

Abstract
Background
Over 60 Asian and European families with cortical myoclonic tremor and epilepsy have been reported under various names. Cerebellar changes may be part of the syndrome. In this study, we report the neuropathology findings in a new Dutch familial cortical myoclonic tremor with epilepsy case and review the literature on this syndrome.
Methods
Neuropathological investigations were performed for a third case of the Dutch pedigree. In addition, we searched the literature for pedigrees meeting the criteria for benign familial myoclonic tremor and epilepsy.
Results
Our third Dutch case showed cerebellar Purkinje cell changes and a normal cerebral cortex. The pedigrees described show phenotypical differences, cerebellar symptoms and cerebellar atrophy to a variable degree. Japanese pedigrees with linkage to chromosome 8q have been reported with milder disease features than members of Italian pedigrees with linkage to chromosome 2p. French pedigrees (5p) possibly show even more severe and progressive disease, including cognitive changes and cerebellar features.
Discussion
Currently, familial cortical myoclonic tremor is not listed by the International League Against Epilepsy, although it can be differentiated from other epileptic syndromes. Genetic heterogeneity and phenotypical differences between pedigrees exist. Cerebellar changes seem to be part of the syndrome in at least a number of pedigrees.
Keywords: Cortical myoclonus, tremor, epilepsy, cerebellar, benign, hereditary
Introduction
Since the first patients with distal myoclonic jerks and epileptic seizures with a non-progressive course were described in the 1980s in Japan, about 60 families with familial cortical myoclonic tremor and epilepsy (FCMTE) have been reported around the world under various names (ADCME, BAFME, FAME, Crt Tr, FCMT, FEME, FMEA, HTE, FCTE; Table 1). This familial syndrome is characterized by distal tremulous movements and generalized seizures. Inheritance is autosomal dominant. Seizures usually manifest after the onset of the distal tremulous movements. These tremulous movements are in fact small high-frequency myoclonic jerks, which probably have a cortical origin. Antiepileptic drugs (AEDs) reduce the number of seizures and diminish the tremulous movements. A gene has not been identified yet, and linkage analysis showed different loci in different pedigrees, or showed exclusion of linkage to known loci.1–15 Pathophysiology is not entirely clear. In addition to features of cortical functional changes, cerebellar signs and cerebellar pathological changes were described in the Dutch pedigree.14,16–18 Here, we describe neuropathology findings in a third, novel Dutch FCMTE case (Study I), and, in addition to our previous review and the recent review of Striano and colleagues,19 review the literature on this syndrome (Study II). The neuropathology and the differences between the pedigrees are discussed in the general discussion. We hypothesize that cerebellar features are part of the syndrome. Furthermore, we argue that this disease is a distinct syndrome that can be differentiated from other epileptic syndromes. To date, FCMTE has not officially been recognized by the International League Against Epilepsy (ILAE).20 A subclassification might be necessary if phenotypical differences are strongly associated with genetic differences.
Table 1. Familial Cortical Tremor/Myoclonus Syndromes with Benign Course.
| ADCME | Autosomal dominant cortical myoclonus and epilepsy |
| BAFME | Benign adult familial myoclonic epilepsy |
| Crt Tr | Cortical tremor |
| FAME | Familial adult myoclonic epilepsy |
| FCMT | Familial cortical myoclonic tremor |
| FCMTE | Familial cortical myoclonic tremor with epilepsy |
| FCTE | Familial cortical tremor with epilepsy |
| FEME | Familial essential myoclonus and epilepsy |
| FMEA | Familial benign myoclonus epilepsy of adult onset |
| HTE | Heredofamilial tremor and epilepsy |
Study I: Neuropathology
Case report
A Dutch woman, a member of the Dutch FCMTE pedigree described earlier,14,16–18,21–23 died at the age of 50 years from pulmonary metastases from colorectal carcinoma (Figure 1, patient III:10; Table 2). Post-mortem examination was performed with informed consent. She had suffered from progressive irregular trembling of the limbs provoked by movement from the age of 38 years, and tonic–clonic generalized seizures from the age of 42 years. She was treated with valproic acid and carbamazepine, which diminished the tremulous movements. She also had visual complaints and memory problems. Past medical history revealed Crohn's disease and type 2 diabetes mellitus in addition to bowel cancer. During neurological examinations, high-frequency myoclonic jerks of the hands were seen. Also, a downbeat nystagmus was noticed. An electroencephalogram (EEG) showed some focal changes over the centrotemporal areas. Electromyography (EMG), recorded over wrist flexors and extensors and the first dorsal interosseus muscles, showed short irregular bursts with a peak frequency around 16 Hz. The presence of a long latency reflex (LLR) and giant sensory evoked potential (g-SEP) were consistent with the diagnosis of FCTME. Magnetic resonance imaging (3 T MRI) at age 42 was reported to show some slight cerebellar vermal atrophy.
Figure 1. Pedigree of the Dutch Family with Familial Cortical Myoclonic Tremor with Epilepsy (FCMTE).

▪, affected male; •, affected female; •, possibly affected; □,○, no established diagnosis; /, diseased; PA, pathological investigations performed; *, one or more additional functional tests performed. The described patient is indicated by an arrow.
Table 2. Clinical and Electrophysiological Features of Relatives of the Dutch Familial Cortical Myoclonic Tremor with Epilepsy Pedigree, in Whom Additional Tests Were Performed16–18, 22, 23, 53.
| FCMTE | Age | Gender | Symptoms (age at onset) |
AEDs | Electrophysiology |
Additional Tests Performed |
|||||||||
| Tremor | Myocl Seiz | GTCS | EEG | g-SEP | LLR | PA | Polymyography | Coherence | fMRI | TMS | Eye move | ||||
| II:3 | 811 | F | 40 | 44? | 44 | VPA, PHT | ± | n.d. | n.d. | + | n.d. | n.d. | n.d. | n.d. | n.d. |
| II:11 | 681 | F | + | ? | + | PB, CBZ | s-w | n.d. | n.d. | + | n.d. | n.d. | n.d. | n.d. | n.d. |
| III:1 | 60 | M | 45 | – | 52 | VPA, CBZ | s-w | + | – | n.d. | + | + | + | + | + |
| III:5 | 57 | M | 30 | 30 | 43 | OCB, CZP | s-w | – | ? | n.d. | + | + | + | + | + |
| III:102 | 501 | F | 38 | – | 42 | VPA, CZP | ± | + | + | + | + | + | + | + | + |
| III:19 | 51 | M | 19 | 20 | 20 | PB, VPA, CBZ | s-w | n.d. | n.d. | n.d. | + | + | + | n.d. | n.d. |
| III:20 | 54 | M | 12 | 13? | 31 | VPA, CZP | ? | n.d. | n.d. | n.d. | + | + | + | n.d. | n.d. |
| IV:1 | 41 | M | 22 | – | – | – | normal | + | + | n.d. | + | + | + | + | + |
| IV:2 | 39 | F | 20 | – | – | – | normal | + | + | n.d. | + | + | + | + | + |
| IV:8 | 27 | F | 12 | – | – | – | normal | + | + | n.d. | + | + | + | + | + |
| IV:9 | 21 | F | + | – | – | – | n.d. | + | + | n.d. | n.d. | n.d. | n.d. | n.d. | + |
Abbreviations: AEDs, antiepileptic drugs; CBZ, clobazam; Coherence, corticomuscular and intermuscular coherence analysis; CZP, clonazepam; EEG, electroencephalogram; Eye move, eye movement recordings; F, female; FCMTE, familial cortical myoclonic tremor with epilepsy; fMRI, functional magnetic resonance imaging; g-SEP, giant sensory evoked potential; GTCS, generalized tonic–clonic seizure; LLR, long latency reflex; M, male; Myocl Seiz, myoclonic seizure; n.d., not done; OCB, oxcarbamazepine; PA, neuropathology post-mortem; PB, phenobarbital; PHT, phenytoin; s-w, spike-wave complexes; TMS, transcranial magnetic stimulation; VPA, valproic acid.
+, present or performed; –, not observed or none; ±, some, not specific; ?, not known.
Age of death.
Current case.
Tissue preparation and immunocytochemistry
Post-mortem examination was carried out.16, 23 The brain was fixed in 10% buffered formalin solution for 3 weeks. After fixation, tissue blocks from all brain regions were embedded in paraffin. Sections 6 µm thick were processed for conventional staining, including hematoxylin and eosin, Nissl, Bielschowsky, and Klüver–Barrerastaining. Several blocks of the cerebellar and cerebral cortex were used for calbindin, neurofilament, and phosphorylated tau immunohistochemistry, carried out as previously described.16, 23
Neuropathological findings
General post-mortem findings performed elsewhere included abnormalities consistent with a previous history of colorectal cancer with extensive surgery, pulmonary metastases, pyelonephritis, and splenomegaly. The brain, with a weight of 1,020 g, showed slight atrophy of the frontoparietal areas. Macroscopically, no other abnormalities were seen; in particular, there was no cerebellar atrophy or vermal atrophy; or abnormalities in the substantia nigra, basal ganglia, white matter, or cortex. Microscopically, a generally normal cerebral cortex was seen, without cell loss. With the stains used in our study, microscopy showed normal layering of the cerebral cortex without prominent cell loss. Non-specific reactive changes, including activation of microglia, were observed in both gray and white matter. There were no neurodegenerative changes. Amyloid depositions were absent, and there was no tau-positivity. The basal ganglia, thalamus, claustrum, and brainstem also showed some reactive changes but were otherwise normal. The cerebellar cortex showed clear loss of Purkinje cells (Figure 2A), and silver stain demonstrated the presence of empty baskets (Figure 2B). The remaining Purkinje cells often displayed abnormal morphology with numerous radial dendritic sprouts (Figure 2C–E) and reduced dendritic arborization was observed in the molecular layer (Figure 2D). Other abnormalities included swollen dendrites in the molecular layer (Figure 2F) and torpedoes (Figure 2G). The dentate nucleus showed loss of neuronal cells and an increased number of glial cells. Diffuse microglia activation was seen in the white matter. The granular layer showed no abnormalities.
Figure 2. Histological Findings in the Cerebellum.
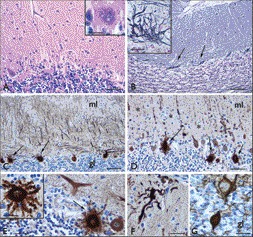
(A) Hematoxylin and eosin: loss of Purkinje cells ; insert, cell with abnormal morphology and unclear outlines of the cytoplasmic membrane. (B) Bielschowski silverstaining: empty baskets (arrows and insert in B). (C–F) Calbindin staining: presence of numerous Purkinje cells with radial sprouting (arrows in C–E and insert in E), reduced and abnormal dendritic arborization of Purkinje cells in the molecular layer (ml; asterisks in D), with swelling of dendritic arborizations (F). (G) Neurofilament staining: swellings of Purkinje cell axons (torpedoes) within the granular layer (gl). Scale bars: A, D: 80 µm; B–C: 160 µm; E–F: 40 µm; G and insert in B: 30 µm; inserts in A and E: 20 µm.
Study II: Literature search
Methods
For this review we searched PubMed using the following terms: cortical tremor, cortical myoclonus, epilepsy, benign course, autosomal dominant, adult onset, and familial. Included were cases of pedigrees who met the following diagnostic criteria: 1) distal action and postural tremor/fine myoclonus; 2) generalized tonic–clonic seizures or electrophysiological features of cortical reflex myoclonus (g-SEP and LLR); and 3) a family history of tremor/epilepsy consistent with autosomal dominant inheritance and no other cause of tremor, epilepsy, and additional symptoms like ataxia, parkinsonism, dementia, etc.
Results
A total of 44 publications describing 56 pedigrees fulfilled the search criteria, with eight new pedigrees2,3,6,10–13 since our previous review.17 Pedigrees consisted of 939 persons, of which 486 (52%) were affected. Clinical and neurophysiological data and results of linkage analysis are summarized in Table 3.
Table 3. Described Pedigrees with Core Disease Characteristics.
| Descent | Genetics | Origin, # Family | Clinical Features |
Electrophysiology |
Structural |
Additional Symptoms, Other Findings | Summary | ||||||
| Tremor, Myoclonus |
Seizures |
Seizure Type | JLA | g-SEP | LLR | EEG | Imaging (cases) | PA (cases) | |||||
| Age at Onset (mean) | |||||||||||||
| Asian | 8q | Japanese, 57, 8 | 18–45 maj >30 | ? | GTC | + | + | + | G-PSW, PPR, PSW, PMR | atr (3) n.a. (>14) | n.d. | – | Classical phenotypeAge at onset at adulthoodInfrequent seizuresNo other neurological signsElectrophysiological abnormalities |
| n.d. | Japanese, 3526–28,32,33,36,37,61,63–66 | 16–70 maj >25 | 17–54 maj >30 | GTC, Ph | + | + | + | PSW, PPR, SW, Sp | atr (3)n.a. (25)inf (11) | n.a. (4) | Rare: nightblindness, behavioral arrest | ||
| Excl 2p, 8q | Chinese, 12 | 5–?(34) | ? | GTC, M | n.d. | n.d. | n.d. | M, SW, PSWSlow waves13/? presymptomatic changes detected | n.d. | n.d. | Schizophrenia in family | Classical phenotype with earlier age of onset in the youngest generations | |
| European | 2p | Italian, 71,4,6,9–11 | 11–50maj >20 | 12- 59maj >25 | G, GTC, Ph, CP, M | + | + | + | Sp, SW, PPR, PMR, GPA, PSW, SW | atr (3)n.a. (27) | n.d. | Visuospatial impairment; Eyelid twitching; Voice tremor; Cognitive impairment;TMS cortical hyperexcitability, normal sensorimotor integration | Symptoms appear earlierComplex partial seizuresMild cognitive impairment |
| Also in presymptomatic 3/711Absent in 1 pedigree11 | |||||||||||||
| n.d. | Italian, 138 | 12–57 | 5–18 | GTC, Abs | + | + | + | Sp, SW, PPR | atr (2) | n.d. | – | ||
| Turkish, 135 | 29–? | 30 | GTC | n.d. | n.d. | n.d. | G-Sp, SW, PPR | n.a. (1) | n.d. | Migraine | |||
| 5p | French, 13, 15, 30 | 10–47 (30.8) | 24–41(29.1) | GTC, Ph, CM, PS | + | + | + | Sp, PPR, PS | n.d. | n.d. | Progression in gait symptoms; Dysarthria; Ophthalmic migraine; sensitivity to exercise;GTC preceding M (5/16); | Later onsetNo cognitive impairmentGait disordersIndication of progression | |
| Excl 2p, 8q | Spanish, 15 | 30–60 (41) | 30–67 (44.6) | GTC | + | + | + | G-PSW | n.a. (5) | n.d. | – | Childhood onsetPyramidal signsCerebellar dysfunctionFrequent seizuresCognitive impairmentProgression in symptoms | |
| Dutch, 114, 16, 17, 22, 23 | 12–45 (23.5) | 13–44 (43) | GTC, M, Ph | – | + | + | SW, PPR | atr (2)n.a. (2) | + (3) | TMS cortical hyperexcitability; nystagmus slight cognitive decline | |||
| Italian, 113 | 3–12 | 23–34 | GTC, CMPh | + | + | + | SW, PMR, Sp | n.d. | n.d | Prominent photic induced myoclonus and epilepsy; changing symptoms with age; Mild axial ataxia; behavioral disorder | |||
| South African, 212Intermarriage between original inhabitants and European settlers | 13–31 (20.9) | ? | GTC | + | + | + | Abnormal background,PSW, Sp | atr (8)n.a. (2) | + (1) | Frequent seizures; cognitive impairment; signs of pyramidal and cerebellar dysfunction, progression in symptoms | |||
Abbreviations: Abs, absence; atr , atrophy; CM, cortical myoclonus; CP, complex partial; EEG, electroencephalography; excl, excluded; G, generalized; GPA, generalized paroxysmal activity; G-SEP, giant sensory evoked potential; GTC, generalized tonic-clonic; inf, infarct; JLA, jerk locked averaging; LLR, long latency reflex; maj, majority; M, myoclonic; n.a., no abnormalities; n.d., not done; PA, pathology; Ph, photosensitivity; PMR, photomyoclonic response; PPR, photoparoxysmal responses; PSW, polyspike-wave complexes; PS, partial seizures; Sp, spikes; SW, spike-wave complexes; TMS, transcranial magnetic stimulation.
+, abnormal; –, normal; # family, number of described families; ?, not known;
Reports differed largely on several aspects. Often, only a selection of patients was described per pedigree, and no standardized clinical scales were used to rate symptoms. Additional investigations differ with respect to the type and methods used, and also results are reported or interpreted differently. These factors hamper straightforward comparisons between pedigrees, making it difficult to make generalized statements or to identify specific traits per pedigree.
General findings in cortical tremor pedigrees
Looking at all pedigrees in general, age of onset ranged from 3 to 70 years. Although the tremulous movements of the upper limbs are often the first symptom in FCMTE syndromes, the majority of patients seek medical attention because of epileptic or myoclonic attacks. Epilepsy was the first symptom presenting in approximately 10% of patients. Severity of myoclonus was not always related to seizure frequency. Electroencephalography has been reported to show generalized (poly)spikes and waves, photoparoxysmal responses, and photomyogenic responses (Table 3). Polymyography, usually of muscles of the upper limbs, shows arrhythmic high-frequency burst-like discharges of very short duration (<50 ms), typical for cortical myoclonus.24 Other electrophysiological characteristics include features of cortical reflex myoclonus, such as a g-SEP and a LLR, pointing to cortical hyperexcitability.17 The cortical origin of the tremulous movements was confirmed with EEG-EMG coherence18 and EMG-functional (f)MRI studies.22 Presymptomatic electrophysiological abnormalities including a g-SEP have been reported in three of seven asymptomatic family members (9–18 years) of an Italian pedigree.10 Hitomi and colleagues25 observed increasing g-SEP amplitudes with aging. g-SEPs and long latency reflexes are not detected in all patients, and it has been reported that antiepileptic drugs (AEDs) can normalize these electrophysiological abnormalities.11, 17 Antiepileptic drugs, especially clonazepam and valproic acid, were generally reported to diminish the tremulous movements and epileptic seizures.1,5,8,14,26–28 More recent data suggest a beneficial antimyoclonic effect of levetiracetam.29, 30 Other AEDs, like clobazam, phenytoin, and carbamazepine, have been reported to be effective, usually in polydrug regimens. Gabapentin may precipitate myoclonus status.31
Neuropathological studies
Reports on pathological findings in FCMTE are still scarce. The pathological findings of the third Dutch case reported here are in line with the previously reported findings. In the Dutch pedigree, post-mortem studies in two patients revealed extensive cerebellar Purkinje cell degeneration and rather typical Purkinje cell changes and no or limited changes of the sensorimotor cortex.16, 23 In a family member of a South African pedigree (South African pedigree, Table 3),12 the pathological investigations showed Purkinje cell loss, dentate atrophy, and neuronal loss, and gliosis in the olives and pallidum. Two studies, which were written in Japanese, did not carry out immunohistochemistry. To the best of our knowledge they reported no symptom-related neuropathological abnormalities in four cortical tremor patients.32, 33
Linkage studies, genetic heterogeneity
To date, no causative gene has been identified for this genetically heterogeneous disorder. Currently, linkage to three chromosomes has been reported. Five Japanese pedigrees showed linkage to chromosome 8q23.3-q24.11 (FAME 1).7, 8 Seven Italian pedigrees showed linkage to 2p (FAME 2).1, 4, 6, 9, 11, 19 Madia and colleagues6 provided evidence of an extended common founder haplotype in five Italian families. Linkage to 5p (FAME 3) has been found in one French pedigree.3, 15, 30 A South African pedigree resulting from intermarriage between people of European descent and original inhabitants with myoclonus and epilepsy, was reported as FAME 3 after exclusion of linkage to 8q and 2p.12 Also, in a Chinese pedigree2 linkage to the known loci were excluded. An additional locus on 7p has been suggested.34
Phenotypical heterogeneity
Symptoms possibly differ between the pedigrees with linkage to 8q (Japanese), 2p (Italian), and 5p (French). In Japanese pedigrees (classical phenotype), the age of onset is around the third decade, tremor is followed by infrequent seizures, possible photosensitivity is present, and mental retardation is absent (Table 3). Symptoms in European pedigrees seem to appear earlier (Table 3). In addition to the classical symptoms, pedigrees show atypical phenomena.3,4,12–15,23,30,35–37 Clinical and electrophysiological features suggest widespread involvement of cortical and subcortical areas, cerebellar involvement, and more progressive disease in the non-Japanese pedigrees. Involvement of areas outside the motor cortex is suggested by cognitive decline, reported in an increasing number of papers.4, 11, 12, 14, 38 A decline in intelligence was observed, especially in patients with more severe epilepsy. It is not clear if the deterioration can be solely explained by the use of AEDs. Other (possible) cortical features include complex partial seizures, intractable seizures, and mental retardation. Other findings, sometimes in isolated pedigrees, include night blindness,36 motionless state,37 migraine,35 visuospatial impairment,11 sensitivity to glucose deprivation/exercise as an aggravating factor, ophthalmic migraine, and gait disorders.3, 15, 30 Transcranial magnetic stimulation (TMS), which was only performed in European pedigrees, showed cortical functional changes.11, 23 The TMS results of Suppa and colleagues11 suggested hyperexcitability of the primary motor cortex, but normal sensorimotor integration.
Several findings specifically point to cerebellar involvement in non-Japanese pedigrees. In the French pedigree, mild ataxia and gait disorders have been reported.15, 30 Furthermore, cerebellar involvement is suggested by (downbeat) nystagmus that has been observed to increase with hyperventilation in the Dutch pedigree.23 Dysarthria has been reported in the South African and French pedigrees.12, 30 Pathological investigations in the Dutch and South African pedigrees point to cerebellar involvement, as described above.12, 16, 23 Also, imaging studies revealed cerebellar atrophy in a few affected family members.11, 12, 14 Proton MR spectroscopy, measuring chemical composition of the brain, was able to detect cerebellar neuronal metabolic dysfunction in an Italian pedigree, which led to the conclusion that the cerebellum is a prominent site of dysfunction.39
Although the syndrome is generally considered to be benign, progression of symptoms has been reported in Italian13 and French3 pedigrees. Coppola and colleagues40 investigated the long-term evolution in three Italian pedigrees and concluded slight age-dependent progression. Additional neuropsychiatric and neuropsychological dysfunctions were presented in long-term evolution. Furthermore, a fairly progressive course was reported in the South African pedigree with European ancestors.12 Carr and co-workers12 described a progressive course in their pedigree and EEG abnormalities, including abnormal background activity and abundant polyspike discharges. It has been suggested that the disorder described by Carr and co-workers should be placed within the group of progressive epilepsies.41 The phenotype, however, does not fit in the spectrum of progressive myoclonus epilepsies. Ikeda and colleagues42 observed in four Japanese families that an earlier onset age of cortical tremor was determined in three out of four pedigrees in the next generation. These observations suggest clinical anticipation.
General discussion
All pedigrees share evidence of a cortical origin for tremulous movements. The tremulous movements are reported to be stimulus sensitive, and electrophysiological investigations revealed cortical hyperexcitability. Clonazepam and valproic acid diminish the tremulous movements. The number of epileptic seizures is decreased and electrophysiological features are normalized by these drugs. In the Dutch pedigree, coherence analysis points to a cortical drive of the tremulous movements, and in the EMG-fMRI study, the tremulous movements correlated with activity of the sensorimotor cortex. Cortical hyperexcitability determined in TMS studies was thought to occur through alterations of the inhibitory neurotransmitter gamma-aminobutyric acid,43, 44 although alternative explanations might be possible.
Here, we report the neuropathological findings in a third case of the Dutch pedigree. The post-mortem investigations, including immunohistochemistry, reveal cerebellar degeneration. Previous studies in the Dutch pedigree and a single case of the South African pedigree are in line with these findings. The Purkinje cell changes in these patients seem to be quite unique. The cerebellar changes show similarities to changes described in spinocerebellar ataxias (SCAs), especially SCA6, a pure cerebellar syndrome.45 Also, there are some similarities with essential tremor (ET).46 Patients with ET also may show Purkinje cell changes. However, the findings are not identical. Furthermore, these findings seem to differ from the pathological changes known in the progressive myoclonic epilepsies and epilepsy.47 It can be concluded that these pathology findings in FCMTE can be differentiated from these other disorders.
The pathological findings indicate an association between cerebellar changes and cortical myoclonic tremor and epilepsy that does not seem to be coincidental. Cerebellar symptoms and signs have been reported in several cortical tremor pedigrees, including dysarthria, mild ataxia, and eye movement abnormalities.3,10–12, 15, 16, 22, 23 Furthermore, imaging studies report cerebellar atrophy. However, it can be questioned whether cerebellar changes in cortical myoclonic tremor are primary, secondary, or just coexisting.
The association between cortical myoclonus and cerebellar changes is well known and has already been pointed out by Hunt,50 and has been described in celiac disease.48–51 Cortical myoclonus, and also epilepsy, in combination with cerebellar pathology, have been hypothesized to be a result of primary cerebellar pathology by dysfunction of the cerebello-thalamo-cortical loop.51, 52 Reduced cerebellar output due to Purkinje cell abnormalities project to the sensorimotor and frontal cortices via the thalamus, resulting in cortical myoclonus and epileptic attacks. Results of an fMRI study were in line with this hypothesis, showing decreased cerebellar activity during intentional hand tapping.22
An alternative hypothesis for the cortical hyperexcitability in combination with cerebellar Purkinje cell changes is that they are both caused by a common factor, leading to cortical functional changes on the one hand, and degenerative cerebellar changes on the other hand. It has been suggested that FCMTE, like other idiopathic epilepsies with autosomal dominant inheritance, is a channelopathy.54–57 Night blindness and migraine were reported in three different cortical tremor pedigrees, and both familial night blindness and familial hemiplegic migraine are associated with mutation in a calcium channel.36, 58 Furthermore, the pathological findings showed remarkable similarities to changes described in SCA6, a pure cerebellar syndrome caused by a CACNA1A mutation.45, 59 The eye movement abnormalities in FCMTE patients also showed striking similarities with those seen in SCA6 patients.
The neuropathological changes of the cerebellum have also been hypothesized to be secondary, fulfilling a compensatory role to overrule the involuntary tremulous movements. In the Dutch FCMTE pedigree, however, a downbeat nystagmus could be observed in very young patients who had a very subtle tremor and who did not meet the criteria for FCMTE at that time. Another hypothesis that can explain the changes of the cerebellum to be secondary is that the changes are due to an effect of AEDs. However, this hypothesis seems unlikely as the neuropathology findings are fairly uniform over different pedigrees, independent from the AEDs used. Furthermore, although Purkinje cell loss has been reported with some AEDs, the observed morphological changes have not been reported and usually are not generalized.60
It is yet not clear what the role of the cerebellum is in FCMTE. Cerebellar involvement seems to vary between pedigrees, and is present in European families in particular. Even though cerebellar symptoms are often limited or subtle, structural abnormalities, including pathological findings and imaging, have been reported throughout the pedigrees. FCMTE-like syndromes share general disease characteristics that are clinically and electrophysiologically distinct; however they are still not recognized by the ILAE. A subclassification might be necessary if phenotypical differences are strongly associated with genetic differences.
The heterogeneity of the phenotype has been emphasized before. There seem to be clinical differences between pedigrees related to genetic background: the disease in Japanese pedigrees being more benign, and the European pedigrees showing more additional symptoms and a (slightly) more progressive course. Caution must be taken, however, with respect to this conclusion: as in most reports, clinical scales (if used at all) and additional investigations differ largely. In the first reports (Japan) the benign nature of the syndrome was stressed. In subsequent reports describing European pedigrees, a progression of symptoms was noted, but also in Japan, progressive disease has been reported during long-term follow-up.8, 61 Thus, differences between pedigrees might (partly) be biased. On the other hand, linkage to 8q has solely been described in pedigrees of Japanese descent. In Italian pedigrees with linkage to 2p, a founder effect was reported, and linkage to 5p was reported in pedigrees outside Italy. Therefore, differences between pedigrees might be based on different genetic backgrounds. It is, however, likely that the genes are closely related or share functions. Several approaches to identify the gene, such as analysis for mutations in known genes that cause disorders with similar symptoms, like dentatorubral-pallidoluysian atrophy, showed no results,62 and future studies are needed.
Conclusion
There is little doubt that the cerebellum is involved in the pathophysiology of FCMTE. Cerebellar involvement seems to be a part of the syndrome: cerebellar symptoms and cerebellar atrophy are reported across pedigrees to a variable degree. The exact relationship between the cortical hyperexcitability, cerebellum, and its pathogenesis remains unclear. In contrast to the ubiquitous evidence for a cortical origin of the tremulous movements in FCMTE, the role of the cerebellum has not fully been clarified yet. Clinically and electrophysiologically, FCMTE is well-defined, and can be differentiated from other neurological disorders despite its heterogeneity. FCMTE includes core symptoms that mainly present with myoclonic jerks and a spectrum of additional symptoms that differ between pedigrees, and it is not merely an epilepsy syndrome. Furthermore, FCMTE is known to be benign; however, several progressive cases have been described. Progressive symptoms reported in new cases and in earlier age at onset within pedigrees might indicate genetic anticipation. Progression is less severe than in progressive myoclonic epilepsy. FCMTE seems to be a distinct syndrome; nevertheless, it is not currently listed by the ILAE. Systematic clinical, functional, neuropathological, and especially genetic studies will not only help to further classify this rare disorder, but also to gain insight into the course of the disease, and to optimize treatment. Also, insight into the pathophysiology of this rare condition with autosomal dominant inheritance might elucidate other, more common disorders with cortical myoclonus or epilepsy.
Acknowledgments
We would like to thank Dr. M. Nap, Pathologist, Atrium Medical Center Heerlen, The Netherlands, for performing the body autopsy and isolating brain tissue for the further investigations described in this paper.
Footnotes
Funding: None.
Financial Disclosures: None.
Conflict of Interest: The authors report no conflict of interest.
References
- 1.De Falco FA, Striano P, De Falco A, et al. Benign adult familial myoclonic epilepsy: genetic heterogeneity and allelism with ADCME. Neurology. 2003;60:1381–1385. doi: 10.1212/01.WNL.0000055874.24000.4A. [DOI] [PubMed] [Google Scholar]
- 2.Deng FY, Gong J, Zhang YC, et al. Absence of linkage to 8q23.3-q24.1 and 2p11.1-q12.2 in a new BAFME pedigree in China: indication of a third locus for BAFME. Epilepsy Res. 2005;65:147–152. doi: 10.1016/j.eplepsyres.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 3.Depienne C, Magnin E, Bouteiller D, et al. Familial cortical myoclonic tremor with epilepsy: the third locus (FCMTE3) maps to 5p. Neurology. 2010;74:2000–2003. doi: 10.1212/WNL.0b013e3181e396a8. [DOI] [PubMed] [Google Scholar]
- 4.Guerrini R, Bonanni P, Patrignani A, et al. Autosomal dominant cortical myoclonus and epilepsy (ADCME) with complex partial and generalized seizures: a newly recognized epilepsy syndrome with linkage to chromosome 2p11.1-q12.2. Brain. 2001;124:2459–2475. doi: 10.1093/brain/124.12.2459. [DOI] [PubMed] [Google Scholar]
- 5.Labauge P, Amer LO, Simonetta-Moreau M, et al. Absence of linkage to 8q24 in a European family with familial adult myoclonic epilepsy (FAME). Neurology. 2002;58:941–944. doi: 10.1212/WNL.58.6.941. [DOI] [PubMed] [Google Scholar]
- 6.Madia F, Striano P, Di BC, et al. Benign adult familial myoclonic epilepsy (BAFME): evidence of an extended founder haplotype on chromosome 2p11.1-q12.2 in five Italian families. Neurogenetics. 2008;9:139–142. doi: 10.1007/s10048-008-0118-4. [DOI] [PubMed] [Google Scholar]
- 7.Mikami M, Yasuda T, Terao A, et al. Localization of a gene for benign adult familial myoclonic epilepsy to chromosome 8q23.3-q24.1. Am J Hum Genet. 1999;65:745–751. doi: 10.1086/302535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Plaster NM, Uyama E, Uchino M, et al. Genetic localization of the familial adult myoclonic epilepsy (FAME) gene to chromosome 8q24. Neurology. 1999;53:1180–1183. doi: 10.1212/WNL.53.6.1180. [DOI] [PubMed] [Google Scholar]
- 9.Striano P, Chifari R, Striano S, et al. A new benign adult familial myoclonic epilepsy (BAFME) pedigree suggesting linkage to chromosome 2p11.1-q12.2. Epilepsia. 2004;45:190–192. doi: 10.1111/j.0013-9580.2004.39903.x. [DOI] [PubMed] [Google Scholar]
- 10.Striano P, Madia F, Minetti C, Striano S, Zara F. Electroclinical and genetic findings in a family with cortical tremor, myoclonus, and epilepsy. Epilepsia. 2005;46:1993–1995. doi: 10.1111/j.1528-1167.2005.00346.x. [DOI] [PubMed] [Google Scholar]
- 11.Suppa A, Berardelli A, Brancati F, et al. Clinical, neuropsychological, neurophysiologic, and genetic features of a new Italian pedigree with familial cortical myoclonic tremor with epilepsy. Epilepsia. 2009;50:1284–1288. doi: 10.1111/j.1528-1167.2008.01976.x. [DOI] [PubMed] [Google Scholar]
- 12.Carr JA, van der Walt PE, Nakayama J, et al. FAME 3: a novel form of progressive myoclonus and epilepsy. Neurology. 2007;68:1382–1389. doi: 10.1212/01.wnl.0000260063.46425.7e. [DOI] [PubMed] [Google Scholar]
- 13.Gardella E, Tinuper P, Marini C, et al. Autosomal dominant early-onset cortical myoclonus, photic-induced myoclonus, and epilepsy in a large pedigree. Epilepsia. 2006;47:1643–1649. doi: 10.1111/j.1528-1167.2006.00636.x. [DOI] [PubMed] [Google Scholar]
- 14.van Rootselaar F, Callenbach PM, Hottenga JJ, et al. A Dutch family with ‘familial cortical tremor with epilepsy’. Clinical characteristics and exclusion of linkage to chromosome 8q23.3-q24.1. J Neurol. 2002;249:829–834. doi: 10.1007/s00415-002-0729-x. [DOI] [PubMed] [Google Scholar]
- 15.Magnin E, Vidailhet M, Depienne C, et al. Familial cortical myoclonic tremor with epilepsy (FCMTE): clinical characteristics and exclusion of linkages to 8q and 2p in a large French family. Rev Neurol (Paris) 2009;165:812–820. doi: 10.1016/j.neurol.2009.05.014. [DOI] [PubMed] [Google Scholar]
- 16.van Rootselaar AF, Aronica E, Jansen Steur EN, Rozemuller-Kwakkel JM, de Vos RA, Tijssen MA. Familial cortical tremor with epilepsy and cerebellar pathological findings. Mov Disord. 2004;19:213–217. doi: 10.1002/mds.10662. [DOI] [PubMed] [Google Scholar]
- 17.van Rootselaar AF, van Schaik IN, van den Maagdenberg AM, Koelman JH, Callenbach PM, Tijssen MA. Familial cortical myoclonic tremor with epilepsy: a single syndromic classification for a group of pedigrees bearing common features. Mov Disord. 2005;20:665–673. doi: 10.1002/mds.20413. [DOI] [PubMed] [Google Scholar]
- 18.van Rootselaar AF, Maurits NM, Koelman JH, et al. Coherence analysis differentiates between cortical myoclonic tremor and essential tremor. Mov Disord. 2006;21:215–222. doi: 10.1002/mds.20703. [DOI] [PubMed] [Google Scholar]
- 19.Striano P, Zara F, Striano S. Autosomal dominant cortical tremor, myoclonus and epilepsy: many syndromes, one phenotype. Acta Neurol Scand. 2005;111:211–217. doi: 10.1111/j.1600-0404.2005.00385.x. [DOI] [PubMed] [Google Scholar]
- 20.Panayiotopoulos CP. Syndromes of idiopathic generalized epilepsies not recognized by the International League Against Epilepsy. Epilepsia. 2005;46((Suppl 9)):57–66. doi: 10.1111/j.1528-1167.2005.00314.x. [DOI] [PubMed] [Google Scholar]
- 21.Bour LJ, van Rootselaar AF, Koelman JH, Tijssen MA. Oculomotor abnormalities in myoclonic tremor: a comparison with spinocerebellar ataxia type 6. Brain. 2008;131:2295–2303. doi: 10.1093/brain/awn177. [DOI] [PubMed] [Google Scholar]
- 22.van Rootselaar AF, Maurits NM, Renken R, et al. Simultaneous EMG-functional MRI recordings can directly relate hyperkinetic movements to brain activity. Hum Brain Mapp. 2008;29:1430–1441. doi: 10.1002/hbm.20477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Rootselaar AF, van der Salm SM, Bour LJ, et al. Decreased cortical inhibition and yet cerebellar pathology in ‘familial cortical myoclonic tremor with epilepsy’. Mov Disord. 2007;22:2378–2385. doi: 10.1002/mds.21738. [DOI] [PubMed] [Google Scholar]
- 24.Hallett M, Chadwick D, Marsden CD. Cortical reflex myoclonus. Neurology. 1979;29:1107–1125. doi: 10.1212/WNL.29.8.1107. [DOI] [PubMed] [Google Scholar]
- 25.Hitomi T, Ikeda A, Kondo T, et al. Increased cortical hyperexcitability and exaggerated myoclonus with aging in benign adult familial myoclonus epilepsy. Mov Disord. 2011 doi: 10.1002/mds.23653. [DOI] [PubMed] [Google Scholar]
- 26.Ikeda A, Kakigi R, Funai N, Neshige R, Kuroda Y, Shibasaki H. Cortical tremor: a variant of cortical reflex myoclonus. Neurology. 1990;40:1561–1565. doi: 10.1212/WNL.40.10.1561. [DOI] [PubMed] [Google Scholar]
- 27.Okuma Y, Shimo Y, Shimura H, et al. Familial cortical tremor with epilepsy: an under-recognized familial tremor. Clin Neurol Neurosurg. 1998;100:75–78. doi: 10.1016/S0303-8467(98)00003-1. [DOI] [PubMed] [Google Scholar]
- 28.Terada K, Ikeda A, Mima T, et al. Familial cortical myoclonic tremor as a unique form of cortical reflex myoclonus. Mov Disord. 1997;12:370–377. doi: 10.1002/mds.870120316. [DOI] [PubMed] [Google Scholar]
- 29.Striano P, Manganelli F, Boccella P, Perretti A, Striano S. Levetiracetam in patients with cortical myoclonus: a clinical and electrophysiological study. Mov Disord. 2005;20:1610–1614. doi: 10.1002/mds.20530. [DOI] [PubMed] [Google Scholar]
- 30.Bourdain F, Apartis E, Trocello JM, et al. Clinical analysis in familial cortical myoclonic tremor allows differential diagnosis with essential tremor. Mov Disord. 2006;21:599–608. doi: 10.1002/mds.20725. [DOI] [PubMed] [Google Scholar]
- 31.Striano P, Coppola A, Madia F, et al. Life-threatening status epilepticus following gabapentin administration in a patient with benign adult familial myoclonic epilepsy. Epilepsia. 2007;48:1995–1998. doi: 10.1111/j.1528-1167.2007.01198.x. [DOI] [PubMed] [Google Scholar]
- 32.Inazuki G, Naito H, Ohama E, et al. [A clinical study and neuropathological findings of a familial disease with myoclonus and epilepsy—the nosological place of familial essential myoclonus and epilepsy (FEME)]. Seishin Shinkeigaku Zasshi. 1990;92:1–21. [PubMed] [Google Scholar]
- 33.Yokochi M, Takaoka S, Kawano H, et al. [A 77-year-old woman with myoclonus and epilepsy]. No To Shinkei. 1993;45:1173–1185. [PubMed] [Google Scholar]
- 34.Striano P, Robbiano A, Zara F, Striano S. Familial cortical tremor and epilepsy: a well-defined syndrome with genetic heterogeneity waiting for nosological placement in the ILAE classification. Epilepsy Behav. 2010;19:669. doi: 10.1016/j.yebeh.2010.09.032. [DOI] [PubMed] [Google Scholar]
- 35.Saka E, Saygi S. Familial adult onset myoclonic epilepsy associated with migraine. Seizure. 2000;9:344–346. doi: 10.1053/seiz.2000.0402. [DOI] [PubMed] [Google Scholar]
- 36.Manabe Y, Narai H, Warita H, et al. Benign adult familial myoclonic epilepsy (BAFME) with night blindness. Seizure. 2002;11:266–268. doi: 10.1053/seiz.2001.0606. [DOI] [PubMed] [Google Scholar]
- 37.Morita S, Miwa H, Kondo T. [A case of the familial essential myoclonus and epilepsy presenting behavioral arrest]. No To Shinkei. 2003;55:345–348. [PubMed] [Google Scholar]
- 38.Elia M, Musumeci SA, Ferri R, et al. Familial cortical tremor, epilepsy, and mental retardation: a distinct clinical entity? Arch Neurol. 1998;55:1569–1573. doi: 10.1001/archneur.55.12.1569. [DOI] [PubMed] [Google Scholar]
- 39.Striano P, Caranci F, Di BR, Tortora F, Zara F, Striano S. (1)H-MR spectroscopy indicates prominent cerebellar dysfunction in benign adult familial myoclonic epilepsy. Epilepsia. 2009;50:1491–1497. doi: 10.1111/j.1528-1167.2008.01900.x. [DOI] [PubMed] [Google Scholar]
- 40.Coppola A, Santulli L, Del GL, et al. Natural history and long-term evolution in families with autosomal dominant cortical tremor, myoclonus, and epilepsy. Epilepsia. 2011;52:1245–1250. doi: 10.1111/j.1528-1167.2011.03017.x. [DOI] [PubMed] [Google Scholar]
- 41.Striano P, Striano S, Zara F. Re: Fame 3: a novel form of progressive myoclonus and epilepsy. Neurology. 2008;70:85–86. doi: 10.1212/01.wnl.0000295707.90283.e6. [DOI] [PubMed] [Google Scholar]
- 42.Ikeda A, Kurihara S, Shibasaki H. Possible anticipation in BAFME: three generations examined in a Japanese family. Mov Disord. 2005;20:1076–1077. doi: 10.1002/mds.20558. [DOI] [PubMed] [Google Scholar]
- 43.Ziemann U, Lonnecker S, Steinhoff BJ, Paulus W. Effects of antiepileptic drugs on motor cortex excitability in humans: a transcranial magnetic stimulation study. Ann Neurol. 1996;40:367–378. doi: 10.1002/ana.410400306. [DOI] [PubMed] [Google Scholar]
- 44.Ziemann U. TMS and drugs. Clin Neurophysiol. 2004;115:1717–1729. doi: 10.1016/j.clinph.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 45.Yang Q, Hashizume Y, Yoshida M, et al. Morphological Purkinje cell changes in spinocerebellar ataxia type 6. Acta Neuropathol (Berl) 2000;100:371–376. doi: 10.1007/s004010000201. [DOI] [PubMed] [Google Scholar]
- 46.Louis ED, Faust PL, Vonsattel JP. Purkinje cell loss is a characteristic of essential tremor. Parkinsonism Relat Disord. 2011;17:406–409. doi: 10.1016/j.parkreldis.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shahwan A, Farrell M, Delanty N. Progressive myoclonic epilepsies: a review of genetic and therapeutic aspects. Lancet Neurol. 2005;4:239–248. doi: 10.1016/S1474-4422(05)70043-0. [DOI] [PubMed] [Google Scholar]
- 48.Bhatia KP, Brown P, Gregory R, et al. Progressive myoclonic ataxia associated with coeliac disease. The myoclonus is of cortical origin, but the pathology is in the cerebellum. Brain. 1995;118:1087–1093. doi: 10.1093/brain/118.5.1087. [DOI] [PubMed] [Google Scholar]
- 49.Finelli PF, McEntee WJ, Ambler M, Kestenbaum D. Adult celiac disease presenting as cerebellar syndrome. Neurology. 1980;30:245–249. doi: 10.1212/WNL.30.3.245. [DOI] [PubMed] [Google Scholar]
- 50.Hunt JR. Dyssynergica cerebellaris myoclonica – primary atrophy of the dentate system. Brain. 1921;44:490–538. doi: 10.1093/brain/44.4.490. [DOI] [Google Scholar]
- 51.Tijssen MA, Thom M, Ellison DW, et al. Cortical myoclonus and cerebellar pathology. Neurology. 2000;54:1350–1356. doi: 10.1212/WNL.54.6.1350. [DOI] [PubMed] [Google Scholar]
- 52.Breton P, Bizot JC, Buee J, De LM. I. Brain neurotoxicity of Penitrem A: electrophysiological, behavioral and histopathological study. Toxicon. 1998;36:645–655. doi: 10.1016/S0041-0101(97)00084-6. [DOI] [PubMed] [Google Scholar]
- 53.van Rootselaar AF, Renken R, de Jong BM, Hoogduin JM, Tijssen MA, Maurits NM. fMRI analysis for motor paradigms using EMG-based designs: a validation study. Hum Brain Mapp. 2007;28:1117–1127. doi: 10.1002/hbm.20336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gutierrez-Delicado E, Serratosa JM. Genetics of the epilepsies. Curr Opin Neurol. 2004;17:147–153. doi: 10.1097/00019052-200404000-00011. [DOI] [PubMed] [Google Scholar]
- 55.Kaneko S, Iwasa H, Okada M. Genetic identifiers of epilepsy. Epilepsia. 2002;43 Suppl 9:16–20. doi: 10.1046/j.1528-1157.43.s.9.5.x. [DOI] [PubMed] [Google Scholar]
- 56.Mulley JC, Scheffer IE, Petrou S, Berkovic SF. Channelopathies as a genetic cause of epilepsy. Curr Opin Neurol. 2003;16:171–176. doi: 10.1097/00019052-200304000-00009. [DOI] [PubMed] [Google Scholar]
- 57.Zhuchenko O, Bailey J, Bonnen P, et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet. 1997;15:62–69. doi: 10.1038/ng0197-62. [DOI] [PubMed] [Google Scholar]
- 58.Ophoff RA, Terwindt GM, Vergouwe MN, Frants RR, Ferrari MD. Familial hemiplegic migraine: involvement of a calcium neuronal channel. Neurologia. 1997;12:31–37. [PubMed] [Google Scholar]
- 59.Zwingman TA, Neumann PE, Noebels JL, Herrup K. Rocker is a new variant of the voltage-dependent calcium channel gene Cacna1a. J Neurosci. 2001;21:1169–1178. doi: 10.1523/JNEUROSCI.21-04-01169.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crooks R, Mitchell T, Thom M. Patterns of cerebellar atrophy in patients with chronic epilepsy: a quantitative neuropathological study. Epilepsy Res. 2000;41:63–73. doi: 10.1016/S0920-1211(00)00133-9. [DOI] [PubMed] [Google Scholar]
- 61.Okino S. Familial benign myoclonus epilepsy of adult onset: a previously unrecognized myoclonic disorder. J Neurol Sci. 1997;145:113–118. doi: 10.1016/S0022-510X(96)00245-6. [DOI] [PubMed] [Google Scholar]
- 62.Kuwano A, Takakubo F, Morimoto Y, et al. Benign adult familial myoclonus epilepsy (BAFME): an autosomal dominant form not linked to the dentatorubral pallidoluysian atrophy (DRPLA) gene. J Med Genet. 1996;33:80–81. doi: 10.1136/jmg.33.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Okuma Y, Shimo Y, Hatori K, Hattori T, Tanaka S, Mizuno Y. Familial cortical tremor with epilepsy. Parkinsonism Rel Disord. 1997;3:83–87. doi: 10.1016/S1353-8020(97)00001-1. [DOI] [PubMed] [Google Scholar]
- 64.Oguni E, Hayashi A, Ishii A, Mizusawa H, Shoji S. A case of cortical tremor as a variant of cortical reflex myoclonus. Eur Neurol. 1995;35:63–64. doi: 10.1159/000117093. [DOI] [PubMed] [Google Scholar]
- 65.Kudo J, Kudo T, Yamauchi T. [Seven families with heredofamilial tremor and epilepsy]. Rinsho Shinkeigaku. 1984;24:1–8. [PubMed] [Google Scholar]
- 66.Nagayama S, Kishikawa H, Yukitake M, Matsui M, Kuroda Y. [A case of familial myoclonus showing extremely benign clinical course]. Rinsho Shinkeigaku. 1998;38:430–434. [PubMed] [Google Scholar]