

Abstract
Renal collecting ducts play a critical role in acid–base homeostasis by establishing steep transepithelial pH gradients necessary for the almost complete reabsorption of bicarbonate and the effective secretion of ammonium into the urine. The mechanisms of urine acidification in collecting ducts involve active, electrogenic hydrogen (H+) secretion and, less importantly, potassium (K+)-H+ exchange. Deranged renal acidification and the inability to lower urine pH are hallmarks of distal tubular acidosis and often result from inborn errors of metabolism involving vacuolar H+-ATPase subunits in the collecting ducts. Three factors regulate H+-ATPase activity in intercalated cells of collecting ducts: the acid–base status, angiotensin II, and aldosterone. Most effects of aldosterone involve activation of the mineralocorticoid receptor and genomic changes in transcription and protein synthesis. Here we demonstrate a nongenomic pathway of vacuolar H+-ATPase activation in intercalated cells of isolated mouse outer medullary collecting ducts (OMCD). In vitro exposure of isolated outer medullary collecting ducts to aldosterone (10 nM) for times as short as 15 min increases vacuolar H+-ATPase activity ≈2- to 3-fold. Neither inhibition of mineralocorticoid receptors nor of transcription and protein synthesis prevented aldosterone-induced stimulation of H+-ATPase. Incubation with colchicine, however, abolished the stimulatory effect of aldosterone, suggesting a role of the microtubular network for H+-ATPase stimulation. Immunohistochemistry in kidneys from aldosterone-injected mice showed increased apical H+-ATPase staining in OMCD-intercalated cells. The stimulatory effect of aldosterone was associated with a transient rise in intracellular Ca2+ and required intact PKC. Thus, rapid nongenomic modulation of vacuolar H+-ATPase activity in OMCD-intercalated cells by aldosterone may play an additional role in hormonal control of systemic acid–base homeostasis.
The kidney plays a major role in the control of systemic acid–base homeostasis by reabsorbing filtered bicarbonate, generating new bicarbonate, and secreting protons largely bound to buffers such as phosphate and ammonia. The final step of urinary acidification occurs along the cortical and outer medullary collecting duct largely through the action of vacuolar H+-ATPases in type A intercalated cells (1, 2). H+/K+-ATPases also contribute to H+ secretion (3, 4). The activity of vacuolar H+-ATPases in the collecting duct is regulated by the systemic acid–base and electrolyte status and by hormones, most prominently aldosterone and, angiotensin II (1, 2). Angiotensin II stimulates several proximal tubule proton- and bicarbonate-transporting systems, such as Na+/H+ exchange, 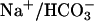 cotransport, and vacuolar H+-ATPases, and urinary acidification in the collecting duct (5–7). Aldosterone has both indirect and direct effects on proton secretion. In the connecting segment and cortical collecting duct, aldosterone stimulates the electrogenic vacuolar H+-ATPase through increased reabsorption of Na+ in principal cells, which renders the tubule lumen more negative and thus facilitates proton diffusion into the lumen (1). In the outer medullary collecting duct, aldosterone also stimulates proton secretion, but this effect persists in the absence of Na+ absorption, suggesting direct activation of vacuolar H+-ATPase activity (8). Long-term stimulation of acidification by aldosterone has been described in rabbit and rat outer medullary collecting ducts (9–11). The regulation of ion-transport processes by aldosterone involves genomic changes in transcription and synthesis of transport proteins, such as subunits of the epithelial Na+ channel ENaC or the Na+/K+-ATPase (12–14). However, increasing evidence demonstrates nongenomic actions of steroid hormones, such as aldosterone, vitamin D3, and cortisone in both epithelial and nonepithelial tissues (15–17). These hormone effects are characterized by rapid onset of action, independence of known cytosolic steroid receptors, and transcriptional events. In the present experiments we define such a pathway of nongenomic stimulation of urinary acidification in intercalated cells of renal tubules by aldosterone.
cotransport, and vacuolar H+-ATPases, and urinary acidification in the collecting duct (5–7). Aldosterone has both indirect and direct effects on proton secretion. In the connecting segment and cortical collecting duct, aldosterone stimulates the electrogenic vacuolar H+-ATPase through increased reabsorption of Na+ in principal cells, which renders the tubule lumen more negative and thus facilitates proton diffusion into the lumen (1). In the outer medullary collecting duct, aldosterone also stimulates proton secretion, but this effect persists in the absence of Na+ absorption, suggesting direct activation of vacuolar H+-ATPase activity (8). Long-term stimulation of acidification by aldosterone has been described in rabbit and rat outer medullary collecting ducts (9–11). The regulation of ion-transport processes by aldosterone involves genomic changes in transcription and synthesis of transport proteins, such as subunits of the epithelial Na+ channel ENaC or the Na+/K+-ATPase (12–14). However, increasing evidence demonstrates nongenomic actions of steroid hormones, such as aldosterone, vitamin D3, and cortisone in both epithelial and nonepithelial tissues (15–17). These hormone effects are characterized by rapid onset of action, independence of known cytosolic steroid receptors, and transcriptional events. In the present experiments we define such a pathway of nongenomic stimulation of urinary acidification in intercalated cells of renal tubules by aldosterone.
Materials and Methods
Animals. C57BL/6J mice (The Jackson Laboratory) (male, 25–30 g) were maintained on standard chow and had access to drinking water ad libitum. All studies were approved by the Yale Animal Care and Use Committee and the Swiss Veterinäramt, Zurich. To test for the effect of aldosterone on vacuolar H+-ATPase abundance and localization, mice were injected i.p. either with aldosterone (150 μg/kg of body weight), diluted in 0.9% NaCl, or with vehicle (ethanol) alone (18). Animals were killed 30 min after injection for Western blotting or for perfusion-fixing for immunohistochemistry.
Tubule Preparation and Intracellular pH Measurements. Segments of the outer medullary collecting ducts (OMCDs) were chosen because of the large abundance of tubule cells with demonstrated H+-ATPase activity (19). Intracellular pH (pHi) was monitored in single type A intercalated cells in isolated OMCDs that were prepared, with a small modification, as described (20). In brief, mice were anesthetized and perfused through the left ventricle with a phosphate/sucrose buffer (140 mM sucrose/140 mM NaH2PO4, pH 7.4) with collagenase (Sigma). Kidneys were harvested, transferred to a dissection microscope, coronal slices 1–2 mm in thickness were prepared, and the outer medulla was dissected. Outer medulla pieces were transferred to a digestion buffer and incubated at 37°C for 30 min, washed several times with ice-cold Hepes buffer, and stored on ice until further use. OMCDs were identified under the dissection microscope and transferred onto glass coverslips prepared with the biological adhesive Cell-Tak. Glass coverslips were mounted on an inverted microscope into a perfusion chamber (≈3 ml/min flow rate, 37°C) with a feedback heater at 37°C. OMCDs were incubated with the pH-sensitive dye BCECF (1 μM, Molecular Probes) or the Ca2+-sensitive dye FURA-2 (2 μM, Molecular Probes) for 15 min and washed to remove all nondeesterified dye. pHi was monitored by using the pH-sensitive dye BCECF and calibrated with the high K+/nigericin technique as described (6, 20). All experiments were performed in the absence of bicarbonate. The initial solution was a Hepes-buffered Ringer solution (125 mM NaCl/3mMKCl/1 mM CaCl2/1.2 mM MgSO4/2mMKH2PO4/32.2 mM Hepes, pH 7.4). Cells were acidified by using the NH4Cl (20 mM) prepulse technique and washed into a Na+-free solution (Na+ was replaced by equimolar concentrations of N-methyl-d-glucamine). The rate of H+-ATPase activity was determined as the concanamycin-sensitive pHi alkalinization rate in the absence of Na+. Rates were calculated over the same range of pHi (6.60–6.75) for all cells studied. All chemicals were of highest purity and were purchased from Sigma. Chemicals were added to the perfusion solutions from stock solutions at the given concentrations for each experimental protocol. All data were tested for significance with the unpaired Student t test, and only results with P values of <0.05 were considered as statistically significant.
Immunohistochemistry. C57BL/6J mice were anesthetized with ketamine and perfused through the left ventricle with PBS followed by paraformaldehyde-lysine-periodate (PLP) fixative (21). Kidneys were removed and fixed overnight at 4°C by immersion in PLP. Kidneys were washed three times with PBS, and 5-μm cryosections were cut after cryoprotection with 2.3 M sucrose in PBS for at least 12 h. Immunostaining was carried out as described (22). Sections were incubated with 1% SDS for 5 min, washed three times with PBS, and incubated with PBS containing 1% BSA for 15 min before the primary antibody. The primary antibodies [rabbit anti-a4 serum (20), 1:1,000, and goat anti-human AQP-2 (Santa Cruz Biotechnology), 1:200] were both diluted in PBS and applied either for 75 min at room temperature or overnight at 4°C. Sections were then washed twice for 5 min with high NaCl PBS (PBS plus 2.7% NaCl), once with PBS, and incubated with the secondary antibodies [donkey anti-rabbit 586 and donkey anti-goat 488 (Molecular Probes) at 1:1,000 and 1:200, respectively] for 1 h at room temperature. Sections were again washed twice with high NaCl PBS and once with PBS before mounting with VectaMount (Vector Laboratories). Sections were viewed with a Leica confocal laser scanning microscope. Pictures were processed (overlays) by using photoshop (Adobe Systems, Mountain View, CA). For cell counts, kidneys from three animals for each treatment were used and 5–10 pictures from at least two sections from each kidney taken. Cells were counted as being positive either for a4 (intercalated cells) or for AQP-2 (principal cells). Intercalated cells were further classified based on the predominant subcellular distribution of a4 immunostaining as described (23, 24).
Results
In the absence of bicarbonate, the initial intracellular pH (pHi) of single type A intercalated cells in isolated superfused OMCDs from mice on standard diet was 7.20 ± 0.01 (n = 133, 12 tubules). Removal of Na+ from the bath led to intracellular acidification, which was further enhanced after an NH4Cl prepulse (20 mM NH4Cl) to pH 6.62 ± 0.01. Such changes in cell pH have been described in other preparations (20). In the continued absence of Na+, a slow intracellular alkalinization of 0.040 ± 0.002 pHi units/min was observed (Fig. 1A). This alkalinization was mainly due to H+ extrusion by the vacuolar H+-ATPase, because application of the specific vacuolar H+-ATPase inhibitor concanamycin (200 nM; ref. 25) reduced the rate of alkalinization by 80.0 ± 12.2%, to 0.008 ± 0.001 pHi units/min (Table 1 and Fig. 1D). Adding Na+ to the bath led to a rapid alkalinization to the initial control pHi values. It is reasonable to conclude that this final step of pH recovery was due to Na+/H+ exchange (20).
Fig. 1.

Effect of aldosterone on vacuolar H+-ATPase activity. Aldosterone (10 nM) stimulated the Na+-independent pHi recovery of OMCD-intercalated cells after 15 min of preincubation (A–C). (A and B) Original pHi tracings from single intercalated cells in isolated mouse OMCD. (C) Summary of the data from all experiments. Of the Na+-independent pHi recovery 80 ± 12% is due to the activity of the vacuolar H+-ATPase and can be blocked by the specific V-H+-ATPase inhibitor concanamycin (200 nM). The increase in H+ extrusion after incubation with aldosterone is mainly due to the stimulation of vacuolar H+-ATPase as demonstrated by its sensitivity to concanamycin (D). Aldo, aldosterone; Conc., concanamycin.
Table 1. Summary of intracellular pH (pHi) measurements in single intercalated cells in mouse OMCD fragments.
| Initial pHi | Maximal aciditication, pHi | Na+ independent pHi recovery, ΔpH/min | Final pHi | No. of cells (OMCD fragments) | |
|---|---|---|---|---|---|
| Control | 7.20 ± 0.01 | 6.62 ± 0.01 | 0.040 ± 0.002 | 7.13 ± 0.01 | 133 (12) |
| Concanamycin | 7.22 ± 0.01 | 6.63 ± 0.01 | 0.008 ± 0.001* | 7.07 ± 0.01 | 179 (10) |
| Aldosterone, 1 nM | 7.21 ± 0.01 | 6.61 ± 0.01 | 0.045 ± 0.001 | 7.07 ± 0.01 | 32 (3) |
| Aldosterone, 10 nM | 7.20 ± 0.01 | 6.61 ± 0.01 | 0.093 ± 0.004* | 7.07 ± 0.01 | 262 (20) |
| Aldosterone + concanamycin | 7.19 ± 0.01 | 6.61 ± 0.01 | 0.019 ± 0.001*,** | 7.09 ± 0.01 | 223 (12) |
| Spironolactone | 7.22 ± 0.02 | 6.61 ± 0.01 | 0.038 ± 0.004 | 6.99 ± 0.02 | 31 (3) |
| Aldosterone + spironolactone | 7.21 ± 0.01 | 6.62 ± 0.01 | 0.091 ± 0.004* | 7.19 ± 0.01 | 132 (9) |
| Actinomycin | 7.18 ± 0.01 | 6.62 ± 0.01 | 0.043 ± 0.003 | 7.00 ± 0.02 | 49 (3) |
| Aldosterone + actinomycin | 7.26 ± 0.01 | 6.60 ± 0.01 | 0.071 ± 0.003* | 7.07 ± 0.01 | 102 (8) |
| Cycloheximide | 7.23 ± 0.01 | 6.60 ± 0.01 | 0.033 ± 0.003 | 6.95 ± 0.01 | 56 (5) |
| Aldosterone + cycloheximide | 7.22 ± 0.01 | 6.60 ± 0.01 | 0.080 ± 0.002* | 7.13 ± 0.04 | 169 (9) |
| Colchicine | 7.20 ± 0.01 | 6.62 ± 0.01 | 0.037 ± 0.002 | 7.10 ± 0.02 | 55 (4) |
| Aldosterone + colchicine | 7.22 ± 0.01 | 6.62 ± 0.04 | 0.035 ± 0.001 | 7.15 ± 0.01 | 146 (8) |
| Aldosterone + chelerythrine | 7.28 ± 0.01 | 6.52 ± 0.02 | 0.030 ± 0.003*** | 7.21 ± 0.02 | 29 (3) |
| Chelerythrine | 7.27 ± 0.02 | 6.47 ± 0.04 | 0.027 ± 0.003 | 7.22 ± 0.03 | 15 (3) |
The statistical mean ± SEM are given. If not stated otherwise substances were used at the following concentrations: aldosterone, 10 nM; concanamycin, 200 nM; spironolactone, 10 μM; actinomycin, 10 μM; cycloheximide, 40 μg/ml; and colchicine, 10 μM. *, results significantly different from control; **, significantly different from concanamycin alone; ***, significantly different from 10 nM aldosterone (P < 0.001).
Preincubation of OMCDs with 10 nM aldosterone at 37°C increased the Na+-independent alkalinization rate 2- to 3-fold, to 0.093 ± 0.004 pHi units/min (Table 1 and Fig. 1 B and C). Again, inhibition of vacuolar H+-ATPase with concanamycin reduced the rate of alkalinization to 0.019 ± 0.001 pHi units/min (Table 1 and Fig. 1D). This reduction indicates that increased H+ extrusion after aldosterone was mainly due to stimulation of vacuolar H+-ATPase activity. The residual H+ extrusion in the presence of concanamycin, however, was also increased and suggests that other transport pathways, such as H+/K+-ATPase, could be stimulated. Incubation of OMCDs with 1 nM aldosterone failed to increase the Na+-independent pHi recovery rate significantly (Table 1 and Fig. 1C). The aldosterone concentrations used occur in humans under conditions such a dietary Na+ restriction or hyperkalemia, which are associated with increased urinary H+ secretion (26). In addition, the concentration is in the same range where many nongenomic effects of aldosterone have been observed for several tissue preparations (16, 27).
The time course of stimulation of vacuolar H+-ATPase activity within 15 min of application suggests a “nonclassical,” nongenomic pathway of action. To further investigate the role of the genomic pathway that depends on mineralocorticoid receptor-mediated changes in transcription and protein synthesis (12), inhibitors of this pathway were used. Blocking of the mineralocorticoid receptors with spironolactone (10 μM) had no effect on the Na+-independent pHi recovery (Fig. 2A and Table 1). It also did not prevent the stimulatory effect of 10 nM aldosterone (Fig. 2 A and Table 1). Moreover, inhibition of transcription or of protein synthesis with actinomycin D (10 μM) and cycloheximide (40 μg/ml) [preincubation of OMCDs (15 min) before application of aldosterone (10 nM)] did not prevent the stimulatory effect of aldosterone on Na+-independent H+ extrusion. The stimulation, however, was slightly and significantly reduced. In control OMCD, actinomycin D had no effect on basal rates of H+ extrusion, whereas cycloheximide alone decreased vacuolar H+-ATPase significantly (Fig. 2B and Table 1). These results demonstrate that the stimulatory effect of aldosterone on vacuolar H+-ATPase activity is mainly mediated by a nongenomic pathway.
Fig. 2.

Stimulation of vacuolar H+-ATPase activity by aldosterone does not require the mineralocorticoid receptor, transcription, or protein synthesis. Parallel incubation of OMCDs with aldosterone (Aldo, 10 nM) and the mineralocorticoid receptor antagonist spironolactone (Spiro, 10μM) for 15 min did not prevent the stimulation of vacuolar H+-ATPase activity in intercalated cells (A). The inhibition of transcription or protein synthesis by preincubation with actinomycin D (Actin., 10 μM) or cycloheximide (Cyclo., 40 μg/ml) for 15 min also failed to prevent the stimulatory effect of aldosterone (B). *, results significantly different from aldosterone alone; #, results significantly different from control.
Good evidence exists that trafficking of vacuolar H+-ATPases plays an important role in the regulation of their activity in the plasma membrane of renal cells (1, 28–30). We tested the role of such trafficking in the aldosterone-induced stimulation of vacuolar H+-ATPase activity in OMCD-intercalated cells. Disruption of the microtubular network by preincubating tubule segments with colchicine (10 μM) for 15 min had no effect on H+ extrusion in control intercalated cells (Fig. 3 and Table 1). Preincubation with colchicine, however, completely prevented stimulation of vacuolar H+-ATPase activity by aldosterone (Fig. 3 and Table 1).
Fig. 3.

The stimulatory effect of aldosterone (Aldo) depends on an intact microtubular network. Disruption of the microtubular network by preincubation with colchicine (10 μM) for 10 min before exposure of aldosterone had no effect on the Na+-independent pHi recovery but completely prevented the stimulatory effect of aldosterone.
The effect of aldosterone on vacuolar H+-ATPase protein subcellular localization was also examined (Fig. 4). Mice were given i.p. injections of vehicle alone (0.9% NaCl) or of aldosterone (150 μg/kg of body weight) and examined after 30 min by using immunohistochemistry. The a4 subunit was used for immunohistochemistry because it has been shown to form an integral part of the vacuolar H+-ATPase. Moreover it has recently been shown that the a4 subunit is targeted to the membrane of A type intercalated cells in response to changes in the acid–base status (24). Immunohistochemical examination showed that intercalated cells of control mice contained the a4 subunit (ATP6V0A4) of the vacuolar H+-ATPase both in the apical membrane and in a subapical cytosolic compartment as described (24). In contrast, mice injected with aldosterone demonstrated enhanced staining of the luminal membrane associated with reduced cytosolic staining (Fig. 5). In control animals 32.5 ± 2.2% of OMCD-intercalated cells showed a predominant apical localization, whereas in aldosterone-injected animals the percentage of intercalated cells was increased to 77.2 ± 5.4%. These results are summarized in Table 2. Thus, aldosterone requires an intact microtubular network for the stimulation of vacuolar H+-ATPase activity in OMCD-intercalated cells.
Fig. 4.

Aldosterone alters the subcellular localization of vacuolar H+-ATPases containing the a4 isoform in mouse kidney medulla in vivo. Mice were injected either with 150 μg of aldosterone per kg of body weight or with the vehicle alone i.p. and were killed after 30 min. Immunostaining against the ATP6V0A4 (a4) subunit (red) of the vacuolar H+-ATPase and against AQP-2 (green) was performed. In control animals a4 immunoreactivity was found on the luminal membrane and in the cytosolic subapical compartment (A and B) in intercalated cells in the OMCD. In aldosterone-treated animals a4 staining appeared to be more luminal and less cytosolic (C and D), suggesting that vacuolar H+-ATPases are trafficked into membrane on stimulation by aldosterone.
Fig. 5.

Aldosterone (Aldo) requires PKC for stimulation of vacuolar H+-ATPase activity in intercalated cells. Preincubation for 10 min with the PKC inhibitor chelerythrine (Chel, 1 μM) prevented the aldosterone-induced stimulation of vacuolar H+-ATPase activity.
Table 2. Subcellular localization of the a4 vacuolar H+-ATPase subunit in single intercalated cells in the OMCD.
| IC, % | PC, % | Apical, % | Subapical, % | Diffuse, % | Cells, n | |
|---|---|---|---|---|---|---|
| Control | 36.5 ± 2.1 | 63.5 ± 2.1 | 32.5 ± 2.2 | 56.8 ± 1.7 | 10.6 ± 1.9 | 265 |
| Aldosterone | 35.5 ± 1.5 | 64.5 ± 1.5 | 77.2 ± 5.4* | 17.6 ± 4.1* | 5.2 ± 2.0 | 297 |
PC, principal cells; IC, intercalated cells. *, Significantly different from control (P < 0.001). Intercalated cells were classified according to the predominant intracellular staining of the a4 subunit. Aldosterone injection resulted in a percentual increase in intercalated cells with a predominantly apical staining.
Previous studies examining the nongenomic effects of aldosterone have implicated a variety of second messengers including Ca2+ and PKC (15, 16). To examine the role of intracellular Ca2+ and PKC in mediating the observed changes in H+-ATPase activity, we carried out measurements of intracellular Ca2+ by using FURA-2. Addition of aldosterone illicited a transient increase in intracellular Ca2+ from 71.7 ± 7.1 nM to 96.2 ± 9.4 nM (n = 4). Furthermore, incubation with a specific inhibitor of PKC (chelerythrine, 1 μM) abolished the stimulatory effect of aldosterone on H+-ATPase activity (see Fig. 5 and Table 1). Chelerythrine had no effect on alkalinization in the absence of aldosterone.
Discussion
A large body of evidence supports the view that mineralocorticoids play an important role in modulating net excretion of acid. These effects are exerted largely on such distal nephron segments as the cortical and medullary collecting ducts. Whereas adrenalectomy interferes with net acid excretion, aldosterone has been shown to correct the inability to establish steep urine-to-blood pH gradients (31). Furthermore, aldosterone increases the activity of vacuolar H+-ATPases in rat and rabbit cortical and medullary collecting ducts, and these effects are mimicked in isolated turtle bladder (32). Evidence that this mineralocorticoid is necessary for appropriate urinary acidification is also derived from patients with inborn or acquired aldosterone deficiency syndromes (distal renal tubular acidosis type IV) or in animal models with adrenalectomy or pharmacological blockade of the aldosterone pathway (10, 33–36). In general, it is accepted that sustained effects of mineralocorticoids on acid secretion involve genomic mechanisms.
The main result of the present study was the identification of a nongenomic pathway for the regulation of vacuolar H-ATPase activity in the terminal nephron segment. Several nongenomic effects of aldosterone have been described (17, 37–39). These involve rapid modulations of epithelial Na and K channel activity and of Na/H exchange (40, 41). Representative examples are stimulation of Na+/H+ exchanger by aldosterone in colon crypt cells, the Madin–Darby canine kidney cell line, and vascular smooth muscle cells (40, 42, 43). Nongenomic actions have also been observed for other steroid hormones, such as glucocorticoids, progesterone, and estrogen, and for androgens and vitamin D3 (16, 27).
The nongenomic effects of aldosterone have been shown to be mediated by various distinct intracellular signaling cascades that appear to be tissue- or cell-specific (16, 27). In our experiments, the nongenomic stimulatory effect of aldosterone required PKC and was associated with a rapid transient increase of intracellular Ca2+. A similar effect was reported in cultured cortical collecting duct cells where aldosterone transiently increased intracellular calcium, a response that could be prevented by inhibition of PKC (38). Our experiments show that an intact microtubular network is also necessary for the nongenomic targeting of the vacuolar H+-ATPase to the apical membrane by aldosterone. Taken together these results suggest that PKC-dependent phosphorylation may be an important element of regulation of vacuolar H+-ATPase. This regulation could involve modification of either microtubule or pump assembly. PKC has been shown to activate apical membrane Na+/H+ exchangers in proximal tubule cells by altering the state of phosphorylation (44).
In conclusion, the present studies provide evidence for a mechanism of stimulation of H+-ATPase by aldosterone. It involves a rapid nongenomic steroid effect on pHi regulation and expands the spectrum of mineralocorticoid effects on epithelia.
Acknowledgments
We thank Dr. Steve Hebert for helpful comments concerning the manuscript. This study was supported by Swiss National Foundation Grant 31-68318.02 (to C.A.W.) and National Institutes of Health Grants DK17433 (to G.G.) and DK14669, DK50230, and DK17433 (to J.P.G.).
Abbreviation: OMCD, outer medullary collecting duct.
References
- 1.Brown, D. & Breton, S. (2000) in The Kidney: Physiology and Pathophysiology, eds. Seldin, D. W. & Giebisch, G. (Lippincott Williams & Wilkins, Philadelphia), pp. 171-191.
- 2.Hamm, L. L. & Alpern, R. J. (2000) in The Kidney: Physiology and Pathophysiology, eds. Seldin, D. W. & Giebisch, G. (Lippincott Williams & Wilkins, Philadelphia), pp. 1935-1979.
- 3.Silver, R. B. & Soleimani, M. (1999) Am. J. Physiol. 276, F799-F811. [DOI] [PubMed] [Google Scholar]
- 4.Jaisser, F. & Beggah, A. T. (1999) Am. J. Physiol. 276, F812-F824. [DOI] [PubMed] [Google Scholar]
- 5.Geibel, J., Giebisch, G. & Boron, W. F. (1990) Proc. Natl. Acad. Sci. USA 87, 7917-7920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wagner, C. A., Giebisch, G., Lang, F. & Geibel, J. P. (1998) Proc. Natl. Acad. Sci. USA 95, 9665-9668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baum, M., Quigley, R. & Quan, A. (1997) Am. J. Physiol. 273, F595-F600. [DOI] [PubMed] [Google Scholar]
- 8.Stone, D. K., Seldin, D. W., Kokko, J. P. & Jacobson, H. R. (1983) J. Clin. Invest. 72, 77-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DuBose, T. D., Jr., & Caflisch, C. R. (1988) J. Clin. Invest. 82, 1624-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garg, L. C. & Narang, N. (1988) Kidney Int. 34, 13-17. [DOI] [PubMed] [Google Scholar]
- 11.Mujais, S. K. (1987) J. Lab. Clin. Med. 109, 34-39. [PubMed] [Google Scholar]
- 12.Verrey, F. (1999) Am. J. Physiol. 277, F319-F327. [DOI] [PubMed] [Google Scholar]
- 13.Verrey, F., Hummler, E., Schild, L. & Rossier, B. (2000) in The Kidney: Physiology and Pathophysiology, eds. Seldin, D. W. & Giebisch, G. (Lippincott Williams & Wilkins, Philadelphia), pp. 1441-1471.
- 14.Stockand, J. D. (2002) Am. J. Physiol. 282, F559-F576. [DOI] [PubMed] [Google Scholar]
- 15.Losel, R. M., Falkenstein, E., Feuring, M., Schultz, A., Tillmann, H. C., Rossol-Haseroth, K. & Wehling, M. (2003) Physiol. Rev. 83, 965-1016. [DOI] [PubMed] [Google Scholar]
- 16.Losel, R. & Wehling, M. (2003) Nat. Rev. Mol. Cell. Biol. 4, 46-55. [DOI] [PubMed] [Google Scholar]
- 17.Boldyreff, B. & Wehling, M. (2003) J. Steroid Biochem. Mol. Biol. 85, 375-381. [DOI] [PubMed] [Google Scholar]
- 18.MacDonald, P., MacKenzie, S., Ramage, L. E., Seckl, J. R. & Brown, R. W. (2000) J. Endocrinol. 165, 25-37. [DOI] [PubMed] [Google Scholar]
- 19.Yip, K. P., Tsuruoka, S., Schwartz, G. J. & Kurtz, I. (2002) Am. J. Physiol. 283, F1098-F1104. [DOI] [PubMed] [Google Scholar]
- 20.Wagner, C. A., Lukewille, U., Valles, P., Breton, S., Brown, D., Giebisch, G. H. & Geibel, J. P. (2003) Pflugers Arch. 446, 623-632. [DOI] [PubMed] [Google Scholar]
- 21.McLean, I. W. & Nakane, P. K. (1974) J. Histochem. Cytochem. 22, 1077-1083. [DOI] [PubMed] [Google Scholar]
- 22.Wagner, C. A., Finberg, K. E., Stehberger, P. A., Lifton, R. P., Giebisch, G. H., Aronson, P. S. & Geibel, J. P. (2002) Kidney Int. 62, 2109-2117. [DOI] [PubMed] [Google Scholar]
- 23.Bastani, B., Purcell, H., Hemken, P., Trigg, D. & Gluck, S. (1991) J. Clin. Invest. 88, 126-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stehberger, P., Schulz, N., Finberg, K. E., Karet, F. E., Giebisch, G., Lifton, R. P., Geibel, J. P. & Wagner, C. A. (2003) J. Am. Soc. Nephrol. 14, 3027-3038. [DOI] [PubMed] [Google Scholar]
- 25.Dröse, S. & Altendorf, K. (1997) J. Exp. Biol. 200, 1-8. [DOI] [PubMed] [Google Scholar]
- 26.Pratt, J. H., Rebhun, J. F., Zhou, L., Ambrosius, W. T., Newman, S. A., Gomez-Sanchez, C. E. & Mayes, D. F. (1999) Hypertension 34, 315-319. [DOI] [PubMed] [Google Scholar]
- 27.Losel, R., Feuring, M. & Wehling, M. (2002) J. Steroid Biochem. Mol. Biol. 83, 167-171. [DOI] [PubMed] [Google Scholar]
- 28.Schwartz, G. J. & Al-Awqati, Q. (1985) J. Clin. Invest. 75, 1638-1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwartz, G. J. & Al-Awqati, Q. (1986) Annu. Rev. Physiol. 48, 153-161. [DOI] [PubMed] [Google Scholar]
- 30.Tsuruoka, S. & Schwartz, G J. (1997) J. Clin. Invest. 99, 1420-1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilcox, C. S., Cemerikic, D. A. & Giebisch, G. (1982) Kidney Int. 21, 546-556. [DOI] [PubMed] [Google Scholar]
- 32.Al-Awqati, Q., Norby, L. H., Mueller, A. & Steinmetz, P. R. (1976) J. Clin. Invest. 58, 351-358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kornandakieti, C. & Tannen, R. L. (1984) Kidney Int. 25, 629-635. [DOI] [PubMed] [Google Scholar]
- 34.Arruda, J. A. & Kurtzman, N. A. (1980) Am. J. Physiol. 239, F515-F523. [DOI] [PubMed] [Google Scholar]
- 35.Henger, A., Tutt, P., Riesen, W. F., Hulter, H. N. & Krapf, R. (2000) J. Lab. Clin. Med. 136, 379-389. [DOI] [PubMed] [Google Scholar]
- 36.Lifton, R. P., Gharavi, A. G. & Geller, D. S. (2001) Cell 104, 545-556. [DOI] [PubMed] [Google Scholar]
- 37.Alzamora, R., Marusic, E. T., Gonzalez, M. & Michea, L. (2003) Endocrinology 144, 1266-1272. [DOI] [PubMed] [Google Scholar]
- 38.Harvey, B. J. & Higgins, M. (2000) Kidney Int. 57, 1395-1403. [DOI] [PubMed] [Google Scholar]
- 39.Wehling, M. (1997) Annu. Rev. Physiol. 59, 365-393. [DOI] [PubMed] [Google Scholar]
- 40.Gekle, M., Golenhofen, N., Oberleithner, H. & Silbernagl, S. (1996) Proc. Natl. Acad. Sci. USA 93, 10500-10504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Urbach, V., Van Kerkhove, E., Maguire, D. & Harvey, B. J. (1997) J. Physiol. 491, 111-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Winter, D. C., Schneider, M. F., O'Sullivan, G. C., Harvey, B. J. & Geibel, J. P. (1999) J. Membr. Biol. 170, 17-26. [DOI] [PubMed] [Google Scholar]
- 43.Ebata, S., Muto, S., Okada, K., Nemoto, J., Amemiya, M., Saito, T. & Asano, Y. (1999) Kidney Int. 56, 1400-1412. [DOI] [PubMed] [Google Scholar]
- 44.Weinman, E. J., Dubinsky, W. & Shenolikar, S. (1989) Kidney Int. 36, 519-525. [DOI] [PubMed] [Google Scholar]