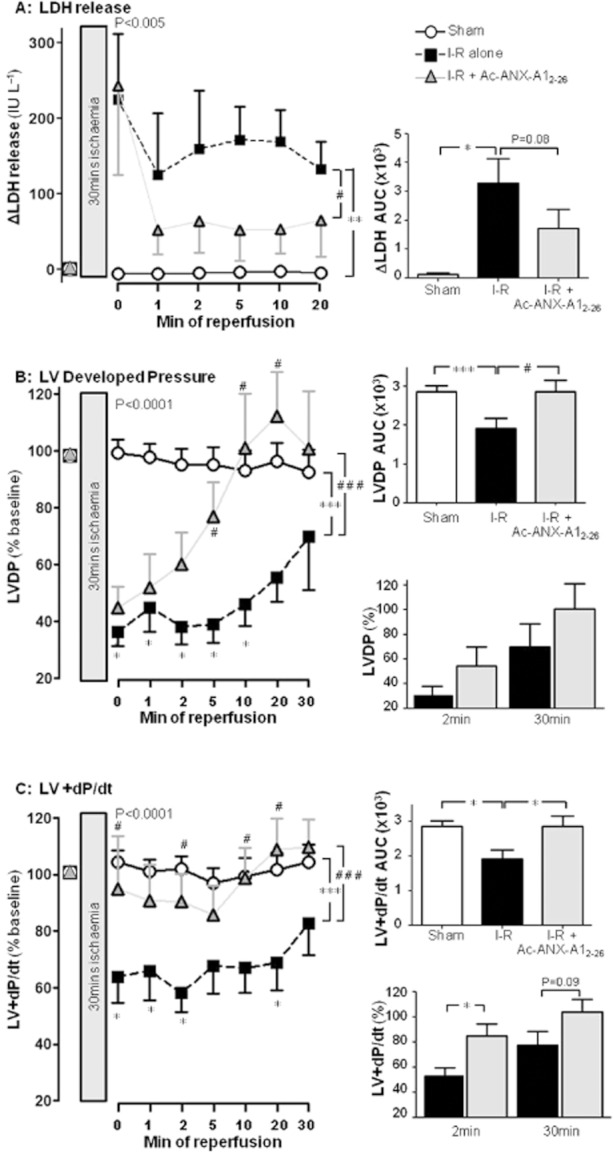
Ac-ANX-A12–26 (0.3 μM) addition at the onset of reperfusion attenuates I–R-induced impairment of myocardial viability and LV systolic function in rat isolated heart ex vivo. (A) Ac-ANX-A12–26 decreases the I–R-stimulated release of LDH from the myocardium (P < 0.005, untreated I–R, n = 16 vs. sham n = 4, and P < 0.05 untreated I–R vs. Ac-ANX-A12–26–treated I–R, n = 15, on two-way anova). The untreated I–R group also had a significantly increased myocardial release of LDH (as assessed by AUC analysis; P < 0.05), a trend that was absent in the presence of Ac-ANX-A12–26 (P = 0.08, right panel). (B) Ac-ANX-A12–26 accelerates recovery of LVDP up to 30 min post reperfusion (P < 0.001 untreated I–R, n = 15 vs. sham n = 9, and P < 0.001 vs. Ac-ANX-A12–26–treated I–R, n = 17, on two-way anova). The untreated I–R group also had a significantly impaired recovery of LVDP on AUC analysis (P < 0.001), which was prevented by Ac-ANX-A12–26 (P < 0.05, upper inset). Ac-ANX-A12–26–induced recovery of LVDP after 2 or 30 min reperfusion is shown in the lower inset (both P = NS). (C) Ac-ANX-A12–26 accelerates recovery of LV + dP/dt up to 30 min post reperfusion (P < 0.001 untreated I–R vs. sham, and P < 0.001 vs. Ac-ANX-A12–26–treated I–R, by two-way anova). The untreated I–R group had a significantly impaired recovery of LV + dP/dt on AUC analysis (P < 0.05), which was prevented by Ac-ANX-A12–26 (upper inset). Ac-ANX-A12–26 significantly improved recovery of LV + dP/dt after 2 min reperfusion (P < 0.05, lower inset). *P < 0.05 and ***P < 0.001 untreated I–R versus sham; #P < 0.05 and ##P < 0.001 Ac-ANX-A12–26–treated I–R versus untreated I–R rat hearts at each time point on SNK respectively.
Figure 3.
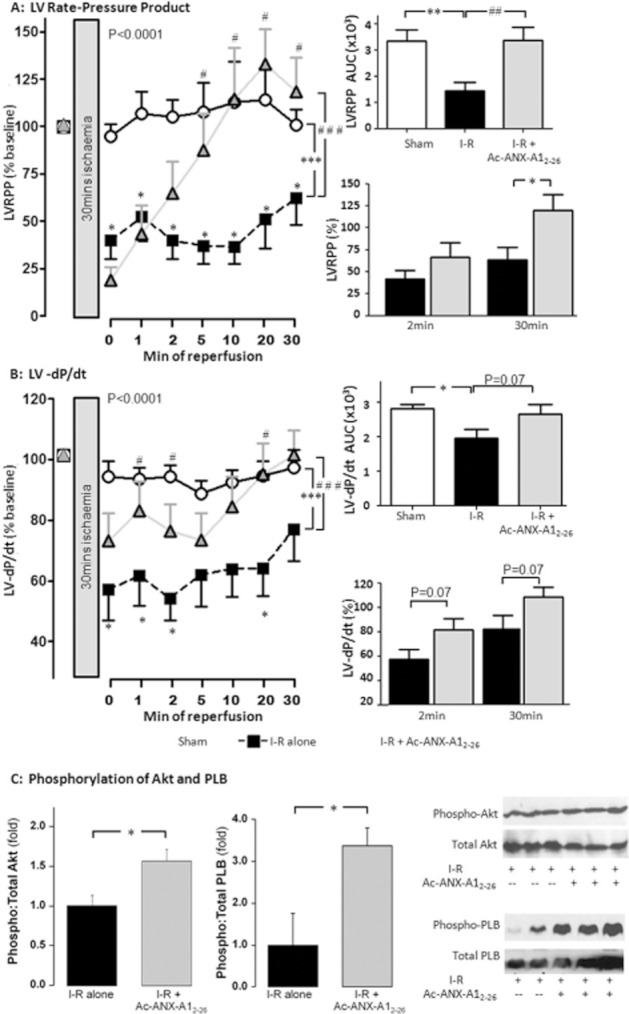
Ac-ANX-A12–26 (0.3 μM) attenuates I–R-induced impairment of (A) LVRPP and (B) LV-dP/dt up to 30 min post reperfusion in the rat isolated heart ex vivo. Recovery of RPP and LV-dP/dt on AUC analysis is shown on the upper inset of each panel, and relative recovery of both parameters after 2 and 30 min reperfusion on the lower insets. (C) ANX-A12–26 also increases phosphorylation of the cell survival kinase, Akt (P < 0.05 ANX-A12–26 I–R, n = 14 vs. untreated I–R rat hearts n = 10, left-hand panel) and of the SERCA regulator, phospholamban (P < 0.05 ANX-A12–26 I–R, n = 4 vs. untreated I–R rat hearts, n = 3, right-hand panel), in rat hearts subjected to I–R. *P < 0.05, **P < 0.005 and ***P < 0.001 untreated I–R versus sham; #P < 0.05, ##P < 0.01 and ###P < 0.001 Ac-ANX-A12–26–treated I–R versus untreated I–R rat hearts at each time point on SNK respectively.
Table 1.
Effect of I–R, in the absence and presence of Ac-ANX-A12–26 (added at the onset of reperfusion), on release of the cardiac enzyme CK in the adult rat heart ex vivo
| Min post reperfusion | ||||||||
|---|---|---|---|---|---|---|---|---|
| Parameter | Treatment | 0 | 1 | 2 | 5 | 10 | 20 | n |
| CK (IU·L−1, P < 0.05) | I–R | 32 ± 8 | 32 ± 16 | 21 ± 9 | 17 ± 5 | 21 ± 6 | 17 ± 7 | 6 |
| I–R + Ac-ANX-A12–26 | 9 ± 5 | 7 ± 4 | 9 ± 6 | 8 ± 6 | 6 ± 4 | 3 ± 2 | 5 | |
Table 2.
Effect of Ac-ANX-A12–26 (added to the perfusion buffer after one hour of drug-free perfusion) on LV function in the normoxic adult rat heart ex vivo
| Min post addition of Ac-ANX-A12–26 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Parameter | Treatment | 0 | 1 | 2 | 5 | 10 | 20 | 30 | AUC | n |
| LVDP (% baseline, P = NS) | sham | 99 ± 5 | 98 ± 5 | 95 ± 6 | 95 ± 6 | 93 ± 7 | 96 ± 7 | 92 ± 8 | 2850 ± 190 | 9 |
| Ac-ANX-A12–26 | 121 ± 8 | 111 ± 14 | 111 ± 14 | 121 ± 15 | 124 ± 15 | 128 ± 12 | 127 ± 14 | 3720 ± 404 | 4 | |
| LVRPP (% baseline, P = NS) | sham | 93 ± 7 | 110 ± 15 | 107 ± 12 | 113 ± 20 | 117 ± 29 | 118 ± 22 | 102 ± 9 | 3340 ± 443 | 6 |
| Ac-ANX-A12–26 | 127 ± 9 | 116 ± 13 | 117 ± 15 | 130 ± 15 | 134 ± 16 | 136 ± 12 | 131 ± 9 | 3950 ± 441 | 4 | |
| LV + dP/dt (% baseline, P < 0.05) | sham | 99 ± 4 | 96 ± 4 | 96 ± 4 | 91 ± 4 | 94 ± 5 | 96 ± 6 | 99 ± 6 | 2860 ± 167 | 8 |
| Ac-ANX-A12–26 | 117 ± 7 | 113 ± 11 | 111 ± 11 | 119 ± 12 | 123 ± 12 | 125 ± 10 | 121 ± 11 | 3650 ± 312 | 4 | |
| LV-dP/dt (% baseline, P < 0.005) | sham | 94 ± 5 | 93 ± 4 | 94 ± 4 | 89 ± 4 | 93 ± 4 | 95 ± 5 | 97 ± 6 | 2810 ± 119 | 8 |
| Ac-ANX-A12–26 | 117 ± 3 | 115 ± 10 | 113 ± 9 | 119 ± 9 | 122 ± 9 | 123 ± 4 | 117 ± 7 | 3600 ± 188 | 4 | |
| LDH (IU·L−1, P < 0.05) | sham | −6 ± 4 | −6 ± 6 | −5 ± 4 | −4 ± 4 | −3 ± 4 | −5 ± 3 | ND | 130 ± 45 | 4 |
| Ac-ANX-A12–26 | −19 ± 7 | −33 ± 8 | −32 ± 7 | −39 ± 5 | −44 ± 6 | −13 ± 13 | ND | 670 ± 130 | 4 | |
To explore the contribution of FPRs to the cardioprotective actions of Ac-ANX-A12–26, we used the non-selective FPR-antagonist Boc2, the FPR1-selective antagonist CsH and the FPR2-selective antagonist QuinC7, added to the perfusion buffer at the onset of reperfusion concomitantly with Ac-ANX-A12–26. Our results clearly demonstrate that Ac-ANX-A12–26-mediated preservation of both LV viability (LDH release, Figure 4A) and LV function (LVDP, LV + dP/dt, RPP and LV-dP/dt; Figures 4B, C and 5A and B, respectively) were prevented by Boc2. A similar blunting of the cardioprotective actions of Ac-ANX-A12–26 was evident with CsH. In contrast, QuinC7 failed to attenuate the Ac-ANX-A12–26-induced cardioprotection on all of these variables, with the exception of LV-dP/dt (Figures 4 and 5), although both the FPR1- and FPR2-selective antagonists tended to blunt Ac-ANX-A12–26-induced Akt phosphorylation (Figure 5E). Moreover, FPR1 antagonism tended to blunt both the early and later stages of Ac-ANX-A12–26-induced cardioprotection. In contrast, a modest inhibition of Ac-ANX-A12–26 cardioprotection was evident with FPR2 antagonism, but this effect was relatively short-lived (evident at 2 min of reperfusion).
Figure 4.
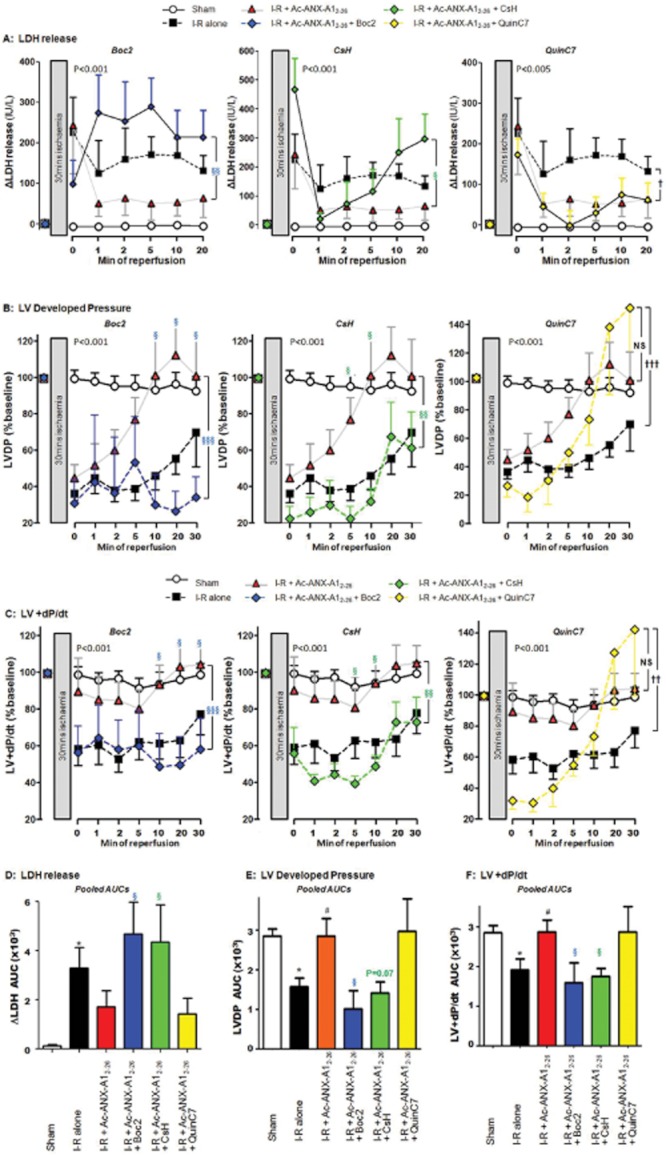
Role of FPR subtypes in Ac-ANX-A12–26 cardioprotection in the rat isolated heart ex vivo, on the time course of protection in the presence of Ac-ANX-A12–26 (0.3 μM), alone or in the presence of the non-selective FPR-antagonist Boc2 (10 μM), the FPR1-selective antagonist CsH (1 μM), or the FPR2-selective antagonist QuinC7 (10 μM) during reperfusion. (A) Both Boc2 and CsH (both P < 0.05, n = 6) significantly inhibited Ac-ANX-A12–26–induced protection of myocardial LDH release, but QuinC7 had no effect (P = NS, n = 6). Similar attenuation of the Ac-ANX-A12–26 effect by Boc2 and CsH, but not QuinC7, was also evident on recovery of both (B) LVDP and (C) LV + dP/dt. The AUC results for Ac-ANX-A12–26 cardioprotection ± FPR antagonists are also shown, on (D) LDH release, (E) LVDP and (F) LV + dP/dt. Sham n = 9, untreated I–R, n = 15 and Ac-ANX-A12–26–treated I–R, n = 17. *P < 0.05 untreated I–R versus sham; #P < 0.05 Ac-ANX-A12–26–treated I–R versus untreated I–R rat hearts; §P < 0.05, §§P < 0.01 and §§§P < 0.001 antagonist + Ac-ANX-A12–26–treated I–R versus Ac-ANX-A12–26–treated I–R and †P < 0.05, ††P < 0.01 and †††P < 0.001 antagonist + Ac-ANX-A12–26–treated I–R versus untreated I–R rat hearts respectively.
Figure 5.
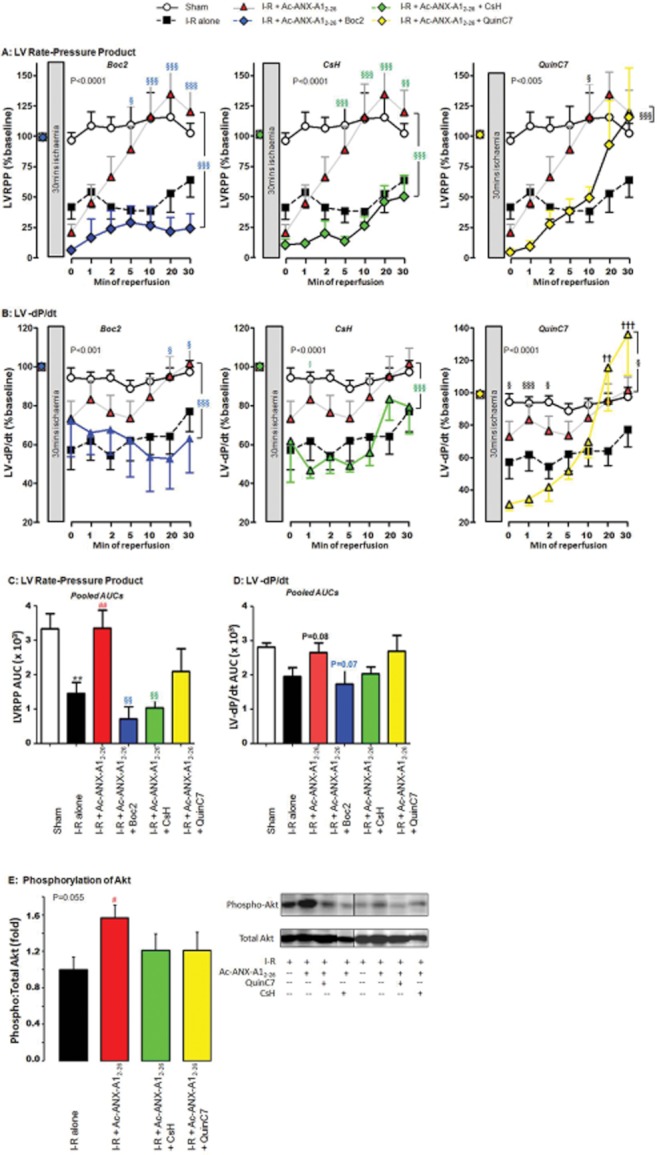
FPR antagonism attenuates the cardioprotective effects of Ac-ANX-A12–26 on recovery of both (A) RPP and (B) LV-dP/dt (sham n = 9, untreated I–R n = 15, Ac-ANX-A12–26–treated I–R n = 17 and all antagonist groups n = 6). Recovery of (C) RPP and (D) LV-dP/dt on AUC analysis is also shown, as well as (E) Ac-ANX-A12–26-induced phosphorylation of Akt (all n = 5). *P < 0.05 and **P < 0.005 untreated I–R versus sham; ##P < 0.01 Ac-ANX-A12–26–treated I–R versus untreated I–R rat hearts, §P < 0.05, §§P < 0.005 and §§§P < 0.001 antagonist + Ac-ANX-A12–26–treated I–R versus Ac-ANX-A12–26–treated I–R and ††P < 0.01 and †††P < 0.001 antagonist + Ac-ANX-A12–26–treated I–R versus untreated I–R rat hearts respectively.
Ac-ANX-A12–26 protects adult wild-type mouse hearts from I–R injury
Male and female wild-type mice were well-matched on baseline characteristics, although a trend for female mice to be slightly younger and smaller was observed (Table 3). In the wild-type mouse heart perfused under constant pressure, baseline LVDP was 126 ± 6 mmHg and heart rate (HR) was 368 ± 17 beats min-1, before ischaemia or normoxic time control. There were no differences in baseline cardiac parameters between hearts from male and female mice (Table 3), or in the time course of recovery of LV function on reperfusion (Table 4); hence, mice of either sex were studied.
Table 3.
Baseline characteristics of annexin-A1 wild-type and ANX-A1−/− mice prior to I–R ex vivo
| Genotype and sex | Age (weeks) | BW (g) | LVDP (mmHg) | LV + dP/dt (mmHg·s−1) | LV-dP/dt (mmHg·s−1) |
|---|---|---|---|---|---|
| Wild-type male (n = 14) | 21.9 ± 0.7 | 25.2 ± 0.8 | 121 ± 8 | 3757 ± 290 | −2982 ± 171 |
| Wild-type female (n = 13) | 20.1 ± 0.6a | 23.9 ± 0.3b | 132 ± 7 | 3925 ± 273 | −3236 ± 176 |
| ANX-A1−/− male (n = 16) | 20.7 ± 0.3 | 24.9 ± 0.4 | 129 ± 8 | 3966 ± 227 | −3358 ± 211 |
| ANX-A1−/− female (n = 16) | 21.4 ± 0.4 | 24.0 ± 0.3 | 124 ± 5 | 3818 ± 267 | −3159 ± 178 |
| P (genotype) | NS | NS | NS | NS | NS |
| P (sex) | NS | P < 0.05b | NS | NS | NS |
| P (genotype x sex) | P < 0.05 | NS | NS | NS | NS |
On two-way anova with SNK post hoc analysis, male wild-type mice tended to be older than female wild-type mice.
On two-way anova, male mice tended to be heavier than females. SNK post hoc analysis indicated this was largely evident in wild-type mice.
Note the absence of sex and genotype effects on cardiac phenotype at baseline
Table 4.
The extent of LV dysfunction following I–R injury was not affected by sex, in either hearts isolated from annexin-A1 wild-type or ANX-A1−/− mice ex vivo
| LVDP (% baseline, min post reperfusion) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Experimental group | 0 | 1 | 2 | 5 | 10 | 20 | 30 | 40 | AUC x 103 | n |
| Wild-type male I–R | 51 ± 19 | 94 ± 27 | 57 ± 9 | 53 ± 9 | 50 ± 9 | 56 ± 11 | 63 ± 12 | 65 ± 13 | 2.33 ± 0.43 | 7 |
| Wild-type female I–R | 34 ± 9 | 75 ± 13 | 60 ± 7 | 45 ± 9 | 44 ± 13 | 60 ± 9 | 65 ± 8 | 63 ± 5 | 2.27 ± 0.31 | 4 |
| ANX-A1−/− male I–R | 27 ± 4 | 56 ± 85 | 37 ± 6 | 10 ± 35 | 13 ± 35 | 28 ± 6 | 29 ± 7 | 29 ± 75 | 1.00 ± 0.205 | 8 |
| ANX-A1−/− female I–R | 17 ± 5 | 57 ± 9 | 33 ± 9 | 16 ± 5 | 21 ± 7 | 30 ± 6 | 33 ± 5 | 34 ± 5 | 1.13 ± 0.20 | 6 |
Analysis of the time course of recovery of LVDP over 40 min of reperfusion on two-way anova indicates that P(min post reperfusion) < 0.0001; P(experimental group) < 0.0001; P(interaction) = NS. Analysis of AUC for LVDP on two-way anova indicates that P(gender) = NS; P(experimental group) < 0.001; P(interaction) = NS.
On two-way anova with SNK post hoc analysis, hearts from male ANX-A1−/− have poorer recovery from I–R injury than male wild-type mice (P < 0.01). No significant differences were observed between male and female mice of either genotype.
In the wild-type mouse heart, I–R again significantly impaired the recovery of LVDP, LV + dP/dt and LV-dP/dt (Figure 6; all P < 0.001 vs. sham). Ac-ANX-A12–26 (0.3 μM) added to the reperfusion buffer significantly improved recovery of LV function, particularly in the latter stages of reperfusion (20 min onwards), in each of LVDP (Figure 6A), LV + dP/dt (Figure 6B) and LV-dP/dt (Figure 6C). RPP was similarly preserved by Ac-ANX-A12–26 (P = 0.06 vs. untreated I–R). Neither the time-to-ischaemic contracture, nor the extent of contracture, were affected by Ac-ANX-A12–26 (results not shown). These results confirm that Ac-ANX-A12–26 preserves LV function in a second species, and under the more physiologically relevant setting of constant pressure perfusion.
Figure 6.
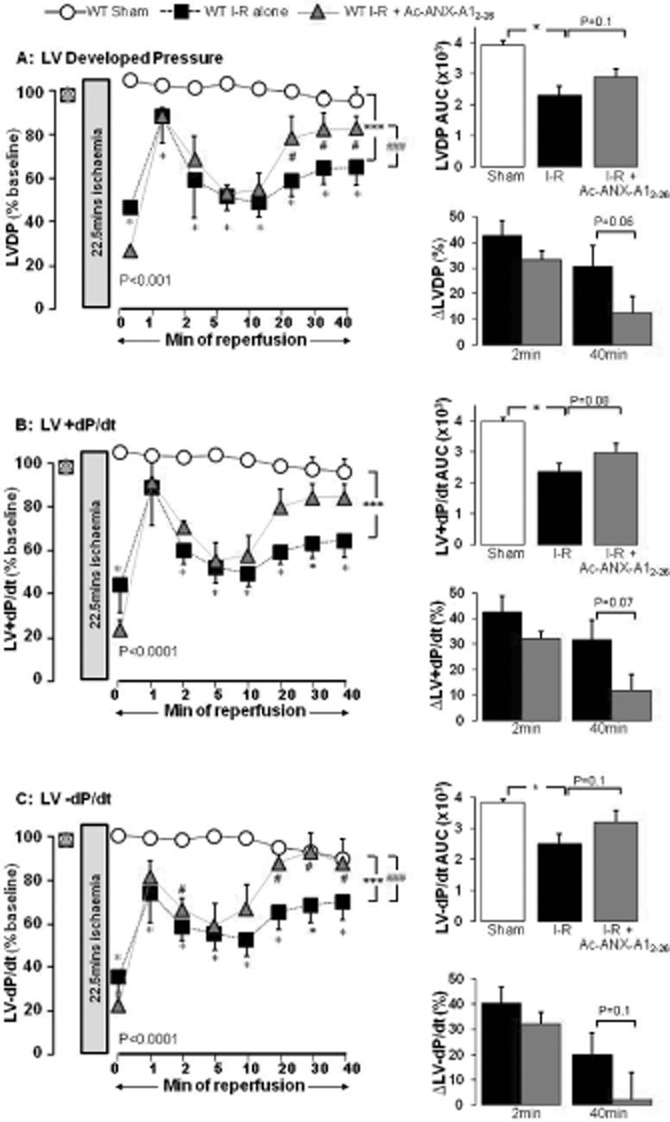
ANX-A12–26 (0.3 μM) addition at the onset of reperfusion attenuates I–R-induced impairment of LV systolic function in the isolated wild-type mouse heart ex vivo. Recovery of LVDP, LV + dP/dt and LV-dP/dt on AUC analysis is shown on the upper inset of each panel, and the relative impairment (i.e. the relative reduction in LV function as % of baseline) of all parameters after 2 and 30 min reperfusion are shown on the lower insets. (A) Ac-ANX-A12–26 accelerates recovery of LVDP up to 40 min post reperfusion (P < 0.001 untreated I–R, n = 11 versus sham n = 10 and P < 0.001 versus ANX-A12–26–treated I–R, n = 6, both on two-way anova). Untreated I–R also significantly impaired recovery of LVDP on AUC analysis (P < 0.05), which tended to be prevented by Ac-ANX-A12–26 (P = 0.1, upper inset). The impairment in recovery of LVDP in untreated versus Ac-ANX-A12–26–treated I–R was not different after 2 min reperfusion (P = NS), but a greater recovery of LVDP in Ac-ANX-A12–26–treated hearts was evident after 40 min reperfusion (P = 0.06, lower inset). (B) Ac-ANX-A12–26 accelerates recovery of LV + dP/dt up to 40 min post reperfusion (untreated I–R, n = 11 P < 0.001 versus sham, n = 10 and P = 0.089 versus ANX-A12–26–treated I–R, n = 6, on two-way anova). Untreated I–R significantly impaired recovery of LV + dP/dt on AUC analysis (P < 0.05), which tended to be prevented by Ac-ANX-A12–26 (P = 0.08, upper inset). Ac-ANX-A12–26 tended to improve recovery of LV + dP/dt after 40 min reperfusion (P = 0.08) but not after 2 min reperfusion (P = NS, lower inset). (C) Ac-ANX-A12–26 accelerates recovery of LV-dP/dt up to 40 min post reperfusion (untreated I–R n = 11 P < 0.001 vs. sham n = 10 and P < 0.001 vs. ANX-A12–26–treated I–R n = 6, both by two-way anova). Untreated I–R significantly impaired recovery of LV-dP/dt on AUC analysis (P < 0.05), which Ac-ANX-A12–26 tended to prevent (P = 0.1, upper inset). Ac-ANX-A12–26 tended to improve recovery of LV-dP/dt after 40 min reperfusion (P = 0.1) but not after 2 min reperfusion (P = NS, lower inset). *P < 0.05 and ***P < 0.001 untreated I–R versus sham and #P < 0.05 and ###P < 0.001 ANX-A12–26–treated I–R versus untreated I–R mouse hearts respectively.
Deficiency of ANX-A1 exaggerates myocardial I–R injury
As outlined in Table 3, no systemic or cardiac phenotype was evident at baseline in ANX-A1−/− mice, regardless of sex. The recovery of LV function in adult ANX-A1−/− mouse hearts from I–R was significantly and markedly impaired compared with ANX-A1+/+ mice. Deficiency of endogenous ANX-A1 resulted in reduced recovery of LVDP, evident even in the earliest stages of reperfusion (Figure 7A). Recovery of LV + dP/dt (Figure 7B), LV-dP/dt (Figure 7C) and RPP (P < 0.001) were all significantly reduced in ANX-A1−/− versus ANX-A1+/+ mouse hearts following ischaemia. There were no differences in the recovery of LV function in male versus female ANX-A1−/− mouse hearts after I–R (Table 4). Furthermore, supplementing the reperfusion buffer with Ac-ANX-A12–26 (0.3 μM) in ANX-A1−/− mouse hearts failed to restore the recovery of LV function following ischaemia (Table 5).
Figure 7.
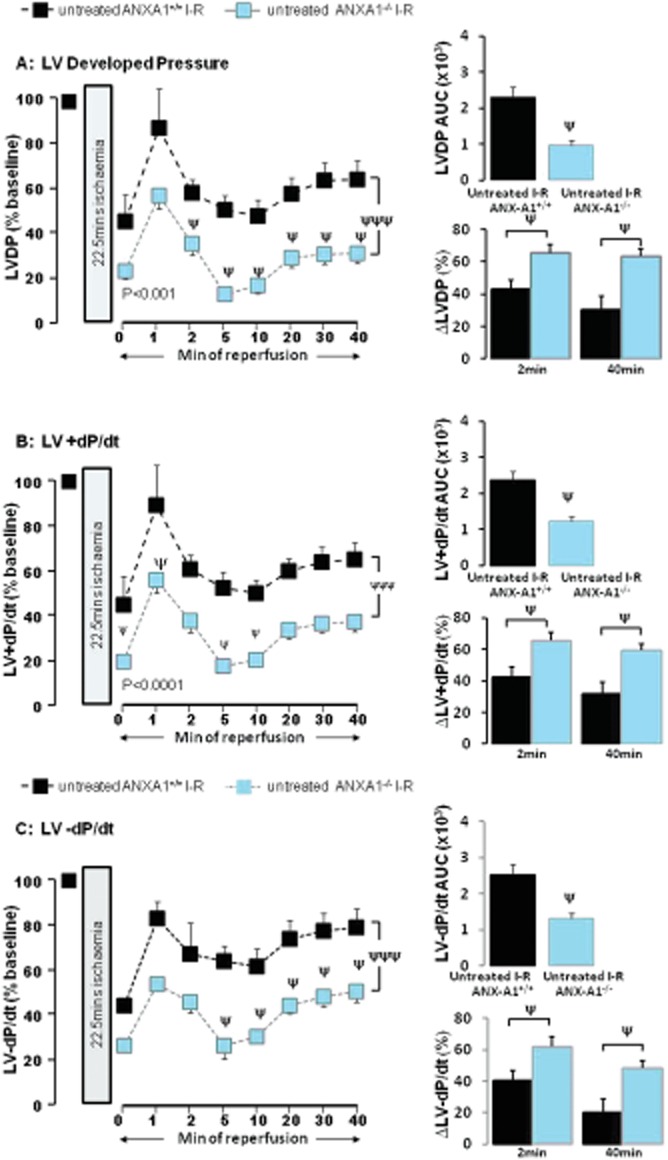
Deficiency of endogenous ANX-A1 exaggerates I–R-induced impairment of LV systolic function in the intact mouse heart ex vivo. Recovery of LVDP, LV + dP/dt and LV-dP/dt on AUC analysis is shown on the upper inset of each panel, and the relative impairment (i.e. the relative reduction in LV function as % of baseline) of all parameters after 2 and 40 min reperfusion are shown on the lower insets. (A) The time course of recovery of LVDP is significantly impaired in ANX-A1−/− (n = 14) versus ANX-A1+/+ mouse hearts (n = 11, P < 0.001 on two-way anova). ANX-A1−/− also significantly exaggerated the impaired recovery of LVDP on AUC analysis (P < 0.05, upper inset), which was evident at both 2 and 40 min reperfusion (both P < 0.05, lower inset). (B) ANX-A1−/− also significantly delayed recovery of LV + dP/dt compared with ANX-A1+/+ mouse hearts (P < 0.0001 on two-way anova), which was also evident on AUC analysis (P < 0.05, upper inset), and at both 2 and 40 min reperfusion (both P < 0.05, lower inset). (C) ANX-A1−/− similarly delayed recovery of LV-dP/dt compared with ANX-A1+/+ mouse hearts (P < 0.001 on two-way anova), which was also evident on AUC analysis (P < 0.05, upper inset), and at both 2 and 40 min reperfusion (both P < 0.05, lower inset). ψP < 0.001 and ψψψP < 0.001 untreated I–R ANX-A1+/+ versus untreated I–R ANX-A1−/− mouse hearts.
Table 5.
Ac-ANX-A12–26 fails to restore recovery of LV function following I–R injury in hearts isolated from both male and female ANX-A1−/− mice ex vivo
| LVDP (% baseline, min post reperfusion) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Experimental group | 0 | 1 | 2 | 5 | 10 | 20 | 30 | 40 | AUC × 103 | n |
| Untreated wild-type I–R | 45 ± 12 | 87 ± 17 | 58 ± 6 | 50 ± 7 | 48 ± 7 | 57 ± 7 | 63 ± 8 | 64 ± 8 | 2.31 ± 0.29 | 11 |
| Ac-ANX-A12–26-treated wild-type I–R | 25 ± 4 | 87 ± 11 | 67 ± 4 | 52 ± 8 | 53 ± 10 | 77 ± 8 | 81 ± 6 | 82 ± 6 | 2.89 ± 0.27 | 6 |
| Untreated ANX-A1−/− I–R | 23 ± 3 | 57 ± 6a | 35 ± 5a | 13 ± 3a | 16 ± 3a | 29 ± 4a | 31 ± 4a | 31 ± 4a | 1.05 ± 0.14a | 14 |
| Ac-ANX-A12–26-treated ANX-A1−/− I–R | 51 ± 32 | 59 ± 7 | 62 ± 16 | 20 ± 5 | 23 ± 5 | 35 ± 8b | 36 ± 7b | 37 ± 7b | 1.36 ± 0.22b | 8 |
Analysis of the time course of recovery of LVDP over 40 min of reperfusion on two-way anova indicates that: P(min post reperfusion) < 0.001; P(experimental group) < 0.001; P(interaction) = NS. Analysis of AUC for LVDP on two-way anova indicates that P(genotype) < 0.0001; P(experimental group) = 0.08; P(interaction) = NS.
P < 0.05 versus untreated wild-type.
P < 0.05 versus Ac-ANX-A12–26-treated wild-type mice.
Effect of ANX-A1 deficiency on myocardial Akt and FPR
As shown in Figure 8A, the exaggerated impairment in LV function in the setting of deficiency of endogenous ANX-A1 was accompanied by reduced phosphorylation of Akt relative to total Akt content (P < 0.05, Figure 8A). Given that our results obtained in Figures 6 strongly implicate a role for at least FPR1 in the cardioprotective actions of Ac-ANX-A12–26, we determined the gene expression of both FPR1 and FPR2. Our results indicate that both receptor subtypes are present in myocardium. FPR1 is selectively down-regulated in ANX-A1−/− hearts, but there was no significant effect on FPR2 expression (Figure 8B).
Figure 8.
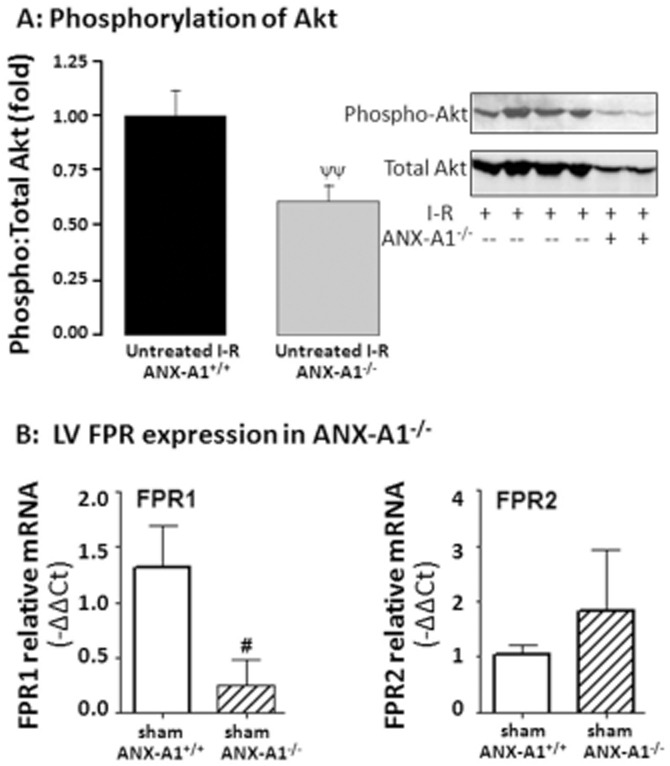
Deficiency of ANX-A1 (A) suppresses phosphorylation of the cell survival kinase Akt following I–R in mouse hearts (both n = 6, ψψP < 0.01) and (B) down-regulates gene expression of FPR1; FPR2 expression is unaffected (n = 4–5 per group).
Discussion
The therapeutic potential of the anti-inflammatory mediator ANX-A1 has been recognized in a range of inflammatory disorders (Hannon et al., 2003; Perretti and Dalli, 2009; Ritchie et al., 2003). Moreover, studies in ANX-A1−/− mice demonstrate that endogenous ANX-A1 serves as a protective brake against inflammation and injury (Hannon et al., 2003; Yang et al., 2004; Ritchie et al., 2005). We now present evidence that ANX-A1 has potent direct protective actions against cardiac dysfunction when administered after an ischaemic insult in the intact heart. Specifically, exogenous Ac-ANX-A12–26 treatment commencing concomitantly with reperfusion significantly potentiated recovery of LV function in the post-ischaemic heart ex vivo. The timing of this cardioprotection is analogous to a ‘pharmacological post-conditioning’. These functional improvements were largely attributed to FPR1 activation by this ligand, based on the greater sensitivity to both the non-selective FPR1/FPR2 antagonist Boc2 and the FPR1-selective antagonist CsH, than observed with the FPR2-selective antagonist QuinC7. Ac-ANX-A12–26 also reduced myocardial necrosis and increased activation of the cell survival kinase, Akt. Given that deficiency of endogenous ANX-A1 exaggerated myocardial I–R injury, we propose that endogenous ANX-A1 contributes to maintaining normal cardiac physiology.
We have demonstrated that Ac-ANX-A12–26 can directly preserve myocardial inotropic responsiveness and cardiomyocyte viability, rather than secondary to any neutrophil-dependent mechanisms, in vitro (Ritchie et al., 2003; 2005). The present study sheds the light, for the first time, on the beneficial impact of ANX-A1 mimetics on the recovery of LV function in the intact heart. Ac-ANX-A12–26 prevented cardiomyocyte injury and preserved LV systolic function (LVDP, LV + dP/dt) and LV diastolic function (LV-dP/dt) in both rat and mouse hearts. The preservation of LV function was significant within 10–20 min of reperfusion, and persisted for the duration of the experiment. Our results suggest that enhanced activation of cardioprotective signalling (e.g. Akt) and/or improved intracellular Ca2+ re-uptake by the sarcoplasmic reticulum (SR, as suggested by the reduced phospholamban brake on the activity of SERCA) may contribute to Ac-ANX-A12–26–induced cardioprotection, which thus targets contributing mechanisms specifically to death and dysfunction at the level of the myocardium (Moens et al., 2005; Murphy and Steenbergen, 2008). Our findings suggest that ANX-A1 analogues represent novel alternative means to salvage recovery of post-ischaemic LV systolic and diastolic function post MI.
Ac-ANX-A12–26 is regarded as a non-subtype selective FPR agonist (Hayhoe et al., 2006; Gavins, 2010; Dufton et al., 2010). Previous studies have focussed predominantly on the anti-inflammatory actions of Ac-ANX-A12–26, which are largely FPR2-dependent (Perretti et al., 2001; Gavins et al., 2003; Maderna et al., 2010; Dufton et al., 2010). These FPR2 actions of Ac-ANX-A12–26 limit early myocardial I–R injury in vivo, as evidenced by reduced infarct size secondary to anti-neutrophil actions (La et al., 2001). Given that the direct cardioprotective actions of exogenous Ac-ANX-A12–26 on the recovery of LV function in the adult male rat heart are inhibited by both the non-selective FPR1/FPR2 antagonist Boc2, as well as by the FPR1-selective antagonist CsH, but are considerably less sensitive to the FPR2-selective antagonist QuinC7, we now largely attribute these actions on the preservation of contractile function to FPR1, rather than FPR2, activation. The protective effects of the FPR2 receptor subtype probably become markedly more important in vivo, via their involvement in inflammatory cell-mediated events.
Systemic deficiency of the parent ANX-A1 protein exaggerates inflammatory responses in vivo (Hannon et al., 2003; Yang et al., 2009). Our findings in the present study reveal a previously unknown additional consequence of ANX-A1 deficiency, an augmented myocardial injury response specifically at the level of cardiac contractile function. Marked aggravation of post-ischaemic impairment of both LV systolic (LVDP, LV + dP/dt) and diastolic function (LVEDP, LV-dP/dt) were observed in ANX-A1−/− mice. This suggests that endogenous ANX-A1 contributes significantly to the maintenance of normal cardiac physiology. Receptor utilization by the full length ANX-A1 protein remains contentious. Although ANX-A1 appears to show FPR2 selectivity relative to Ac-ANX-A12–26 (Hayhoe et al., 2006), roughly 50% of the ANX-A1 effects are lost in FPR1-deficient mice. Furthermore, FPR1-transfected HEK293 cells bind ANX-A1 (Perretti et al., 2001). Some residual protective ANX-A1 actions may thus remain in FPR2-deficient mice (Dufton et al., 2010). It is hence possible that although most evidence suggests that ANX-A1 predominantly binds to FPR2 (Hayhoe et al., 2006), under some settings, FPR1 may also bind ANX-A1 (Dufton and Perretti, 2010). We now confirm that both FPR1 and FPR2 subtypes are present in the heart, as previously suggested (Migeotte et al., 2006). Interestingly, our results suggest that a deficiency of endogenous annexin-A1 down-regulates myocardial FPR1 (but not FPR2) expression, which probably accounts for the marked impairment in recovery of LV contractile function following the ischaemic insult. Indeed this marked down-regulation (by >80%) of LV FPR1 expression in ANX-A1−/− probably accounts for the inability of exogenous Ac-ANX-A12–26 to protect LV contractile function (Table 5). The subtype of FPR responsible for the cardioprotective actions of endogenous ANX-A1 on myocardial function demonstrated in the present study remains to be resolved in vivo. However, our results support the hypothesis that targeting ANX-A1 is a potential strategy to maintain myocardial function in settings of I–R. ANX-A1-based therapeutic strategies may facilitate both direct effects on myocardial function, as well as those downstream of anti-inflammatory actions.
Until relatively recently, the anti-inflammatory and other protective actions of full-length ANX-A1 and Ac-ANX-A12–26 were regarded as virtually interchangeable (Flower and Rothwell, 1994). However, evidence is now emerging that different mechanisms may mediate some components of their actions (Hayhoe et al., 2006; Migeotte et al., 2006; Yang et al., 2009). For example, ANX-A1-mediated up-regulation of glucocorticoid-induced leucine zipper protein is FPR-independent and cannot be rescued by Ac-ANX-A12–26 treatment in vitro (Yang et al., 2009). Perretti and Gavins previously postulated that a component of the cardioprotective actions of Ac-ANX-A12–26 might be mediated by competing with full-length ANX-A1 protein for binding to the (as yet unidentified) enzyme responsible for ANX-A1 catabolism, preserving ANX-A1 bioavailability (La et al., 2001). Such a mechanism would also be absent in ANX-A1−/− mice and could not be restored by Ac-ANX-A12–26. We now demonstrate that the exaggerated impairment of LV function seen in ANX-A1−/− mouse hearts subjected to I–R injury ex vivo also persists in the face of acute Ac-ANX-A12–26 treatment. Our studies thus reveal detrimental consequences of life-long systemic deficiency of ANX-A1-mediated basal myocardial FPR1 and/or FPR2 activation, or suggest FPR-independent myocardial effects of ANX-A1, analogous to those in macrophages (Yang et al., 2009). Elucidating which of these possible phenomena contributed to the exaggerated impairment of LV function post I–R in ANX-A1−/− mouse hearts was beyond the scope of the present study. However, our results clearly demonstrate the protective role of endogenous ANX-A1 in the myocardial response to I–R injury in the intact heart.
Cardiac necrosis, inflammation and dysfunction all increase the risk of ischaemic cardiomyopathy, heart failure and death (Moens et al., 2005); each of these is targeted by ANX-A1 analogues. We thus propose that ANX-A1 analogue-based therapies represent potential cardioprotective agents for treating I–R injury. In addition to their established inhibition of the systemic inflammatory response (D'Amico et al., 2000; La et al., 2001; Gavins et al., 2005; 2006; Hecht et al., 2009), ANX-A1 analogues address damage by acting directly on the myocardium, limiting its ability to contract and relax, as evidenced by our own observations, as well as preventing cardiomyocyte death, an important determinant of outcome from MI (Murphy and Steenbergen, 2008; Peart and Headrick, 2009). Furthermore, this protection is effective when treatment is initiated subsequent to the injury. In conclusion, our results suggest that ANX-A1-based strategies may offer novel potential therapeutic approaches for the treatment of myocardial I–R injury and dysfunction. This could be applied to both planned open heart surgery and angioplasty, unplanned injury during the development of infarction, as well as to injury such as in other cardiomyopathies associated with inflammation. Development of treatment strategies that are effective when initiated subsequent to injury, and target inflammatory cell-dependent and -independent causes of myocardial injury may ultimately reduce progression to heart failure and death in patients suffering MI. The findings presented here warrant future studies exploring the cardioprotective actions of ANX-A1 analogues on LV function in vivo, as well as on late myocardial necrosis (days to weeks post onset of reperfusion). With the development of synthetic, orally-active non-peptide ANX-A1 analogues (Burli et al., 2006), such studies are now possible.
Acknowledgments
This study was supported by the National Heart Foundation of Australia (grant-in-aid G 04 M 1529), the National Health and Medical Research Council of Australia (Senior Research Fellowship to RHR, ID472673) and supported in part by the Victorian Government's Operational Infrastructure Support Program.
Glossary
- ANX-A1
annexin-A1
- CK
creatine kinase
- CsH
cyclosporin H
- FPR
formyl peptide receptors (including FPR1, FPR2 and FPR3 subtypes)
- HR
heart rate
- H–R
hypoxia–reoxygenation
- I–R
ischaemia–reperfusion
- LV
left ventricular
- LVDP
LV developed pressure
- LV ± dP/dt
first derivatives of LV pressure
- LVEDP
LV end-diastolic pressure
- MI
myocardial infarction
- RM
repeated measures
- RPP
rate-pressure product
- SNK
Student–Newman–Keuls
- SERCA
sarco(endo)plasmic reticulum Ca2+-ATPase
- SR
sarcoplasmic reticulum
- Ac-ANX-A12–26
N-terminal-derived ANX-A1 peptide mimetic
Conflict of interest
The authors have no conflicts of interest with respect to this work.
References
- Burli RW, Xu H, Zou XM, Muller K, Golden J, Frohn M, et al. Potent hFPRL1 (ALXR) agonists as potential anti-inflammatory agents. Bioorg Med Chem Lett. 2006;16:3713–3718. doi: 10.1016/j.bmcl.2006.04.068. [DOI] [PubMed] [Google Scholar]
- D'Amico M, Di Filippo C, La M, Solito E, McLean PG, Flower RJ, et al. Lipocortin 1 reduces myocardial ischemia-reperfusion injury by affecting local leukocyte recruitment. FASEB J. 2000;14:1867–1869. doi: 10.1096/fj.99-0602fje. [DOI] [PubMed] [Google Scholar]
- Dreier R, Schmid KW, Gerke V, Riehemann K. Differential expression of annexins I, II and IV in human tissues: an immunohistochemical study. Histochem Cell Biol. 1998;110:137–148. doi: 10.1007/s004180050275. [DOI] [PubMed] [Google Scholar]
- Dufton N, Perretti M. Therapeutic anti-inflammatory potential of formyl-peptide receptor agonists. Pharmacol Ther. 2010;127:175–188. doi: 10.1016/j.pharmthera.2010.04.010. [DOI] [PubMed] [Google Scholar]
- Dufton N, Hannon R, Brancaleone V, Dalli J, Patel HB, Gray M, et al. Anti-inflammatory role of the murine formyl-peptide receptor 2: ligand-specific effects on leukocyte responses and experimental inflammation. J Immunol. 2010;184:2611–2619. doi: 10.4049/jimmunol.0903526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flower RJ, Blackwell GJ. Anti-inflammatory steroids induce biosynthesis of a phospholipase-A2 inhibitor which prevents prostaglandin generation. Nature. 1979;278:456–459. doi: 10.1038/278456a0. [DOI] [PubMed] [Google Scholar]
- Flower RJ, Rothwell NJ. Lipocortin-1 – cellular mechanisms and clinical relevance. Trends Pharmacol Sci. 1994;15:71–76. doi: 10.1016/0165-6147(94)90281-x. [DOI] [PubMed] [Google Scholar]
- Garreffa AM, Woodman OL, Cao AH, Ritchie RH. Sodium nitroprusside protects adult rat cardiac myocytes from cellular injury induced by simulated ischemia – role for a non-cGMP- dependent mechanism of nitric oxide protection. J Cardiovasc Pharmacol. 2006;47:1–8. doi: 10.1097/01.fjc.0000189601.12276.8b. [DOI] [PubMed] [Google Scholar]
- Gavins FNE. Are formyl peptide receptors novel targets for therapeutic intervention in ischaemia-reperfusion injury? Trends Pharmacol Sci. 2010;31:266–276. doi: 10.1016/j.tips.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavins FNE, Yona S, Kamal AM, Flower RJ, Perretti M. Leukocyte antiadhesive actions of annexin 1: ALXR- and FPR-related anti-inflammatory mechanisms. Blood. 2003;101:4140–4147. doi: 10.1182/blood-2002-11-3411. [DOI] [PubMed] [Google Scholar]
- Gavins FNE, Kamal AM, D'Amico M, Oliani SM, Perretti M. Formyl-peptide receptor is not involved in the protection afforded by annexin 1 in murine acute myocardial infarct. FASEB J. 2005;19:100–102. doi: 10.1096/fj.04-2178fje. [DOI] [PubMed] [Google Scholar]
- Gavins FNE, Leoni G, Getting SJ. Annexin 1 and melanocortin peptide therapy for protection against ischaemic-reperfusion damage in the heart. ScientificWorldJournal. 2006;6:1008–1023. doi: 10.1100/tsw.2006.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh SSC, Woodman OL, Pepe S, Cao AH, Qin CX, Ritchie RH. The red wine antioxidant resveratrol prevents cardiomyocyte injury following ischemia-reperfusion via multiple sites and mechanisms. Antioxid Redox Signal. 2007;9:101–113. doi: 10.1089/ars.2007.9.101. [DOI] [PubMed] [Google Scholar]
- Gordon JM, Dusting GJ, Woodman OL, Ritchie RH. Cardioprotective action of CRF peptide urocortin against simulated ischemia in adult rat cardiomyocytes. Am J Physiol. 2003;284:H330–H336. doi: 10.1152/ajpheart.01121.2001. [DOI] [PubMed] [Google Scholar]
- Hannon R, Croxtall JD, Getting SJ, Roviezzo F, Yona S, Paul-Clark MJ, et al. Aberrant inflammation and resistance to glucocorticoids in Annexin 1−/− Mouse. FASEB J. 2003;17:253–255. doi: 10.1096/fj.02-0239fje. [DOI] [PubMed] [Google Scholar]
- Hayhoe RPG, Kamal AM, Solito E, Flower RJ, Cooper D, Perretti M. Annexin 1 and its bioactive peptide inhibit neutrophil-endothelium interactions under flow: indication of distinct receptor involvement. Blood. 2006;107:2123–2130. doi: 10.1182/blood-2005-08-3099. [DOI] [PubMed] [Google Scholar]
- Hecht I, Rong J, Sampaio ALF, Hermesh C, Rutledge C, Shemesh R, et al. A novel peptide agonist of formyl-peptide receptor-like 1 (alx) displays anti-inflammatory and cardioprotective effects. J Pharmacol Exp Ther. 2009;328:426–434. doi: 10.1124/jpet.108.145821. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La M, D'Amico M, Bandiera S, Di Filippo C, Oliani SM, Gavins FNE, et al. Annexin 1 peptides protect against experimental myocardial ischemia-reperfusion: analysis of their mechanism of action. FASEB J. 2001;15:2247–2256. doi: 10.1096/fj.01-0196com. [DOI] [PubMed] [Google Scholar]
- Li XM, Ma YT, Yang YN, Zhang JF, Chen BD, Liu F, et al. Ischemic postconditioning protects hypertrophic myocardium by ERK1/2 signaling pathway: experiment with mice. Zhonghua Yi Xue Za Zhi. 2009;89:846–850. [PubMed] [Google Scholar]
- Maderna P, Cottell DC, Toivonen T, Dufton N, Dalli J, Perretti M, et al. FPR2/ALX receptor expression and internalization are critical for lipoxin A4 and annexin-derived peptide-stimulated phagocytosis. FASEB J. 2010;24:4240–4249. doi: 10.1096/fj.10-159913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C. Guidelines for reportingexperiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migeotte I, Communi D, Parmentier M. Formyl peptide receptors: a promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev. 2006;17:501–519. doi: 10.1016/j.cytogfr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Moens AL, Claeys MJ, Timmermans JP, Vrints CJ. Myocardial ischemia/reperfusion-injury, a clinical view on a complex pathophysiological process. Int J Cardiol. 2005;100:179–190. doi: 10.1016/j.ijcard.2004.04.013. [DOI] [PubMed] [Google Scholar]
- Morand EF, Hall P, Hutchinson P, Yang YH. Regulation of annexin I in rheumatoid synovial cells by glucocorticoids and interleukin-1. Mediators Inflamm. 2006;2006:73831–73836. doi: 10.1155/MI/2006/73835. Article ID 73835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peart JN, Headrick JP. Clinical cardioprotection and the value of conditioning responses. Am J Physiol. 2009;296:H1705–H1720. doi: 10.1152/ajpheart.00162.2009. [DOI] [PubMed] [Google Scholar]
- Perretti M, Dalli J. Exploiting the Annexin A1 pathway for the development of novel anti-inflammatory therapeutics. Br J Pharmacol. 2009;158:936–946. doi: 10.1111/j.1476-5381.2009.00483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perretti M, Croxtall JD, Wheller SK, Goulding NJ, Hannon R, Flower RJ. Mobilizing lipocortin 1 in adherent human leukocytes downregulates their transmigration. Nat Med. 1996;2:1259–1262. doi: 10.1038/nm1196-1259. [DOI] [PubMed] [Google Scholar]
- Perretti M, Getting SJ, Solito E, Murphy PM, Gao JL. Involvement of the receptor for formylated peptides in the in vivo anti-migratory actions of annexin 1 and its mimetics. Am J Pathol. 2001;158:1969–1973. doi: 10.1016/S0002-9440(10)64667-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perretti M, Chiang N, La M, Fierro IM, Marullo S, Getting SJ, et al. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A(4) receptor. Nat Med. 2002;8:1296–1302. doi: 10.1038/nm786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie RH, Sun XL, Bilszta JL, Gulluyan LM, Dusting GJ. Cardioprotective actions of an N-terminal fragment of annexin-1 in rat myocardium in vitro. Eur J Pharmacol. 2003;461:171–179. doi: 10.1016/s0014-2999(03)01314-1. [DOI] [PubMed] [Google Scholar]
- Ritchie RH, Gordon JM, Woodman OL, Cao AH, Dusting GJ. Annexin-1 peptide Anx-12-26 protects adult rat cardiac myocytes from cellular injury induced by simulated ischaemia. Br J Pharmacol. 2005;145:495–502. doi: 10.1038/sj.bjp.0706211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondepierre F, Bouchon B, Papon J, Bonnet-Duquennoy M, Kintossou R, Moins N, et al. Proteomic studies of B16 lines: involvement of Annexin A1 in melanoma dissemination. Biochim Biophys Acta. 2009;1794:61–69. doi: 10.1016/j.bbapap.2008.09.014. [DOI] [PubMed] [Google Scholar]
- Stenfeldt A-L, Karlsson J, Wennerås C, Bylund J, Fu H, Dahlgren C. Cyclosporin H, Boc-MLF and Boc-FLFLF are antagonists that preferentially inhibit activity triggered through the formyl peptide receptor. Inflammation. 2007;30:224–229. doi: 10.1007/s10753-007-9040-4. [DOI] [PubMed] [Google Scholar]
- Tawa M, Fukumoto T, Yamashita N, Ohkita M, Ayajiki K, Okamura T, et al. Postconditioning improves postischemic cardiac dysfunction independently of norepinephrine overflow after reperfusion in rat hearts: comparison with preconditioning. J Cardiovasc Pharmacol. 2010;55:6–13. doi: 10.1097/FJC.0b013e3181bfb1c1. [DOI] [PubMed] [Google Scholar]
- Venardos KM, Zatta AJ, Marshall T, Ritchie RH, Kaye DM. L-arginine transport contributes to the pathogenesis of myocardial ischemia-reperfusion injury. J Cell Biochem. 2009;108:156–168. doi: 10.1002/jcb.22235. [DOI] [PubMed] [Google Scholar]
- Vinten-Johansen J, Zhao ZQ, Zatta AJ, Kin H, Halkos ME, Kerendi F. Postconditioning – a new link in nature's armor against myocardial ischemia-reperfusion injury. Basic Res Cardiol. 2005;100:295–310. doi: 10.1007/s00395-005-0523-x. [DOI] [PubMed] [Google Scholar]
- Yang YH, Morand EF, Getting SJ, Paul-Clark M, Liu DL, Yona S, et al. Modulation of inflammation and response to dexamethasone by annexin 1 in antigen-induced arthritis. Arthritis Rheum. 2004;50:976–984. doi: 10.1002/art.20201. [DOI] [PubMed] [Google Scholar]
- Yang YH, Aeberli D, Dacumos A, Xue JR, Morand EF. Annexin-1 regulates macrophage IL-6 and TNF via glucocorticoid-induced leucine zipper. J Immunol. 2009;183:1435–1445. doi: 10.4049/jimmunol.0804000. [DOI] [PubMed] [Google Scholar]
- Zhou CH, Zhang S, Nanamori M, Zhang YY, Liu Q, Li N, et al. Pharmacological characterization of a novel nonpeptide antagonist for formyl peptide receptor-like 1. Mol Pharmacol. 2007;72:976–983. doi: 10.1124/mol.107.037564. [DOI] [PubMed] [Google Scholar]