

Abstract
Animal studies have linked perinatal bisphenol A (BPA) exposure to altered DNA methylation, but little attention is given to analyzing multiple physiologically relevant doses. Utilizing the viable yellow agouti (Avy) mouse, we examine the effects of developmental exposure through maternal diet to 50 ng BPA/kg (n = 14 litters), 50 μg BPA/kg (n = 9 litters), or 50 mg BPA/kg (n = 13 litters) on global and candidate gene methylation at postnatal day 22. Global methylation analysis reveals hypermethylation in tail tissue of a/a and Avy/a offspring across all dose groups compared with controls (n = 11 litters; P < 0.02). Analysis of coat color phenotype replicates previous work showing that the distribution of 50 mg BPA/kg Avy/a offspring shifts toward yellow (P = 0.006) by decreasing DNA methylation in the retrotransposon upstream of the Agouti gene (P = 0.03). Maternal exposure to 50 μg or 50 ng BPA/kg, however, results in altered coat color distributions in comparison with control (P = 0.04 and 0.02), but no DNA methylation effects at the Agouti gene are noted. DNA methylation at the CDK5 activator-binding protein (CabpIAP) metastable epiallele shows hypermethylation in the 50 μg BPA/kg offspring, compared with controls (P = 0.02). Comparison of exposed mouse liver BPA levels to human fetal liver BPA levels indicates that the three experimental exposures are physiologically relevant. Thus, perinatal BPA exposure affects offspring phenotype and epigenetic regulation across multiple doses, indicating the need to evaluate dose effects in human clinical and population studies.
Keywords: epigenetics, DNA methylation, bisphenol A, viable yellow agouti (Avy) mouse, developmental origins of disease
INTRODUCTION
A growing body of work supports the developmental origins of health and disease hypothesis, in which chemical and nutritional exposures early in development influence chronic disease outcomes in adulthood [Barker, 2004; Bateson et al., 2004]. Epigenetic modifications, such as DNA methylation and histone modifications, established early in development can shape susceptibility to disease, resulting in diverse phenotypes among genetically identical individuals [Rakyan et al., 2002]. For example, metastable epialleles are genes that are variably expressed due to epigenetic modifications established early in development, thus making them vulnerable targets to environmental disruption during gestation [Rakyan et al., 2002; Waterland and Jirtle, 2004]. A handful of murine metastable epialleles have been identified (Avy, AxinFu, CabpIAP) in which the activity of a contraoriented intracisternal A particle (IAP) retrotransposon controls expression of an adjacent gene (Fig. 1) [Duhl et al., 1994; Vasicek et al., 1997; Ruvinsky et al., 2001; Rakyan et al., 2002; Druker et al., 2004]. Importantly, DNA methylation patterns at these metastable epialleles have been shifted following maternal exposure to nutritional and environmental factors, including bisphenol A (BPA) [Cooney et al., 2002; Waterland and Jirtle, 2003; Dolinoy et al., 2006, 2007; Kaminen-Ahola et al., 2010].
Fig. 1.
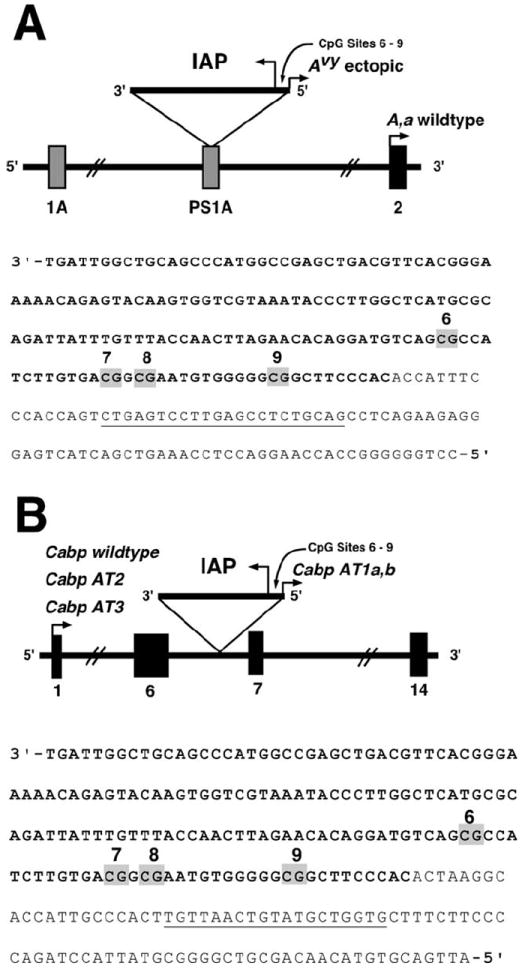
Avy and CabpIAP loci (A) The Avy allele contains a contra-oriented IAP insertion within pseudoexon 1A (PS1A) of the Agouti gene. A cryptic promoter (short arrowhead labeled Avy ectopic) drives constitutive ectopic Agouti expression. Transcription of the Agouti gene normally initiates from a developmentally regulated hair-cycle specific promoter in exon 2 (short arrowhead labeled A, a wildtype). The location of the bisulfite-converted genomic reverse primer for amplifying the Avy IAP is underlined. (B) The CabpIAP metastable epiallele contains a contra-oriented IAP insertion within intron 6 of the murine CDK5 activator-binding protein (Cabp) gene, resulting in short aberrant transcripts originating from the 5′-LTR of the IAP (short arrowhead labeled Cabp AT1a,b). Aberrant transcripts also originate at the normal transcription start site (short arrowhead labeled Cabp wildtype) and truncate 5′ of the IAP insertion (Cabp AT2 and AT3). Normal Cabp transcription covers 14 exons, resulting in a 2-kb transcript. The location of the bisulfite-converted genomic reverse primer for amplifying the CabpIAP locus is underlined.
Accumulating work suggests that early BPA exposure increases susceptibility for adverse phenotypic outcomes via epigenetic mechanisms. BPA is a chemical used for the industrial manufacturing of polycarbonate plastics and epoxy resins. There are multiple routes of BPA exposure including ingestion, dermal absorption, and inhalation due to its widespread use in commercial products such as food and beverage containers, baby bottles, dental sealants, and receipt paper [Vandenberg et al., 2007]. Recurrent exposure to BPA is evident from detectable levels present in greater than 92% of the United States population [Calafat et al., 2008]. As an endocrine active compound, BPA can exert estrogenic activity by interfering with estrogen receptors alpha and beta, and estrogen related receptor gamma even at low exposure levels [vom Saal et al., 2006]. Additionally, BPA has an antagonistic effect on thyroid hormone signaling [Moriyama et al., 2002; Rubin et al., 2009].
Early exposure to BPA may promote chronic disease development such as prostate and breast cancer, type 2 diabetes, and obesity as well as impaired brain development and behavior by altering the developing epigenome [Jirtle and Skinner, 2007; Kundakovic and Champagne, 2011]. For example, early developmental exposure to 10 μg BPA/kg BW/day decreased methylation of the phosphodiesterase type 4 variant 4 gene in prostate cancer cells in adult male rats [Ho et al., 2006; Prins et al., 2008]. Additionally, in utero exposure to 5 mg BPA/kg BW in CD-1 mice on days 9–16 of pregnancy decreased methylation in the promoter region of Hoxa10, a gene involved in uterine organogenesis [Bromer et al., 2010]. Previously, our group reported a shift toward DNA hypomethylation at the viable yellow agouti (Avy) and CDK5 activator-binding protein (CabpIAP) metastable epialleles (Figs. 1A and 1B) in offspring exposed to a relatively high dosage of BPA (50 mg BPA/kg diet) perinatally [Dolinoy et al., 2007]. Moreover, restoration of normal methylation patterns occurred with maternal supplementation of genistein or methyl donors such as folate, choline, betaine, and vitamin B12 [Dolinoy et al., 2006, 2007].
Previous epigenetics studies involving BPA have not captured the full range of human physiologically relevant exposure levels, and most attempts to elucidate the effects on the epigenome following environmental manipulations, including perinatal exposure to BPA, have been restricted in dose-response assessment. Thus, utilizing the viable yellow agouti (Avy) mouse model, we examined global and candidate gene methylation patterns following perinatal exposure at three dosages (50 ng, μg, and mg of BPA/kg diet). The murine Avy allele resulted from the random insertion of a murine IAP retrotransposon into the 5′ end of the Agouti gene (Fig. 1A) [Duhl et al., 1994]. Methylation of CpG sites in and near the Avy IAP correlates inversely with ectopic Agouti expression and varies dramatically among isogenic Avy/a mice, resulting in a wide array of coat colors, ranging from yellow (unmethylated) to pseudoagouti (methylated), and additionally results in adultonset obesity (~10 weeks of age) among low methylated mice [Miltenberger et al., 1997; Morgan et al., 1999]. To our knowledge, this is the first study to utilize the Avy mouse model as an epigenetic biosensor to evaluate maternal exposure to multiple, rather than single dose levels.
METHODS
Animals and Diet
Avy mice were obtained from a colony that has been maintained with sibling mating and forced heterozygosity for the Avy allele for over 220 generations, resulting in a genetically invariant background [Waterland and Jirtle, 2003]. Virgin a/a dams, 6 weeks of age, were randomly assigned to one of four phytoestrogen-free AIN-93G diets (diet 95092 with 7% corn oil substituted for 7% soybean oil; Harlan Teklad, Madison, WI): (1) standard diet (n = 11 litters, 86 total offspring, 39 Avy/a offspring); (2) standard diet supplemented with 50 ng BPA/kg diet (n = 14 litters, 107 total offspring, 48 Avy/a offspring); (3) standard diet supplemented with 50 μg BPA/kg diet (n = 9 litters, 67 total offspring, 32 Avy/a offspring); (4) standard diet supplemented with 50 mg BPA/kg diet (n = 13 litters, 91 total offspring, 45 Avy/a offspring). All diet ingredients were supplied by Harlan Teklad except BPA, which was supplied by NTP (National Toxicology Program, Durham NC). The mg dosage was formulated to be an order of magnitude lower than the dietary administered maximum nontoxic threshold in rodents (200 mg/kg BW/day) [Takahashi et al., 2003], whereas the ng and μg BPA dosages were used to potentially capture the physiologically relevant range of human exposure.
Following 2 weeks on their respective diets, at 8 weeks of age a/a virgin dams were mated with Avy/a males, 7–8 weeks of age. All animals were housed in polycarbonate-free cages and provided ad libitum access to diet and BPA-free water. The dams remained on the assigned diets throughout pregnancy and lactation. At postnatal day 22 (d22), a/a and Avy/a offspring were weighed and tail-tipped. In addition, at d22, a single observer visually classified Avy/a offspring coat color phenotype into one of five categories based on proportion of brown fur: yellow (<5% brown), slightly mottled (between 5 and 40% brown), mottled (~50% brown), heavily mottled (between 60 and 95% brown), and pseudoagouti (>95% brown). Tail tissue was collected for analysis from all offspring.
Animals used in this study were maintained in accordance with the Guidelines for the Care and Use of Laboratory Animals [Institute of Laboratory Animal Resources, 1996] and were treated humanely and with regard for alleviation of suffering. The study protocol was approved by the University of Michigan Committee on Use and Care of Animals.
DNA Isolation and Methylation Analysis
Total genomic DNA was isolated from d22 tail tissue of all a/a and Avy/a offspring using magnetic particle-based methodology and the Maxwell 16® Instrument (Promega Corporation, Madison, WI). The Luminometric Methylation Assay (LUMA), a methylation-sensitive restriction enzyme digest followed by quantitative DNA methylation analysis via pyrosequencing, was utilized to analyze global DNA methylation at CCGG sites throughout the mouse genome using genomic DNA [Karimi et al., 2006]. Approximately 300 ng of genomic DNA was digested with both HpaII, an endonuclease sensitive to CpG methylation, and MspI, an endonuclease insensitive to CpG methylation; both endonucleases cut CCGG sites between the first and second cytosines. EcoRI was used in both restriction enzyme reactions as a normalization reference [Karimi et al., 2006]. Following DNA digestion, samples were pyrosequenced in duplicate to quantify CCGG DNA methylation. The pyrosequencing output provides the incorporation of dCTP, which is directly correlated with DNA methylation. The ratio of digested sites to undigested sites was calculated and represents the percentage of genomic DNA methylation. Duplicates with measurement differences greater than 15% were omitted prior to statistical analysis.
Using the Qiagen Epitect kit automated on the Qiagen QIAcube® purification system, approximately 1 μg of genomic DNA was treated with sodium bisulfite to allow conversion of unmethylated cytosines to uracil, read as thymine during polymerase chain reaction (PCR), whereas the methylated cytosines remain unconverted [Grunau et al., 2001]. Following bisulfite conversion, candidate gene regions of interest were amplified using HotStarTaq master mix (Qiagen Inc., Valencia, CA), forward primer (0.5 pmol) and reverse primer (0.5 pmol) in a 30 μL PCR and resolved by gel electrophoresis. PCR and sequencing primers for assays are listed in Table I. DNA methylation of CpG sites of interest was quantified using PyroMark MD (Qiagen Inc., Valencia, CA) pyrosequencing technology. Percent methylation was computed using PyroMark software, which calculates the degree of methylation as percent 5-methylated cytosines (%5mC) over the sum of methylated and unmethylated cytosines. All samples were run in duplicate using the average CpG site methylation of the duplicates for statistical analysis. Sequences to analyze for pyrosequencing runs are provided in Table I. The four CpG sites studied at the Avy allele are located at nucleotide positions 306, 319, 322, and 334 of GenBank accession number AF540972.1. The four CpG sites studied at the CabpIAP allele are located at nucleotide positions 44, 57, 60, and 72 of GenBank accession number BB842254.
TABLE I.
PCR Primers and Sequences: Primers (5′ to 3′) and sequences to analyze for DNA methylation quantification via pyrosequencing for candidate genes
| Primer/sequence to analyze | Avy Assay | CabpIAP Assay |
|---|---|---|
| Forward PCR primer | ATTTTTAGGAAAAGAGAGTAAGAAGTAAG | ATTATTTTTTGATTGGTTGTAGTTTATGG |
| Reverse PCR prime | CTACAAAAACTCAAAAACTCA | CACCAACATACAATTAACA |
| Sequencing primer | TAGAATATAGGATGTTAG | TAGAATATAGGATGTTAG |
| Sequence to analyze | C/TGTTATTTTGTGAC/TGGC/TGAATGTGGGGGC/TGGTT | C/TGTTATTTTGTGAC/TGGC/TGAATGTGGGGGC/TGGTT |
Statistical Analysis
The influence of gestational BPA exposure on litter size, survival, wean weight, genotypic ratio and sex ratio was evaluated using ANOVA with Bonferroni post-hoc analysis. The distribution of the five coat color phenotypes between each exposure group was analyzed using a Chi-square goodness-of-fit test, with the control coat color distribution representing the expected distribution. All comparisons resulted in cell counts with no more than 20% of cells containing fewer than five observations. The influence of sex, genotype and coat color on mean CpG methylation was measured using ANOVA with Bonferroni corrections. Average CpG methylation within an amplicon, site-specific CpG methylation, and global methylation among the three BPA exposed groups and the control group were evaluated by two-sample hypothesis analysis of means and ANOVA with Bonferroni correction as post-hoc analyses. Statistical significance was defined as P-value < 0.05 for all analyses. Normality of percent methylation was evaluated using histograms and Q-Q plots. Outliers defined as having a studentized residual greater than 2.0 were excluded in the final methylation analysis. The resulting exclusion of outliers consisted of 2 mg exposed subjects for Avy locus methylation analysis. All statistical analyses were completed using SAS software version 9.2 (Cary, NC).
BPA Analysis in Liver
Approximately 300–500 mg of d22 Avy/a mouse liver tissue (n = 8–11 per exposure group) was flash frozen and homogenized to fine powder over a mortar above liquid nitrogen. Homogenized tissue was transferred into a 2 mL polypropylene eppendorf tube and transported overnight on dry ice to the Wadsworth Center (New York State Department of Health, Albany, NY). Five mL of acetonitrile and 5 ng of 13C12-BPA, an internal standard, were added to homogenized tissue, and the mixture was shaken for 30 min. Extraction with acetonitrile was repeated twice and the mixture was centrifuged at 4,500g for 3 min. Combined aliquots were concentrated to near-dryness under a gentle stream of nitrogen and reconstituted with 1.5 mL of 10% dichloromethane in hexane. The sample extract was then loaded onto a Strata® NH2 cartridge (200 mg/3 mL, Phenomenex, Torrance, CA), preconditioned with 5 mL of 80% methanol in acetone and 5 mL of hexane. The cartridge was washed with 5 mL of hexane and eluted with 5 mL of 80% methanol in acetone. The eluate was concentrated to 0.5 mL under a gentle stream of nitrogen resulting in the free BPA fraction.
To determine conjugated BPA, 1 mL of Milli-Q H2O and 1 mL of 2 μL/mL β-glucuronidase (from Helix pomatia, 145,700 units/mL, Sigma, St Louis, MO) were added to the residue from free-BPA aliquot. 13C12-BPA was added and digested at 37°C for 12 hr. The sample was extracted thrice with ethyl acetate (5 + 3.5 + 3.5 mL) and purified by passing through Strata® NH2 cartridge as described above. The final eluate was concentrated to 0.5 mL. A high-performance liquid chromatograph (HPLC) interfaced with API 2000 electrospray triple-quadruple mass spectrometry (ESI-MS/MS; Applied Biosystems, Foster City, CA) was used for the quantification of free and conjugated BPA [Padmanabhan et al., 2008]. Ten microliters of the extract was injected onto an analytical column (Betasil® C18, 100 × 2.1 mm column; Thermo Electron Corporation, Waltham, MA), which was connected to a Javelin® guard column (Betasil® C18, 20 × 2.1 mm). The mobile phase flow rate was 300 μL/min. The mobile phase consisting of methanol and water started at a gradient of 25% methanol to 99% methanol in 4 min and was held for 10 min before reverting to its initial condition. The MS/MS was operated in the electrospray negative ionization mode and optimized to transmit the [M-H]- ion before fragmentation of one or more product ions. Cone voltage and collision energies were 30 and 25 V, respectively. The capillary voltage was 4.5 KV, and desolvation temperature was 400°C. Multiple reaction monitoring transitions monitored were 227 > 212 for BPA, and 239 > 224 for 13C12-BPA.
A procedural blank was analyzed with every 10 samples to check for interferences or laboratory contamination. The limit of quantitation (LOQ) of BPA was 0.1 ng/g. The LOQ was calculated as twice the concentration of the “lowest acceptable calibration standard”; the amount of sample taken for analysis and final extract volume. The mean recovery of 13C12-BPA spiked into samples was 96%. Reported concentrations were corrected for the recoveries of the internal standard (isotope dilution method). BPA standards spiked into selected sample matrices and passed through the entire analytical procedure yielded a mean recovery of 101%. An external calibration curve was prepared by injecting 10 μL of 0.05, 0.1, 0.2, 0.5, 1, 2, 5, 10, 50, and 100 ng/mL standards and the calibration coefficient was 0.99.
In order to compare mouse liver BPA concentrations to physiologically relevant levels in humans, free and conjugated BPA were measured in 51 human fetal liver samples obtained from the NIH-funded (R24 HD000836-47) Birth Defects Research Laboratory fetal tissue bank at the University of Washington, Seattle. These tissues were derived from conceptuses with gestational ages ranging from 74 to 120 days. Information provided with these tissues include gestational age and sex; occasionally race is indicated. The health status of the individuals is unknown but presumed to have been healthy and with no known chromosomal abnormalities. Prior to shipment to the University of Michigan, samples were flash frozen with liquid nitrogen and stored in polycarbonate-free tubing. Fetal liver tissues were processed for BPA levels as described above.
To ensure no contamination was introduced into the sample preparation process, a negative control of BPA-free water was processed identically as the mouse and human samples described above and resulted in free and conjugated BPA levels below the LOQ. For calculation of mean and median BPA concentrations, liver BPA levels below the LOQ were assigned a value of 0.071, which was estimated by dividing the LOQ (0.1 ng/g) by the square root of 2.
RESULTS
Gestational BPA exposure at 50 ng BPA/kg (n = 14 litters, 107 total offspring), 50 μg BPA/kg (n = 9 litters, 67 total offspring), or 50 mg BPA/kg (n = 13 litters, 91 total offspring) diet did not significantly influence litter size (P = 0.84), survival (P = 0.86), genotypic ratio (P = 0.49), or sex ratio (P = 0.16) in comparison to control offspring (n = 11 litters, 86 total offspring; Table II). BPA exposure was, however, significantly associated with lower wean weight of ng exposed a/a and Avy/a offspring (mean weight 8.65 g in ng versus 9.28 g in control; P = 0.03) but not μg or mg exposed offspring (Table II). When d22 body weight analysis is restricted to a/a animals only, the decreased body weight in ng exposed offspring, and not in μg and mg exposed offspring, is still observed (mean weight 8.24 g in ng versus 9.15 g in control; P = 0.01).
TABLE II.
Litter Parameters: Offspring litter size, survival rate, wean weight, genotypic ratio, and sex ratio across exposure groups
| Exposure | N (litter) | Mean no. pups | Pup survival rate | Mean wean weight (g) | Mean percent a/a offspring | Mean percent male offspring |
|---|---|---|---|---|---|---|
| Control | 11 | 7.82 | 0.95 | 9.28 | 55 | 45 |
| mg | 13 | 7.00 | 0.95 | 9.27 | 51 | 51 |
| μg | 9 | 7.44 | 0.91 | 9.33 | 52 | 54 |
| ng | 14 | 7.64 | 0.93 | 8.65* | 55 | 57 |
indicates a P < 0.05 compared with control exposure group.
Global DNA Methylation
Global CCGG DNA methylation levels throughout the mouse genome were measured in Avy/a and a/a offspring d22 tail DNA using the LUMA assay (Table III). Mean methylation did not differ by sex (P = 0.58), genotype (P = 0.43), or coat color (P = 0.93). Offspring exposed to the 50 mg BPA/kg diet demonstrated an average of 59.3% global DNA methylation in comparison to control offspring with measured percent global methylation of 51.6 (n = 71 and 60, respectively; P = 0.01). Offspring exposed to the 50 μg BPA/kg diet also exhibited significantly higher average percent global DNA methylation of 60.3 compared with controls (n = 52 and 60, respectively; P = 0.008). Lastly, the offspring exposed to a 50 ng BPA/kg diet showed significantly higher average percent global methylation of 58.8 compared with controls (n = 74 and 60, respectively; P = 0.02). There were no statistically significant differences in global methylation among the three BPA exposed groups (data not shown).
TABLE III.
Global DNA Methylation: LUMA methylation levels in tail DNA among Avy/a and a/a offspring
| Exposure | N | Mean percent methylation (SD) | P-value (compared with control) |
|---|---|---|---|
| Control | 60 | 51.6 (17.9) | |
| mg | 71 | 59.3 (15.2) | 0.02 |
| μg | 52 | 60.3 (13.4) | 0.007 |
| ng | 74 | 58.8 (9.8) | 0.01 |
Maternal Dietary Exposure Coat Color Shift
The total number of offspring evaluated for coat color shift was among 11 litters from the corn oil control group (n = 39 Avy/a offspring), 13 litters from the mg BPA/kg supplemented group (n = 45 Avy/a offspring), 9 litters from the μg BPA/kg supplemented group (n = 32 Avy/a offspring), and 14 litters from the ng BPA/kg supplemented group (n = 48 Avy/a offspring). Perinatal BPA exposure through maternal diet shifted the coat color distribution of Avy/a offspring in a dose-dependent fashion. Maternal dietary exposure to 50 mg BPA/kg shifted the coat color distribution of genetically identical d22 Avy/a offspring toward yellow in comparison to control offspring (P = 0.006; Fig. 2A). Twenty-nine percent of the 50 mg BPA/kg exposed offspring were classified as yellow coat color compared with only 15% of control offspring. Conversely, maternal dietary exposure to 50 μg BPA/kg resulted in a statistically significant altered coat color distribution in comparison to controls (P = 0.04; Fig. 2B). Twenty-five percent of the 50 μg BPA/kg exposed offspring were classified as pseudoagouti compared with only 12% of the control offspring; however, the 50 μg BPA/kg exposed offspring were also more likely to be classified as slightly mottled (31% compared with 18%). Finally, maternal dietary exposure to 50 ng BPA/kg resulted in increased incidence of slightly mottled and heavily mottled offspring compared with controls (P = 0.02) but did not shift the coat color distribution toward the pseudoagouti or yellow phenotype (Fig. 2C).
Fig. 2.
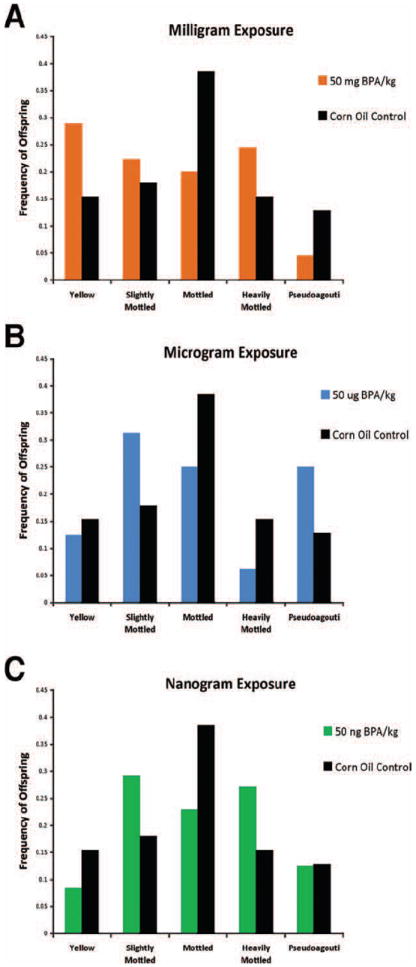
Coat color distribution (A) Coat color phenotype distribution among 50 mg/kg BPA exposed offspring (n = 45) versus corn oil control offspring (n = 38). 50 mg/kg BPA maternal intake demonstrates a shift in offspring coat color toward yellow (P = 0.006). (B) Coat color phenotype distribution among 50 μ*g/kg BPA offspring (n = 32) and versus corn oil control offspring. 50 μ*g/kg BPA maternal intake demonstrates a shift in coat color toward pseudoagouti (P = 0.04). (C) Coat color phenotype distribution among 50 ng/kg BPA offspring (n = 48) versus corn oil control offspring. 50 ng/kg BPA maternal intake demonstrates a shift in offspring coat color toward heavily mottled/pseudoagouti (P = 0.02). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Candidate Gene DNA Methylation at Avy and CabpIAP
Site-specific and average DNA methylation in d22 tail tissue at four CpG sites (sites 6–9) in the cryptic promoter of the Avy IAP was quantified using bisulfite pyrosequencing on Avy/a offspring (Fig. 1A). The mg exposure group (n = 43 Avy/a offspring) demonstrated an average methylation of 24.3% across the four CpG sites in comparison to 35.6% average methylation of the controls (Table IV; n = 38 Avy/a offspring; P = 0.03). Evaluation of each individual CpG site indicated a significant decrease in methylation at sites 6 (P = 0.03), 7 (P = 0.02), and 8 (P = 0.02) in the mg exposed offspring versus control. Methylation at site 9 was marginally significantly lower (P = 0.07). Average methylation across the four CpG sites of the μg (n = 32 Avy/a offspring; P = 0.97) and ng (n = 48 Avy/a offspring; P = 0.79) exposed offspring did not differ in comparison to the control group (Table IV). There were no significant differences in methylation at sites 6–9 of μg (P = 0.96, 0.93, 0.99, and 0.80, respectively) and ng (P = 0.79, 0.60, 0.75, and 0.99, respectively) groups in comparison to the control group.
TABLE IV.
Percent Methylation Summary
| Locus | N | Exposure | Mean percent methylation (SD) across CpG sites 6–9 | Mean percent methylation (SD) site 6 | Mean percent methylation (SD) site 7 | Mean percent methylation (SD) site 8 | Mean percent methylation (SD) site 9 |
|---|---|---|---|---|---|---|---|
| Avy (Avy/a offspring) | 38 | Control | 35.6 (26.4) | 34.5 (26.4) | 35.2 (23.2) | 35.5 (27.2) | 37.3 (30.3) |
| 43 | mg | 24.3 (18.7)** | 23.7 (18.9)** | 24.1 (16.5)** | 22.6 (18.8)** | 26.7 (21.6)* | |
| 32 | μg | 35.9 (27.5) | 34.2 (27.8) | 34.7 (23.7) | 35.4 (27.7) | 39.2 (31.5) | |
| 48 | ng | 34.1 (25.2) | 33.1 (25.5) | 32.6 (21.9) | 33.7 (25.4) | 37.2 (28.8) | |
| CabpIAP (Avy/a and a/a offspring) | 82 | Control | 83.1 (8.2) | 84.6 (8.1) | 81.5 (8.4) | 78.5 (9.7) | 88.0 (9.3) |
| 85 | mg | 83.6 (6.4) | 85.8 (6.4) | 81.4 (6.9) | 79.1 (7.1) | 88.3 (7.7) | |
| 67 | μg | 85.8 (5.6)** | 87.5 (5.4)** | 83.3 (6.6) | 81.8 (6.5)** | 90.6 (6.2)** | |
| 107 | ng | 84.3 (6.0) | 86.0 (5.8) | 82.3 (7.2) | 79.3 (7.6)** | 89.6 (6.1)** |
indicates P < 0.10 compared with control exposure group.
indicates P < 0.05 compared with control group. Methylation status of Avy and CabpIAp loci from tail DNA.
DNA methylation in d22 tail tissue at four CpG sites (sites 6–9) in the CabpIAP metastable epiallele [Druker et al., 2004) was measured in both Avy/a and a/a offspring (Fig. 1B). In comparison to the control group (n = 82 Avy/a and a/a offspring), the mg exposed offspring (n = 85 Avy/a and a/a offspring) showed no difference in average methylation across the four CpG sites (83.1 and 83.6%, respectively; P = 0.64; Table IV). Average methylation across the four CpG sites in the μg exposed offspring (n = 67 Avy/a and a/a offspring), however, was increased compared with controls (85.8 and 83.1%, respectively; P = 0.02). Site-specific methylation showed increased methylation at sites 6, 8, and 9 in the μg exposed group versus the controls (P = 0.01, 0.02, and 0.04, respectively). The ng exposure group (n = 107 Avy/a and a/a offspring) displayed an average methylation across the four CpG sites of 84.3% compared with 83.1% in the control group (P = 0.25). Sites 8 and 9 exhibited statistically significant increased methylation in ng exposed offspring when compared with controls (P = 0.02 and 0.04, respectively).
Liver BPA Measurements
Free and conjugated BPA concentrations were analyzed in a subset of d22 mouse liver samples (~1 pup per litter) from each BPA exposure and control group as well as in 51 human fetal liver samples (Table V). Total BPA (free plus glucuronide-conjugated) concentrations measured in the 50 mg BPA/kg exposed mice ranged from 9.46 to 870 ng/g (mean = 441; median = 472; n = 9). Total BPA in animals exposed to 50 μg BPA/kg ranged from below LOQ to 11.3 ng/g (mean = 2.02; median = 0.56; n = 10). BPA concentrations in the liver from mice exposed to 50 ng BPA/kg ranged from below LOQ to 13.0 ng/g (mean = 2.78; median = 0.31; n = 11). Total BPA in the control group ranged from below LOQ to 11.5 ng/g (mean = 4.26; median = 4.24; n = 10). To compare mouse liver BPA concentrations to physiologically relevant doses in humans, fetal human liver tissues were also analyzed for free and glucuronide-conjugated BPA. Total BPA concentrations in human fetal liver ranged from below LOQ to 96.8 ng/g (mean = 10.8, median = 3.39; n = 51). The overlap between mouse liver BPA levels and human fetal liver BPA levels indicates that the experimental approach used for dietary animal exposure captures a relevant and full range of human BPA exposure.
TABLE V.
BPA Liver Concentrations (ng/g): Measurements from mouse d22 Avy/a offspring tissue liver and human fetal liver tissue ranging from gestational ages 74–120
| Mice fed mg/kg diet (n = 9) | Mice fed μg/kg diet (n = 10) | Mice fed ng/kg diet (n = 11) | Mice fed control diet (n = 10) | Human fetal (n = 51) | |
|---|---|---|---|---|---|
| Range Total BPA (ng/g) | 9.5–870 | <LOQ-11.3 | <LOQ-13 | <LOQ-11.5 | <LOQ-96.8 |
| Mean (SD) free-BPA (ng/g) | 164 (132) | 1.8 (3.5) | 1.8 (2.9) | 3.7 (2.8) | 7.6 (12.2) |
| Mean (SD) conjugated-BPA (ng/g) | 278 (233) | 0.3 (0.3) | 1.0 (1.6) | 0.6 (0.8) | 3.2 (8.0) |
| Mean (SD) total BPA (ng/g) | 441 (338) | 2.0 (3.5) | 2.8 (4.5) | 4.3 (3.5) | 10.8 (18.5) |
| Median Total BPA (ng/g) | 472 | 0.6 | 0.3 | 4.2 | 3.4 |
LOQ = Limit of quantitation (0.1 ng/g).
DISCUSSION
In the current study, we report dose-dependent phenotypic and epigenetic responses following maternal dietary exposure to three levels of BPA. First, we observed a decrease in d22 wean body weight in a/a and Avy/a offspring exposed to 50 ng/kg diet of BPA versus control offspring. This association persists when analysis is restricted to a/a offspring alone, indicating that this effect is not associated with the epigenetically controlled adult onset obesity associated with Avy/a offspring, but rather manifests as a result of perinatal BPA exposure. Both low-birth weight in humans and early BPA exposure in animal models have been correlated to adult onset obesity [Barker, 2004; Heindel et al., 2009]; hence, it will be of interest to further evaluate early BPA exposure as a potential obesogen in adulthood. Body weight differences were not detected in offspring exposed to either 50 μg or 50 mg BPA/kg diet indicating a nonmonotonic dose response of wean weight and, corroborating previous studies using multiple doses of BPA with nonlinear outcomes [Rubin et al., 2001; Honma et al., 2002]. Mechanisms of action supporting nonmonotonic effects BPA, and endocrine disruptors in general, should be further investigated.
Global methylation of the mouse genome assessed using the LUMA assay reveals a significant increase in methylation across all BPA exposure groups in comparison to controls. This assay provides a measure of methylation at CCGG sites throughout the entire genome regardless of location, representing the degree to which the genome is globally methylated. The LUMA assay has been extensively used in analysis of human cancers [Lee et al., 2008; Deneberg et al., 2010; Poage et al., 2011]. There are limited studies, however, exploring environmental and/or nutritional exposures and their impact on global methylation [Gallou-Kabani et al., 2010]. Recently, using a mouse model, Gallou-Kabani et al. [2010] associated maternal high fat diet with decreased placental tissue DNA methylation in female offspring. The global decrease in CCGG methylation was not associated with decreased methylation at LINE-1 or B1 repetitive elements. Gene specific methylation at the Igf2r gene in female offspring exposed to high fat diet, on the other hand, was increased. Thus, it is important to note that the LUMA assay is restricted to methylation of CCGG sequences throughout the genome and is not necessarily representative of environmentally induced local changes at candidate genes or repetitive content derived from transposable elements such as LINE-1 and B1.
We also note dose-dependent shifts in the coat color distribution of genetically identical Avy/a offspring exposed to a 50 mg, μg, or ng/kg diet of BPA perinatally. The coat color distribution of offspring exposed to a 50 mg/kg diet of BPA displays a shift toward the yellow obese phenotype, reproducing our 2007 single dose study results [Dolinoy et al., 2007], whereas the μg dose offspring displays a shift toward the pseudoagouti lean coat color phenotype. Average methylation at the Avy locus of the mg exposed offspring is significantly decreased in comparison to the control group, providing epigenetic validation of the coat color distribution shift. In contrast, average methylation at the Avy locus of the μg exposure group was not statistically significant. An excess of categorization as slightly mottled Avy/a offspring may have offset a hypermethylation response among the μg exposure group when compared with the control group. Increased methylation in μg offspring compared with control offspring was, however, detected at the CabpIAP metastable epiallele, signifying that perinatal exposure to BPA at this dose increases methylation at this particular epigenetically labile locus. Taken together, these results (1) indicate that methylation at more than one locus is variable after perinatal exposure to BPA, (2) strengthen the evidence for nonmonotonic dose-dependent effects of BPA, and (3) provide evidence that variable dose levels of BPA act across different biological pathways [Vandenberg et al., 2009]. Genome-wide methylation and transcriptomics investigation should now be considered in light of this evidence.
Because of BPA’s ubiquitous existence in the environment and the ongoing debate about whether human internal BPA levels pose a health concern [Volkel et al., 2002; Ginsberg et al., 2009], it is of significance that animal studies capture human physiologically relevant exposure levels to determine BPA’s impact on human health outcomes. In the present study, we aimed to achieve physiologically relevant levels by including a high (mg), medium (μg), and low (ng) dose of BPA in the maternal diet. Calafat et al. [2008] reported a range of urinary total BPA (free and conjugated) of 0.4–149 ng/mL representative of individuals 6 years of age or older (n = 2,517) in subjects measured as a part of the 2003–2004 National Health and Nutrition Examination Survey (NHANES). Lang et al. [2008] reported urinary total BPA levels ranging from 3.34 to 4.48 ng/mL in individuals aged 18 years or older who have normal BMI (n = 469) from the 2003–2004 NHANES. Additionally, Padmanabhan et al. [2008] measured a range of 0.5–22.3 ng/mL of circulating free BPA (unconjugated) in maternal blood collected upon delivery.
We report d22 mouse liver BPA measurements ranging from below the limit of quantitation (LOQ) to 870 ng/g across all exposure groups comparable to human fetal liver measurements ranging from below LOQ to 96.8 ng/g. Ideally, liver BPA levels would be measured in fetal mouse tissues and compared with developmentally matched human fetal tissues; however, study design and current analytical requirements preclude this direct comparison. Nonetheless, mouse liver total and free BPA levels among Avy/a offspring exposed to 50 mg BPA/kg diet in our study range from 9.46 to 870 and 2.68 to 390 ng/g, respectively, (Table V) and are comparable to mouse circulating serum BPA concentrations recently reported in adult mice exposed to 100 mg BPA-d6/kg diet ad libitum for a 24-hr period [Sieli et al., 2011]. Sieli et al. [2011] show that in comparison to mice receiving a single oral bolus exposure of 20 mg BPA/kg body weight, animals fed BPA in the diet reach a maximum serum concentration of total and unconjugated (free) BPA at 6 hr of 802 and 18.8 ng/mL, respectively compared with 1 hr in the bolus group. Moreover, the observed serum concentrations following BPA administration in the diet are within the range of human exposure. Within the current study, we also observe a high degree of interindividual variation in mouse liver BPA concentrations within a particular dose group as well as the controls, perhaps reflecting time and metabolism effects associated with recent feeding bouts and/or continued nursing of pups. Unlike single bolus ingestion or injection routes of exposure, dietary exposure through feed results in inherent interindividual variability. It is important to note that we do not see a profound difference in mean or median mouse liver BPA concentrations among control and the two low dose groups; in fact, BPA is detected in some control animals despite housing in BPA-free caging and receiving BPA-free water. A limitation of this study is possible BPA cross-contamination via air given that animals were housed in a single room to minimize environmental heterogeneity that contributes to underlying epigenetic lability.
To our knowledge, this is the first study conducted using the viable yellow agouti epigenetic biosensor to analyze offspring phenotypic and epigenetic effects following multiple dose levels of either an environmental exposure or nutritional agent. Isogenic Avy mice allow for reproducible experiments as seen here with the coat color shift toward yellow in the Avy/a offspring exposed to the 50 mg BPA/kg diet [Dolinoy et al., 2007]. We also took a candidate gene approach limited to metastable epialleles unique to murine models and a global CCGG sequence assay. In order to conduct an unbiased epigenetic analysis, genome-wide methylation experiments must be applied in animal models. Epigenome-wide approaches will generate a template useful for the foundation and understanding of the full effect of BPA on the mouse epigenome. Concurrent studies are needed to assess BPA’s effect on the human epigenome, and whether labile loci between the mouse and human display significant overlap. In understanding the epigenome as a whole, one must also consider other epigenetic mechanisms such as histone modifications and microRNA interference separately and in conjunction with each other, and their sensitivity to environmental disruptions. Recently, DNA methylation and histone modifications have been observed to act in concert with one another at the Avy metastable epiallele [Dolinoy et al., 2010]. Increasing studies focusing on multiple epigenetic mechanisms will strengthen the understanding of environmentally induced alterations to the epigenome.
Acknowledgments
Grant sponsor: NIH; Grant Number: ES017524; Grant sponsor: University of Michigan National Institutes of Environmental Health Sciences (NIEHS) Core Center; Grant Number: P30 ES017885; Grant sponsor: Institutional Training Grant; Grant Number: T32 ES007062; Grant sponsor: University of Washington Birth Defects Research Laboratory; Grant Number: R24 HD000836-47.
Abbreviations
- %5mC
percent 5-methylated cytosine
- Avy
viable yellow agouti
- BPA
bisphenol A
- BW
body weight
- Cabp
CDK5 activator-binding protein
- d22
postnatal day 22
- ESI-MS/MS
electrospray triplequadruple mass spectrometry
- HPLC
high-performance liquid chromatography
- IAP
intracisternal A particle
- LOQ
limit of quantitation
- LUMA
luminometric methylation assay
- NHANES
National Health and Nutrition Examination Survey
- NTP
National Toxicology Program
- PCR
polymerase chain reaction
References
- Barker DJ. The developmental origins of adult disease. J Am Coll Nut. 2004;23:588–595. doi: 10.1080/07315724.2004.10719428. [DOI] [PubMed] [Google Scholar]
- Bateson P, Barker D, Clutton-Brock T, Deb D, D’Udine B, Foley RA, Gluckman P, Godfrey K, Kirkwood T, Lahr MM, McNamara J, Metcalfe NB, Monaghan P, Spencer HG, Sultan SE. Developmental plasticity and human health. Nature. 2004;430:419–421. doi: 10.1038/nature02725. [DOI] [PubMed] [Google Scholar]
- Bromer JG, Zhou Y, Taylor MB, Doherty L, Taylor HS. Bisphenol-A exposure in utero leads to epigenetic alterations in the developmental programming of uterine estrogen response. The FASEB J. 2010;24:2273–2280. doi: 10.1096/fj.09-140533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calafat A, Ye X, Wong L, Reidy J, Needham L. Exposure of the U.S. population to bisphenol A and 4-tertiary-octylphenol: 2003–2004. Environ Health Perspect. 2008;116:39–44. doi: 10.1289/ehp.10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooney CA, Dave AA, Wolff GL. Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. J Nutr. 2002;132:2393–2400. doi: 10.1093/jn/132.8.2393S. [DOI] [PubMed] [Google Scholar]
- Deneberg S, Grovdal M, Karimi M, Jansson M, Nahi H, Corbacioglu A, Gaidzik V, Dohner K, Paul C, Ekstrom TJ, Hellstrom-Lindberg E, Lehmann S. Gene-specific and global methylation patterns predict outcome in patients with acute myeloid leukemia. Leukemia. 2010;24:932–941. doi: 10.1038/leu.2010.41. [DOI] [PubMed] [Google Scholar]
- Dolinoy DC, Wiedman J, Waterland R, Jirtle RL. Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ Health Perspect. 2006;114:567–572. doi: 10.1289/ehp.8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci USA. 2007;104:13056–13061. doi: 10.1073/pnas.0703739104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinoy D, Weinhouse C, Jones T, Rozek L, Jirtle R. Variable histone modifications at the A (vy) metastable epiallele. Epigenetics. 2010;5:637–644. doi: 10.4161/epi.5.7.12892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druker R, Bruxner TJ, Lehrbach NJ, Whitelaw E. Complex patterns of transcription at the insertion site of a retrotransposon in the mouse. Nucl Acids Res. 2004;32:5800–5808. doi: 10.1093/nar/gkh914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhl D, Vrieling H, Miller K, Wolff G, Barsh G. Neomorphic agouti mutations in obese yellow mice. Nat Genet. 1994;8:59–65. doi: 10.1038/ng0994-59. [DOI] [PubMed] [Google Scholar]
- Gallou-Kabani C, Gabory A, Tost J, Karimi M, Mayeur S, Lesage J, Boudadi E, Gross M-S, Taurelle J, Vigé A, Breton C, Reusens B, Remacle C, Vieau D, Ekström TJ, Jais J-P, Junien C. Sex- and diet-specific changes of imprinted gene expression and DNA methylation in mouse placenta under a high-fat diet. PLoS ONE. 2010;5:e14398. doi: 10.1371/journal.pone.0014398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg G, Rice DC. Does rapid metabolism ensure negligible risk from bisphenol A? Environ Health Perspect. 2009;117:1639–1643. doi: 10.1289/ehp.0901010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunau C, Clark S, Rosenthal A. Bisulfite genomic sequencing: Systematic investigation of critical experimental parameters. Nucl Acids Res. 2001;29:E65–5. doi: 10.1093/nar/29.13.e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heindel JJ, vom Saal FS. Role of nutrition and environmental endocrine disrupting chemicals during the perinatal period on the aetiology of obesity. Mol Cell Endocrinol. 2009;304:90–96. doi: 10.1016/j.mce.2009.02.025. [DOI] [PubMed] [Google Scholar]
- Ho S-M, Tang W-Y, Belmonte de Frausto J, Prins GS. Developmental exposure to estradiol and bisphenol A increases susceptibility to prostate carcinogenesis and epigenetically regulates phosphodiesterase Type 4 Variant 4. Cancer Res. 2006;66:5624–5632. doi: 10.1158/0008-5472.CAN-06-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honma S, Suzuki A, Buchanan DL, Katsu Y, Watanabe H, Iguchi T. Low dose effect of in utero exposure to bisphenol A and diethylstilbestrol on female mouse reproduction. Reprod Toxicol. 2002;16:117–122. doi: 10.1016/s0890-6238(02)00006-0. [DOI] [PubMed] [Google Scholar]
- Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminen-Ahola N, Ahola A, Maga M, Mallitt K-A, Fahey P, Cox T, Whitelaw E, Chong S. Maternal ethanol consumption alters the epigenotype and the phenotype of offspring in a mouse model. PLoS Genet. 2010;6:e1000811. doi: 10.1371/journal.pgen.1000811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi M, Johansson S, Ekstrm T. Using LUMA: A luminometricbased assay for global DNA-methylation. Epigenetics. 2006;1:45–48. doi: 10.4161/epi.1.1.2587. [DOI] [PubMed] [Google Scholar]
- Kundakovic M, Champagne FA. Epigenetic perspective on the developmental effects of bisphenol A. Brain Behav Immunity. 2011;25:1084–1093. doi: 10.1016/j.bbi.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang IA, Galloway TS, Scarlett A, Henley WE, Depledge M, Wallace RB, Melzer D. Association of urinary bisphenol A concentration with medical disorders and laboratory abnormalities in adults. JAMA. 2008;300:1303–1310. doi: 10.1001/jama.300.11.1303. [DOI] [PubMed] [Google Scholar]
- Lee J-J, Geli J, Larsson C, Wallin G, Karimi M, Zedenius J, Hg A, Foukakis T. Gene-specific promoter hypermethylation without global hypomethylation in follicular thyroid cancer. Int J Oncol. 2008;33:861–869. [PubMed] [Google Scholar]
- Miltenberger R, Mynatt R, Wilkinson J, Woychik R. The role of the agouti gene in the Yellow Obese Syndrome. J Nutr. 1997;127:1902S–1907S. doi: 10.1093/jn/127.9.1902S. [DOI] [PubMed] [Google Scholar]
- Morgan H, Sutherland H, Martin D, Whitelaw E. Epigenetic inheritance at the agouti locus in the mouse. Nat Genet. 1999;23:314–318. doi: 10.1038/15490. [DOI] [PubMed] [Google Scholar]
- Moriyama K, Tagami T, Akamizu T, Usui T, Saijo M, Kanamoto N, Hataya Y, Shimatsu A, Kuzuya H, Nakao K. Thyroid hormone action is disrupted by bisphenol a as an antagonist. J Clin Endocrinol Metab. 2002;87:5185–5190. doi: 10.1210/jc.2002-020209. [DOI] [PubMed] [Google Scholar]
- Padmanabhan V, Siefert K, Ransom S, Johnson T, Pinkerton J, Anderson L, Tao L, Kannan K. Maternal bisphenol-A levels at delivery: A looming problem? J Perinatol. 2008;28:258–263. doi: 10.1038/sj.jp.7211913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poage GM, Houseman EA, Christensen BC, Butler RA, Avissar-Whiting M, McClean MD, Waterboer T, Pawlita M, Marsit CJ, Kelsey KT. Global hypomethylation identifies loci targeted for hypermethylation in head and neck cancer. Clin Cancer Res. 2011;17:3579–3589. doi: 10.1158/1078-0432.CCR-11-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins GS, Tang W-Y, Belmonte J, Ho S-M. Perinatal exposure to oestradiol and bisphenol A alters the prostate epigenome and increases susceptibility to carcinogenesis. Basic Clinical Pharmacol Toxicol. 2008;102:134–138. doi: 10.1111/j.1742-7843.2007.00166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakyan VK, Blewitt ME, Druker R, Preis JI, Whitelaw E. Metastable epialleles in mammals. Trends Genet. 2002;18:348–351. doi: 10.1016/s0168-9525(02)02709-9. [DOI] [PubMed] [Google Scholar]
- Rubin BS, Soto AM. Bisphenol A: Perinatal exposure and body weight. Mol Cell Endocrinol. 2009;304:55–62. doi: 10.1016/j.mce.2009.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin BS, Murray MK, Damassa DA, King JC, Soto AM. Perinatal exposure to low doses of bisphenol A affects body weight, patterns of estrous cyclicity, and plasma LH levels. Environ Health Perspect. 2001;109:675–680. doi: 10.1289/ehp.01109675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruvinsky A, Flood W, Costantini F. Developmental mosaicism may explain spontaneous reappearance of the Axin(Fu) mutation in mice. Genesis. 2001;29:49–54. doi: 10.1002/1526-968x(200102)29:2<49::aid-gene1004>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Sieli PT, Jašarević E, Warzak DA, Mao J, Ellersieck MR, Liao C, Kannan K, Collet SH, Toutain P-L, vom Saal FS, Rosenfeld CS. Comparison of serum bisphenol A concentrations in mice exposed to bisphenol a through the diet versus oral bolus exposure. Environ Health Perspect. 2011;119:1260–1265. doi: 10.1289/ehp.1003385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi O, Oishi S. Testicular toxicity of dietarily or parenterally administered bisphenol A in rats and mice. Food Chem Toxicol. 2003;41:1035–1044. doi: 10.1016/s0278-6915(03)00031-0. [DOI] [PubMed] [Google Scholar]
- Vandenberg LN, Hauser R, Marcus M, Olea N, Welshons WV. Human exposure to bisphenol A (BPA) Reprod Toxicol. 2007;24:139–177. doi: 10.1016/j.reprotox.2007.07.010. [DOI] [PubMed] [Google Scholar]
- Vandenberg LN, Maffini MV, Sonnenschein C, Rubin BS, Soto AM. Bisphenol-A and the great divide: A review of controversies in the field of endocrine disruption. Endocrine Rev. 2009;30:75–95. doi: 10.1210/er.2008-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasicek T, Zeng L, Guan X, Zhang T, Costantini F, Tilghman S. Two dominant mutations in the mouse fused gene are the result of transposon insertions. Genetics. 1997;147:777–786. doi: 10.1093/genetics/147.2.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkel W, Colnot T, Csanady G, Filser J, Dekant W. Metabolism and kinetics of bisphenol A in humans at low doses following oral administration. Chem Res Toxicol. 2002;15:1281–1287. doi: 10.1021/tx025548t. [DOI] [PubMed] [Google Scholar]
- vom Saal FS, Welshons WV. Large effects from small exposures. II. The importance of positive controls in low-dose research on bisphenol A. Environ Res. 2006;100:50–76. doi: 10.1016/j.envres.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Waterland R, Jirtle R. Transposable elements: Targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003;23:5293–5300. doi: 10.1128/MCB.23.15.5293-5300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterland R, Jirtle R. Early nutrition, epigenetic changes at transposons and imprinted genes, and enhanced susceptibility to adult chronic diseases. Nutrition. 2004;20:63–68. doi: 10.1016/j.nut.2003.09.011. [DOI] [PubMed] [Google Scholar]