

Iron uptake promotes hydrogen peroxide–dependent differentiation of Leishmania promastigotes into infective amastigotes.
Abstract
During its life cycle, Leishmania undergoes extreme environmental changes, alternating between insect vectors and vertebrate hosts. Elevated temperature and decreased pH, conditions encountered after macrophage invasion, can induce axenic differentiation of avirulent promastigotes into virulent amastigotes. Here we show that iron uptake is a major trigger for the differentiation of Leishmania amazonensis amastigotes, independently of temperature and pH changes. We found that iron depletion from the culture medium triggered expression of the ferrous iron transporter LIT1 (Leishmania iron transporter 1), an increase in iron content of the parasites, growth arrest, and differentiation of wild-type (WT) promastigotes into infective amastigotes. In contrast, LIT1-null promastigotes showed reduced intracellular iron content and sustained growth in iron-poor media, followed by cell death. LIT1 up-regulation also increased iron superoxide dismutase (FeSOD) activity in WT but not in LIT1-null parasites. Notably, the superoxide-generating drug menadione or H2O2 was sufficient to trigger differentiation of WT promastigotes into fully infective amastigotes. LIT1-null promastigotes accumulated superoxide radicals and initiated amastigote differentiation after exposure to H2O2 but not to menadione. Our results reveal a novel role for FeSOD activity and reactive oxygen species in orchestrating the differentiation of virulent Leishmania amastigotes in a process regulated by iron availability.
Infection with the protozoan parasite Leishmania impairs the health of millions of people throughout the world. Depending on the Leishmania species, the pathology ranges from self-healing cutaneous lesions to lethal visceralizing disease. In mammals, Leishmania parasites are present as oval-shaped amastigotes lacking a long flagellum, which replicate inside macrophages. After a blood meal, amastigotes ingested by sand flies transform into flagellated and motile promastigotes, which replicate in the insect’s digestive tract (Sacks and Kamhawi, 2001). Several days after the initial blood meal, promastigotes differentiate into infective metacyclic forms (Sacks and Perkins, 1984), which migrate to the esophagus of the sand fly (Sacks and Kamhawi, 2001; Gossage et al., 2003) and are regurgitated into the skin of the host during a blood meal. After uptake by host macrophages, the parasites establish parasitophorous vacuoles (PVs) with properties of phagolysosomes, where differentiation and replication of amastigotes occurs (Antoine et al., 1998). Amastigotes persist in host tissues during chronic infections and propagate the infection from cell to cell and to uninfected sand flies. Thus, amastigotes are the most important Leishmania life cycle forms in the context of human disease. A better understanding of the morphological and metabolic changes associated with promastigote to amastigote differentiation is of critical importance because it can facilitate the development of new and less toxic drugs to treat these infections.
Transformation of noninfective promastigotes into infective amastigotes can be induced axenically in a few Leishmania species by conditions that mimic the intracellular environment, such as elevated temperature and low pH (Bates et al., 1992; Zilberstein and Shapira, 1994; Barak et al., 2005). Several groups have taken advantage of this system to show that promastigote to amastigote differentiation, a process which involves shortening of the flagellum and change from an elongated to spherical shape, is coupled to marked metabolic changes and the development of infectivity for mammals (Saar et al., 1998; Gupta et al., 2001; Debrabant et al., 2004). A shift to a higher rate of energy production was proposed to be required for establishment of Leishmania infections because of the need for driving proton pumps and carriers to counter the steep proton gradient between the acidic phagolysosome lumen and the amastigote cytosol (Tsigankov et al., 2012). Of particular relevance for our current study with Leishmania amazonensis, genome-wide expression profiling in L. mexicana revealed differential gene expression between promastigotes and lesion-derived and axenic amastigote forms (Holzer et al., 2006). A detailed analysis of the transcriptome of L. donovani during promastigote to amastigote differentiation also showed ordered changes in the expression of hundreds of genes, shedding light on pathways that are up- and down-regulated as the parasites undergo this transformation (Saxena et al., 2007). Proteomic studies revealed modulation in protein posttranslational modifications (Rosenzweig et al., 2008a) and showed that during differentiation into amastigotes, L. donovani shifts its main energy source from glucose to fatty acids and amino acids, consistent with an adaptation to intracellular life (Rosenzweig et al., 2008b). Overall, the results from these genome-wide studies suggested that regulation of mRNA levels plays a major role early in the L. donovani promastigote to amastigote differentiation process, whereas translational and posttranslational regulation are more important at a later stage (Lahav et al., 2011).
Despite these advances, it is still unclear how environmental cues are sensed by Leishmania parasites and translated into the cellular differentiation program. An unexpected insight into this question came from studying how the iron-poor environment of macrophage PVs induces expression of the iron transporter LIT1 (Leishmania iron transporter 1) in L. amazonensis amastigotes (Huynh et al., 2006). While characterizing an L. amazonensis strain lacking LIT1, we found that iron transport is a major factor regulating the transition of promastigotes to amastigotes. Iron depletion from the culture medium induces LIT1 expression and is sufficient for triggering promastigote to amastigote differentiation, without a need for a shift in temperature or pH. Our results indicate that iron uptake exerts its role on parasite differentiation by controlling the intracellular redox balance, by modulating iron superoxide dismutase (SOD [FeSOD]) activity and hydrogen peroxide (H2O2) generation.
RESULTS
Expression of the iron transporter LIT1 is induced by low iron availability and controls proliferation of L. amazonensis promastigotes
An earlier study showed that the LIT1 ferrous iron transporter is only detected with antibodies on the surface of L. amazonensis amastigotes after 24 h of intracellular residence in macrophages. LIT1 was not detected on recently internalized amastigotes or on extracellular promastigotes grown in complete culture medium containing a source of iron (Huynh et al., 2006). Interestingly, LIT1 expression occurred earlier in amastigotes within macrophages expressing the iron efflux pump Nramp1. This observation suggested that the low iron PV environment might favor LIT1 expression (Huynh et al., 2006). To directly determine whether iron availability controlled LIT1 expression in promastigotes, we developed a growth media specifically depleted in iron but maintaining all other nutrients, ions, and growth factors necessary to support parasite growth (see Materials and methods).
After 24 h of growth in iron-depleted medium, LIT1 was detected with specific antibodies on the surface of live WT promastigotes in a punctate pattern similar to what was previously observed in intracellular amastigotes (Huynh et al., 2006). As expected, no immunofluorescence signal was detected on Δlit1 promastigotes cultured under identical conditions (Fig. 1 A). Quantitative real-time PCR (qPCR) analysis revealed that LIT1 transcripts increased steadily after iron removal from the medium, reaching approximately sixfold up-regulation after 24 h (Fig. 1 B). The 24-h peak in transcript levels coincided with the time point when the LIT1 protein was detected with antibodies on the surface of extracellular promastigotes (Fig. 1 A).
Figure 1.

Iron depletion induces LIT1 expression and LIT1-dependent inhibition of replication. (A) Immunofluorescence with anti-LIT1 antibodies of live WT and Δlit1 promastigotes after 24-h incubation in iron-depleted medium. The arrows point to promastigotes expressing LIT1 on the cell surface. Bars, 6 µm. (B) qPCR detection of LIT1 transcripts in WT promastigotes at different periods after iron depletion. (C) WT promastigotes grown in regular media to mid-log phase were incubated in iron-depleted medium for 6 h, harvested, and resuspended in iron-depleted medium supplemented or not with 8 µM Fe-NTA, 5 µM hemin, or 8 µM Fe-NTA/5 µM hemin. After 14 h, total RNA was isolated from 108 cells harvested from each culture, and LIT1 transcript levels were determined by qPCR. *, P < 0.0125; **, P < 0.0075 (Student’s t test). (D) WT or Δlit1 promastigotes grown to mid-log in regular medium were used to initiate cultures in iron-depleted medium. The growth rates of WT and Δlit1 strains under iron-depleted conditions were determined by microscopic counting of viable cells at the indicated time points. (B–D) The data represent the mean ± SD of triplicate determinations and are representative of three independent experiments.
When intracellular iron levels were determined using a ferrozine assay, a transient decrease was observed 24 h after iron removal from the medium, followed by a gradual increase after 48 and 72 h (Table 1). These findings suggest that the initial drop in intracellular iron caused by removing iron from the medium is subsequently reversed as a consequence of elevated LIT1 expression, resulting in iron uptake and an overall increase in intracellular iron availability. These data also indicate that iron availability controls expression of LIT1 because transcripts for this iron transporter dropped (Fig. 1 B) as the intracellular iron levels in the parasites increased (Table 1). Importantly, the up-regulation in LIT1 transcripts observed after 6 h of culture in iron-depleted medium was reversed when iron was added back to the medium and the parasites were cultured for an additional 14 h. Either ferric nitrolotriacetate (Fe-NTA) or hemin alone significantly reduced LIT1 transcripts, but full restoration to the low levels seen in parasites kept in complete iron-rich medium required both Fe-NTA and hemin (Fig. 1 C).
Table 1.
Iron content of L. amazonensis cultured in iron-poor medium
| Days of culture in iron-depleted medium | WT | Δlit1 |
| nmol/mg protein | nmol/mg protein | |
| 0 | 8.8 ± 0.7 | 8.7 ± 0.4 |
| 1 | 6.7 ± 0.6 (P = 0.0424)a | 5.5 ± 0.9 (P = 0.0158)a |
| 2 | 9.3 ± 1.9 (P = 0.1978) | 5.8 ± 1.2 (P = 0.0798) |
| 3 | 10.88 ± 1.7 (P = 0.0369)a | 5.9 ± 1.1 (P = 0.0451)a |
Determination of total cellular iron content of L. amazonensis WT and Δlit1 parasites was performed using ferrozine-based quantitation (Riemer et al., 2004). The data represent the mean and SD of five independent experiments.
Indicates significant differences (P ≤ 0.05) from day 0 values (two-tailed Student’s t test).
In regular iron-replete promastigote culture medium, WT and Δlit1 promastigotes grew at similar rates and reached a density of ∼8 × 107–8 parasites/ml during stationary phase (not depicted; Huynh et al., 2006). When grown in iron-depleted medium, the growth rate of WT promastigotes was decreased by ∼40%, with the culture reaching stationary phase at a maximum density of ∼3 × 107 parasites/ml (Fig. 1 D). In sharp contrast, the growth rate of Δlit1 promastigotes was unaffected by the low iron conditions, with the culture reaching a high density of ∼7 × 107 parasites/ml. Surprisingly, instead of entering the plateau characteristic of the stationary phase of growth, Δlit1 promastigote cultures underwent sudden massive death, as indicated by a sharp decrease in the number of viable parasites (Fig. 1 D). These data suggested that expression of the ferrous iron transporter LIT1, which is expressed on the cell surface under conditions of low iron availability in the environment, controls the proliferation of L. amazonensis promastigotes.
Up-regulation of LIT1 induced by an iron-poor environment promotes promastigote to amastigote differentiation
In addition to the growth rate inhibition, removing iron from the culture medium induced a marked morphological change in WT promastigotes. By the fourth day after shift to iron-poor medium, in addition to the elongated and motile promastigote forms, the WT cultures also contained numerous rounded amastigote-like parasites lacking long flagella (comprising ∼55% of the population). These amastigote-like forms became more abundant by day 5, reaching ∼75% of the viable parasites in the culture. In sharp contrast, Δlit1 promastigotes cultured under identical conditions did not accumulate rounded forms (Fig. 2, A and B). Importantly, by days 4 and 5, >70% of the Δlit1 population had lost viability and showed clear signs of degeneration, with the few remaining viable parasites retaining the elongated, flagellated morphology typical of promastigotes (Fig. 2 A).
Figure 2.

Iron depletion induces morphological changes and reduced growth rate in WT but not in Δlit1 promastigotes. WT or Δlit1 promastigotes grown to mid-log phase in regular medium were collected and resuspended in iron-depleted medium at 106 cells/ml. Parasites were examined microscopically after staining with the viability dye FDA. (A) Phase-contrast images showing representative WT and Δlit1 parasites on days 4 and 5 during growth in iron-depleted medium. Bars, 12 µm. (B) Quantification of flagellated or nonflagellated WT or Δlit1 parasites at increasing periods after iron deprivation. (C and D) WT (C) or Δlit1 (D) promastigotes grown to mid-log in regular medium were harvested and resuspended in regular or iron-depleted medium or iron-depleted medium supplemented with 8 µM Fe-NTA. Growth was determined by counting viable parasites at the indicated time points. (B–D) The data represent the mean ± SD of triplicate determinations. (E) Quantification of flagellated or nonflagellated WT parasites under the different growth conditions described in C. The data represent the mean ± SD of determinations of three independent experiments.
Next, we examined whether the delayed growth and morphological change induced by depleting iron could be reversed by adding back several forms of iron chelates to the culture medium. Addition of holo-transferrin and holo-lactoferrin failed to make any noticeable changes in the pattern of WT promastigote growth, whereas addition of hemin alone partially rescued the growth defect (not depicted). However, Fe-NTA added at 8 µM rescued promastigote growth to ∼90% of normal levels after a lag of a few days (Fig. 2 C). Consistent with the inability of Δlit1 promastigotes to transport iron across their plasma membrane (Huynh et al., 2006), addition of 8 µM Fe-NTA to this mutant strain had no effect, with cultures showing the same exponential growth pattern followed by sudden death (Fig. 2 D). The kinetics of transformation of WT promastigotes into amastigote-like forms after the shift to iron-poor medium was also markedly delayed after Fe-NTA supplementation (Fig. 2 E). Thus, the presence of iron in the environment has a dual effect, inhibition of LIT1 expression (Fig. 1 C) and maintenance of the parasites in the promastigote proliferative stage. When LIT1 is expressed under low iron availability conditions, conditions appear to be generated that decrease promastigote replication and promote the onset of differentiation into amastigotes.
To determine whether the round, nonflagellated parasites generated in iron-depleted cultures were actually in the process of differentiating into amastigotes, we examined the expression of several Leishmania stage-specific markers. The amastigote-specific nuclease P4 (Kar et al., 2000) was detected in WT parasites grown in iron-depleted medium for 4 d, and the immunofluorescence signal was particularly strong in the rounded parasites with amastigote-like morphology. In contrast, P4 was not detected in WT parasites cultured in regular iron-containing medium or in Δlit1 promastigotes grown with or without iron (Fig. 3 A). Conversely, expression of the paraflagellar rod protein PFR-1, an integral component of the promastigote flagellum, was markedly inhibited in WT parasites cultured in iron-poor medium, whereas levels of cysteine protease B (CPB), which is primarily expressed in amastigotes (Duboise et al., 1994), increased (Fig. 3 B).
Figure 3.

Iron depletion induces expression of amastigote-specific proteins in WT but not in Δlit1 promastigotes. WT or Δlit1 L. amazonensis promastigotes were grown for 4 d in iron-depleted or regular medium. (A) Immunofluorescence with a monoclonal antibody against the amastigote-specific P4 endonuclease. The arrowheads point to rounded forms expressing P4. The left column shows phase-contrast, the middle column shows DAPI DNA stain (blue), and the right column shows anti-P4 (green). Bars, 10 µm. (B) Immunoblots of parasite lysates (10 µg protein/lane) probed with monoclonal antibodies against the promastigote-specific paraflagellar rod protein (PFR-1) and amastigote–up-regulated CPB. Anti-actin antibodies were used to detect loading controls. All data shown are representative of at least three independent experiments.
To obtain a genome-wide overview of changes in gene expression after iron starvation, we isolated RNA from WT L. amazonensis promastigotes grown for 24 h in medium with and without iron and prepared RNA-seq libraries using a splice leader (SL) primer to amplify the 5′ end of all mRNAs. Analysis of the results revealed that 11 genes were up-regulated by more than threefold in two independent replicates, whereas 21 genes were down-regulated by at least threefold after growth in iron-depleted medium. Another 59 and 109 genes showed consistent twofold up- or down-regulation, respectively (selected examples are shown in Table S2). As expected, genes up-regulated more than threefold include the ferrous iron transporter LIT1 (LmxM.30.3070; Huynh et al., 2006) and the ferric iron reductase LFR1 (LmxM.29.1610; Flannery et al., 2011), whereas several genes that encode iron-dependent proteins were down-regulated (LmxM.34.1540, LmxM.08.0290, and LmxM.18.0510).
The SL RNA-seq results also demonstrated that culture in iron-poor medium increases mRNA levels for several genes known to be up-regulated in the amastigote stage: several members of the amastin gene family (Wu et al., 2000) that encode surface proteins up-regulated in L. major and L. infantum amastigotes (Rochette et al., 2008; Rosenzweig et al., 2008a; Lahav et al., 2011), myo-inositol-1-phosphate synthetase (LmxM.14.1360; Rochette et al., 2008), and an orthologue (LmxM.28.0980) of Ldp27, which is specifically expressed during promastigote to amastigote differentiation in L. donovani (P27, Table S2; Dey et al., 2010). qPCR assays confirmed the marked up-regulation of three amastin-like genes (LmxM.08.0760, LmxM.08.0770, and LmxM.24.1260), whose mRNAs increased 8-, 11-, and 3-fold, respectively, by day 3. Similarly, transcripts of the P27 gene (LmxM.28.0980) showed 10-fold up-regulation by day 3 (Fig. 4). In agreement with an essential role for LIT1 in the differentiation trigger, the levels of these amastigote stage-specific transcripts showed no significant changes in Δlit1 parasites.
Figure 4.

Iron depletion induces expression of amastigote-specific transcripts in WT but not in Δlit1 promastigotes. Total RNA extracted from WT or Δlit1 parasites grown for the indicated periods in iron-depleted medium was analyzed by qPCR using primers for the coding regions of amastin-like genes 1 and 2 (LmxM.08.0760 and LmxM.08.0770), amastin-like 3 surface protein (LmxM.24.1260), P27 (LmxM.28.0980), 60S ribosomal L5 gene (LmxM.34.1900), and eIF-3 subunit (LmxM.36.3880). Day 0 represents the time point when promastigote cultures at mid-log phase were transferred to iron-depleted medium. The data represent the mean ± SD of triplicate determinations and are representative of three independent experiments.
In contrast, both the SL RNA-seq and qPCR results showed a reduction in mRNA for several ribosomal proteins and translation factors (LmxM.36.3880, LmxM.34.1430, LmxM.26.0010, LmxM.36.2360, and LmxM.08_29.0030; Table S2), consistent with the reported down-regulation of the protein translation machinery during amastigote differentiation (Lahav et al., 2011). The eIF-3 subunit mRNA was reduced by more than fivefold after 3 d in iron-depleted medium (Fig. 4), whereas ribosomal 60S L5 transcript levels were reduced by approximately twofold (Fig. 4).
Autophagy plays an important role in Leishmania differentiation (Bates, 2006; Besteiro et al., 2006; Williams et al., 2006, 2009, 2012). The SL RNA-seq results also showed up-regulation of mRNA from several genes (LmxM.09.0150, LmxM.19.0820, and LmxM.19.0870) encoding proteins (ATG8/AUT7/APG8/PAZ2) associated with autophagosome formation during differentiation of L. major, as well as LmxM.29.0270, which encodes the cysteine peptidase (AUT2/APG4/ATG4) responsible for processing ATG8 to its active form (Williams et al., 2009). These results were confirmed by qPCR of mRNA from WT and Δlit1 L. amazonensis promastigotes after exposure to low iron (Fig. 5, A and B). Transcripts for ATG8 and ATG4.1 were both up-regulated six- to sevenfold 3 d after WT promastigotes were shifted to iron-depleted medium. The CPB mentioned above (encoded by LmxM.08.1070 and LmxM.08.1080) is necessary for autophagy and differentiation in L. mexicana (Williams et al., 2006). Both SL RNA-seq (Table S2) and qPCR (Fig. 5 C) indicate it was also up-regulated (by ∼16-fold after 3 d) after growth of WT promastigotes in iron-depleted medium. In contrast, Δlit1 promastigotes, grown under identical conditions, showed no change in transcript levels for these autophagy-related genes (Fig. 5). Collectively, our data suggest that iron uptake mediated by LIT1 expressed under low iron conditions signals initiation of differentiation of L. amazonensis promastigotes into amastigote forms. The signaling pathway that drives differentiation is not activated if iron transport is blocked, as observed in Δlit1 parasites.
Figure 5.
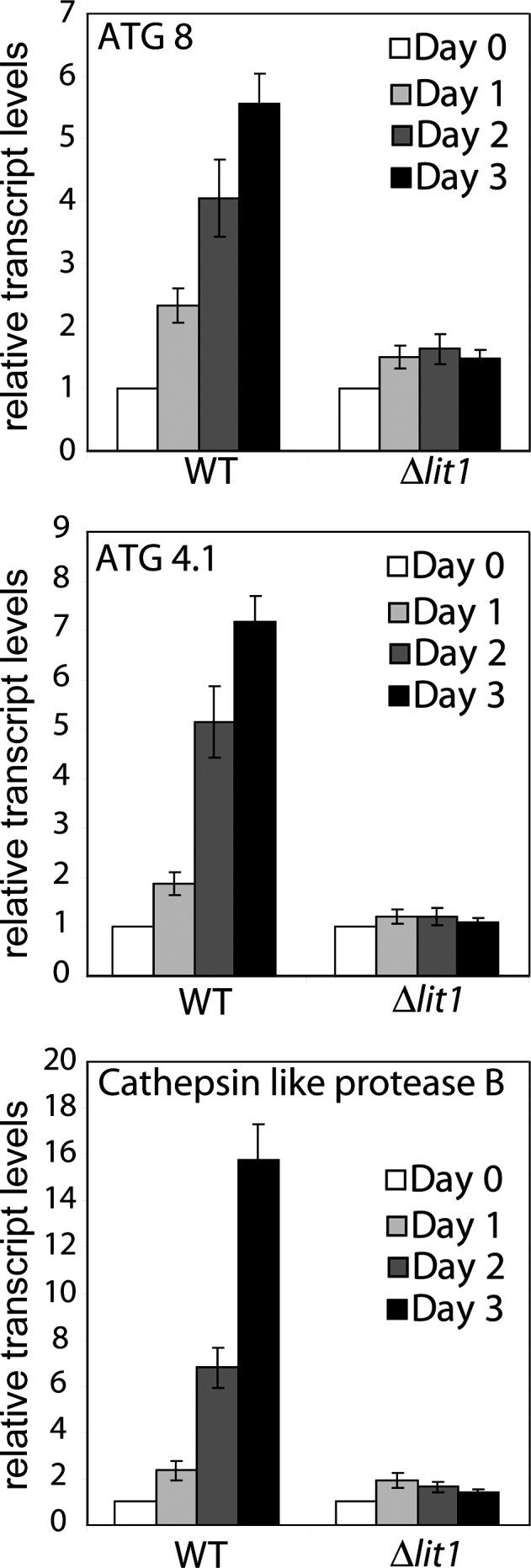
Iron depletion induces expression of autophagy-related gene transcripts in WT but not in Δlit1 promastigotes. Total RNA from WT or Δlit1 parasites grown for the indicated time periods in iron-depleted medium was analyzed by qPCR using primers specific for the coding regions of ATG8 (LmxM.19.0870), ATG4.1 (LmxM.31.3890), and CPB (LmxM.08.1070). The data represent the mean ± SD of triplicate determinations and are representative of three independent experiments.
L. amazonensis FeSOD activity increases before promastigote to amastigote differentiation
In higher eukaryotic cells, reactive oxygen species (ROS) recently emerged as important players in cellular signaling pathways (Owusu-Ansah and Banerjee, 2009; Theopold, 2009; Hamanaka and Chandel, 2010). ROS are produced endogenously from several sources, including the mitochondrial electron transport chain and NADPH oxidases. Reduction in one electron converts molecular O2 into superoxide radical (O2•−), which is dismutated by the enzyme SOD to nonradical H2O2 (Hamanaka and Chandel, 2010). Elevated levels of O2•− can accelerate cell proliferation, as observed in cancer cell lines (Kinnula and Crapo, 2004). In contrast, H2O2 generation has been implicated in the arrest of uncontrolled cell proliferation and initiation of cellular differentiation (Hamanaka and Chandel, 2010; Tsukagoshi et al., 2010; Rigoulet et al., 2011). Considering the increasingly evident role of ROS in determining cell fate, and the fact that kinetoplastid SODs exclusively use iron as an essential cofactor (FeSOD), we examined SOD activity levels during the growth of WT and Δlit1 L. amazonensis promastigotes in iron-poor medium.
Although the FeSOD genes (LmxM.08.0290, LmxM.31.1820, and LmxM.31.1830) were down-regulated at the mRNA level (Table S2), we observed a significant increase in FeSOD activity on the third and fourth day after transfer to iron-depleted medium (Fig. 6 A). This observation coincided with the appearance of amastigote forms in the culture. This observation is very consistent with a previous study in L. donovani, which showed a reduction in FeSODA mRNA during promastigote to amastigote differentiation induced by 37°C/pH 5.5 but an actual increase in this enzyme at the protein level (Lahav et al., 2011).
Figure 6.
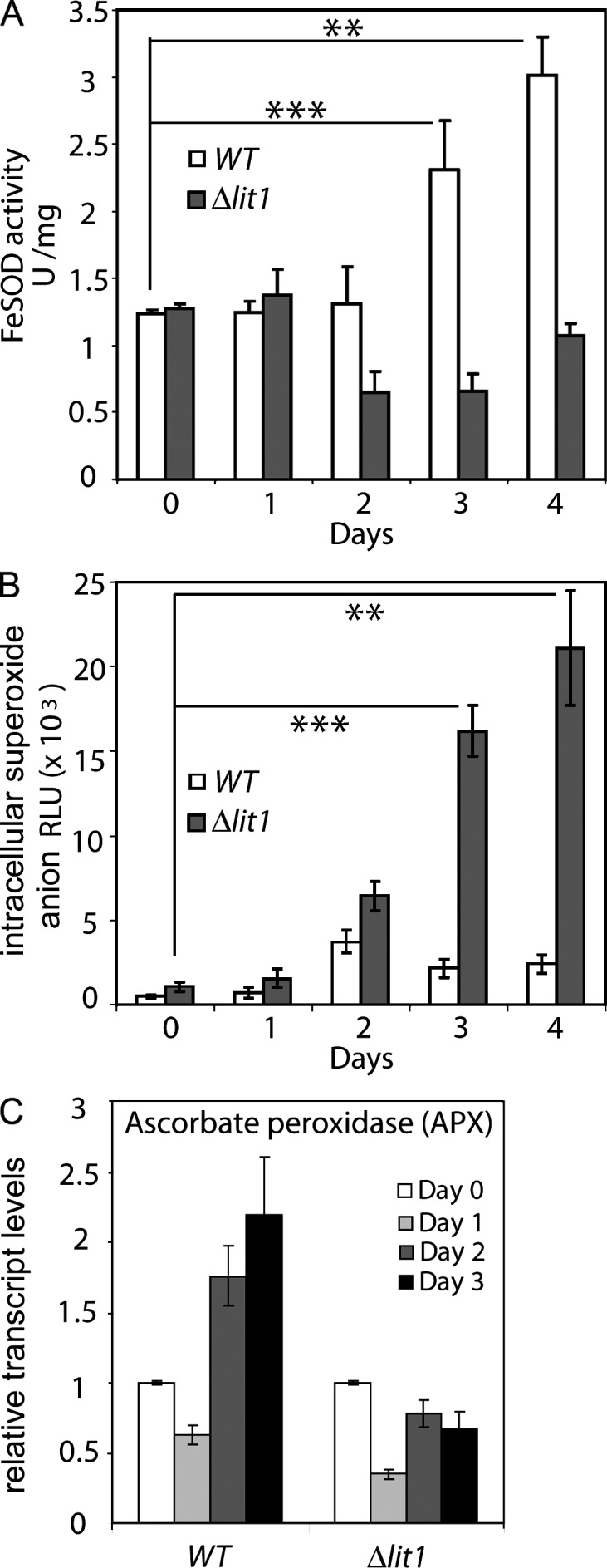
Amastigote differentiation triggered by iron deprivation is preceded by an increase in SOD activity. (A) WT and Δlit1 promastigotes grown in regular media were transferred from regular to iron-depleted medium (day 0) and grown for an additional 4 d (days 1–4), until the majority of the WT parasites exhibited a rounded, nonflagellated morphology. Whole cell extracts prepared at the indicated time points were assayed for detection of SOD activity. ***, P = 0.0043; day 4: **, P = 0.0061 (two-tailed Student’s t test). (B) Intracellular concentrations of superoxide anion were determined in WT and Δlit1 parasites during days 1–4 of iron deprivation. **, P = 0.012; day 4: ***, P = 0.009 (two-tailed Student’s t test). (C) Total RNA extracted from WT or Δlit1 parasites grown for the indicated time points in iron-depleted medium was analyzed by qPCR using primers specific for the coding regions of APX (LmxM.33.0070). (A–C) The data represent the mean ± SD of triplicate determinations and are representative of three independent experiments.
In contrast, in Δlit1 parasites, FeSOD activity decreased to half of day 0 levels by the third day of culture in iron-poor medium, and no increase was observed until sudden death of the whole population by day 4 (Fig. 6 A). Consistent with the low levels of FeSOD activity, in Δlit1, O2•− levels increased progressively with time after iron depletion, reaching values markedly higher than in WT parasites by days 3 and 4 (Fig. 6 B). These data suggest that LIT1-mediated iron uptake increases the levels of FeSOD activity in L. amazonensis promastigotes, a process which may play a role in signaling differentiation of promastigotes into amastigotes through the generation of the diffusible ROS H2O2. Conversely, the inability of Δlit1 parasites to take up iron, which prevents increase in FeSOD activity and H2O2 generation, may explain why this mutant strain fails to respond to iron depletion by differentiating into amastigotes.
Repeated attempts to detect H2O2 in WT promastigotes undergoing differentiation were unsuccessful. Investigating this issue, we found that iron depletion from the culture medium also induced elevated levels of ascorbate peroxidase (APX) by days 3 and 4 in WT but not Δlit1 parasites (Fig. 6 C). APX is a Leishmania enzyme involved in H2O2 detoxification in Leishmania (Pal et al., 2010). Thus, our results suggest that up-regulation of the degradation machinery may prevent H2O2 accumulation to detectable levels in differentiating parasites, as expected from the known tight regulation of ROS involved in signaling pathways.
Role of ROS in signaling promastigote to amastigote differentiation
To directly test a potential role of ROS-mediated signaling in initiating differentiation, we treated log phase L. amazonensis promastigotes cultured in regular, iron-containing media (conditions which inhibit LIT1 expression; Fig. 1 C) with the superoxide-generating drug menadione and with H2O2. Nontoxic concentrations of these agents were determined based on the maximal parasite survival rate after 24 h of exposure, assessed using a sensitive live-dead fluorescent viability assay. Based on our hypothesis, we expected H2O2 to provide the differentiation signal for both WT and Δlit1 parasites even in the absence of iron uptake because the need for FeSOD would be bypassed. In contrast, the superoxide radical–generating drug menadione should only initiate differentiation if the parasites were capable of acquiring iron and up-regulating FeSOD activity to generate H2O2. Consistent with this scenario, both menadione and H2O2 slowed the growth of WT parasites (Fig. 7 A) and promoted the appearance of rounded amastigote-like forms (Fig. 7 B). Morphometric measurements confirmed that a significant fraction of the population of WT parasites exposed to menadione and H2O2 had a decreased body length (Fig. 7 B). Treatment with menadione or H2O2 also induced expression of the amastigote-specific protein P4 and increased levels of transcripts for amastin-like 1 and 2, CPB, and the autophagy-related ATG8 and ATG4.1 genes (Fig. 8, A–C). In contrast, despite a reduction in growth rate (Fig. 7 C), Δlit1 promastigotes treated with menadione showed a marked change in morphology compared with untreated cells (Fig. 7 D) and no significant up-regulation in amastigote-associated gene expression (Fig. 8 B). These findings are in agreement with the inability of Δlit1 parasites to increase expression of active FeSOD (Fig. 6 A). In contrast, H2O2 treatment of Δlit1 parasites led to significant up-regulation of transcripts associated with promastigote to amastigote differentiation, similar to what was observed with WT parasites (Fig. 8 C).
Figure 7.

ROS-generating pro-oxidants inhibit proliferation and induce amastigote differentiation. Mid-log phase WT or Δlit1 promastigotes were resuspended at 4 × 106 cells/ml in regular medium and treated overnight with 0.1% DMSO, 5 µM menadione, or 150 µM H2O2. (A and C) Growth was estimated by microscopic counting of WT (A) and Δlit1 (C) viable cells at 0 and 18 h after treatment. The data represent the mean ± SD of triplicate measurements of three independent experiments. The asterisks represent significant differences between the DMSO-treated and ROS-generating pro-oxidant–treated cultures. WT: **, P = 0.008 (menadione); **, P = 0.0073 (H2O2); Δlit1: **, P = 0.0047 (menadione); **, P = 0.009 (H2O2; two-tailed Student’s t test). (B and D) The longest cell body axes of WT (B) or Δlit1 (D) promastigotes were measured in differential interference contrast (DIC) images at 24 h after treatment with DMSO, menadione, or H2O2. The data represent the body length distribution for the population of parasites after each treatment. Bars, 10 µm.
Figure 8.
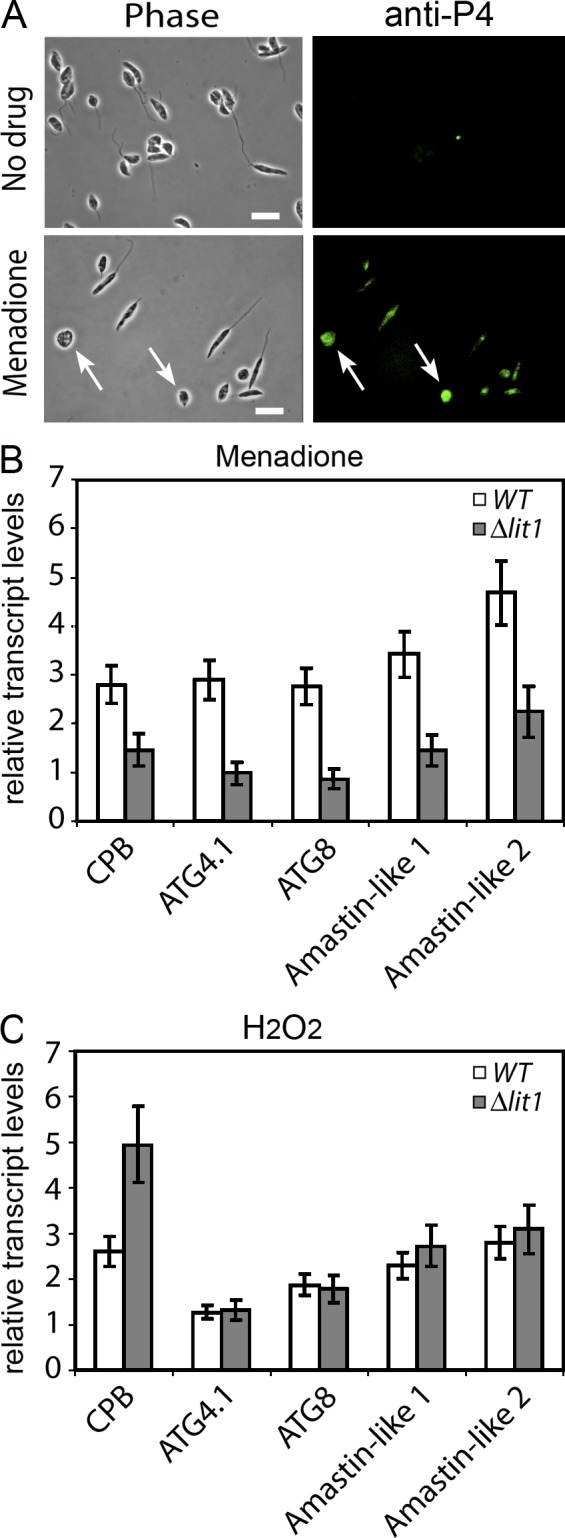
ROS-generating agents induce expression of amastigote-specific markers. (A) Immunofluorescence of WT parasites with monoclonal antibodies against the amastigote-specific marker P4 endonuclease (green) after 18 h of treatment with 5 µM menadione. The left panels show phase-contrast, and the right panels show anti-P4 immunofluorescence. The arrows point to round forms expressing the amastigote marker P4. Bars, 10 µm. (B and C) Total RNA extracted from WT or Δlit1 parasites treated with 5 µM menadione (B) or 150 µM H2O2 (C) for 18 h was analyzed by qPCR using primers specific for the coding regions of genes up-regulated during differentiation to amastigotes. The data represent the mean ± SD of triplicate determinations of three independent experiments.
These results provide strong evidence for a role of ROS in signaling events leading to L. amazonensis promastigote to amastigote differentiation. The fact that addition of H2O2 restored the ability of Δlit1 promastigotes to differentiate into amastigotes directly points to a role for this highly diffusible FeSOD product as a signaling molecule capable of initiating differentiation, in agreement with earlier observations in higher eukaryotes (Rhee, 2006; Sarsour et al., 2009; Rigoulet et al., 2011).
An iron-poor environment or exposure to ROS is sufficient to transform noninfective promastigotes into virulent amastigotes
Log phase promastigotes are insect stages not capable of replicating intracellularly in mammals, whereas amastigotes are fully infective (Sacks and Melby, 2001). Thus, we examined whether the differentiation pathway triggered by iron deprivation or treatment with ROS-generating agents affected L. amazonensis virulence. Both iron-deprived and menadione-treated parasites were able to invade and replicate within BMDMs at a significantly higher efficiency when compared with undifferentiated promastigotes grown in regular iron-containing medium (Fig. 9, A and B). The kinetics of intracellular replication of parasites cultured in iron-depleted medium or exposed to the superoxide-generating drug menadione were indistinguishable from that of amastigotes generated by exposing promastigotes to higher temperature and low pH, the “classical” axenic amastigote differentiation protocol (Bates et al., 1992; Frame et al., 2000; Sacks and Melby, 2001).
Figure 9.

Iron depletion induces differentiation of noninfective promastigotes into virulent amastigotes. The infectivity for macrophages (BMDMs) and mice was compared between WT promastigotes grown in regular medium for 4 d, WT promastigotes grown in iron-depleted medium for 4 d, mid-log phase promastigotes treated with the ROS-generating agent menadione for 18 h [(+) menadione], and axenic amastigotes induced by reduced pH/elevated temperature (axenic amastigotes). (A) Immunofluorescence images of BMDMs derived from BALB/c mice after infections with the indicated parasite populations for 1 or 48 h. Red indicates anti-Leishmania antibodies, green indicates anti-Lamp1, and blue indicates DAPI DNA stain. Bars, 5 µm. (B) BMDMs were infected for 1 h and either fixed immediately (1 h) or further incubated for 24, 48, or 72 h, and the number of intracellular parasites was determined microscopically. The data represent the mean ± SD of triplicate determinations and are representative of more than three independent experiments. (C–E) BALB/c mice were inoculated in the left hind footpad with regular, iron-depleted, or menadione-treated WT parasites. Axenic amastigotes induced by reduced pH/elevated temperature were injected as positive control. (C) Lesion development was determined by weekly caliper measurements. The data correspond to the mean ± SD of values obtained from five individual mice in each group. (D) Representative images showing the extent of lesion formation in each mouse group at 8 wk after challenge. (E) Parasite load in the footpad was determined at 8 wk after challenge. The results represent parasite loads per footpad of individual mice with geometric means and p-values between respective groups (Student’s two-tailed t test).
To determine whether the ability to replicate within BMDMs in culture correlated with a capacity to grow within tissue macrophages and to generate cutaneous lesions, we injected parasites exposed to regular, iron-depleted or menadione-containing medium into mouse footpads and followed lesion development. In parallel, elevated temperature/low pH–induced axenic amastigotes were injected into another group of mice, as a positive control. As expected (Frame et al., 2000), the mice injected with elevated temperature/low pH axenic amastigotes showed a vigorous and progressive lesion development after 2–3 wk of infection (Fig. 9, C and D). Remarkably, promastigotes induced to differentiate into amastigotes through culture in iron-poor medium or by exposure to menadione showed an almost identical pattern of lesion development (Fig. 9, C and D). This enhanced infectivity was confirmed when the parasite load in the infected footpads was quantified after 8 wk (Fig. 9 E). In contrast, mice injected with promastigotes grown in regular iron-containing medium only started to show a slight swelling of the infected footpads 7–8 wk after injection, and the tissue parasite load at 8 wk was >105-fold lower than in the other three groups. The mild and delayed lesion development observed with promastigotes grown in iron-containing medium is likely to have resulted from small numbers of infective metacyclic promastigotes present in the cultures (Sacks and Melby, 2001). The high degree of correlation between the results of our in vitro and in vivo infectivity assays demonstrates that iron uptake–dependent regulation of ROS-mediated signaling plays a central role in triggering the differentiation of noninfective L. amazonensis promastigotes into the highly virulent amastigote forms.
DISCUSSION
Iron uptake by L. amazonensis is mediated by LIT1, a ferrous iron transporter which is required for growth of the amastigotes inside host macrophages (Huynh et al., 2006). In this study we demonstrate for the first time that specifically removing iron from the culture medium induces LIT1 expression in extracellularly grown L. amazonensis promastigotes. Surprisingly, while performing these experiments we discovered that low iron in the environment is a potent trigger for the differentiation of noninfective promastigotes into infective amastigotes. Our data indicate that upon sensing low iron, L. amazonensis promastigotes activate expression of the ferrous iron transporter LIT1 and increase iron uptake, a condition necessary for triggering differentiation. The ability of L. amazonensis to express LIT1 is thus a critical determinant of the capacity of these parasites to respond to an iron-poor environment by initiating differentiation. This conclusion is supported by three lines of evidence: (1) WT promastigotes grown in iron-poor medium up-regulate LIT1 expression and show a reduced growth rate that is followed by differentiation into infective amastigotes; (2) the intracellular iron content of WT promastigotes grown in iron-poor medium drops initially, followed by an increase that reflects the elevated LIT1 expression levels; and (3) LIT1-null mutant promastigotes (Δlit1) that lack the ability to transport Fe2+ (Huynh et al., 2006) show reduced levels of intracellular iron and continue to grow exponentially after iron depletion, until the population dies off without differentiating.
Our results also indicate for the fist time that this iron-regulated differentiation process in L. amazonensis involves ROS production. First, the up-regulation of LIT1 induced by iron deprivation results in increased FeSOD activity within the parasites, immediately before the onset of promastigote to amastigote differentiation. Second, LIT1-null mutants grown in iron-depleted medium contain significantly higher levels of superoxide than WT parasites. Third, the superoxide-generating drug menadione induces differentiation of infective amastigotes in WT but not in LIT1-null mutants. Fourth, exposure to H2O2 triggers promastigote to amastigote differentiation in both WT and LIT1-null mutants. Collectively, our results suggest that increased intracellular iron availability resulting from enhanced iron transport by LIT1 is required for generation of active FeSOD and conversion of superoxide into H2O2, a molecule which is sufficient to trigger a key step in the life cycle of Leishmania, the generation of infective amastigotes.
In addition to the ferrous iron transporter LIT1, earlier work from our laboratory identified LFR1, a membrane-associated ferric reductase from L. amazonensis that converts Fe3+ (the oxidized form present in biological fluids) to Fe2+ (the reduced form that can be translocated across membranes; Flannery et al., 2011). Both LIT1 and LFR1 mRNAs are up-regulated after growth of L. amazonensis promastigotes in iron-depleted medium (Table S2). Null mutants of these genes have intracellular replication and virulence defects that are clearly linked to defective iron acquisition because the intracellular growth of both mutants is rescued by loading macrophage phagolysosomes with iron (Flannery et al., 2011). Interestingly, in addition to impaired replication within macrophages, LFR1-null promastigotes are also defective in their ability to differentiate into infective forms (Flannery et al., 2011). In light of our present findings, the LFR1-null differentiation defect suggests that although L. amazonensis may have alternative, lower affinity pathways for Fe2+ acquisition, defective Fe3+ to Fe2+ conversion severely impairs the parasites’ ability to respond to the low iron within PVs by transforming into infective amastigotes. Intracellular amastigotes have to compete with the macrophage efflux transporter Nramp1 for access to iron, and Nramp1 mutations are a known susceptibility factor for Leishmania infections (Blackwell et al., 2001; Marquis and Gros, 2007). Our present results significantly expand this scenario, by showing that the reduced iron availability within macrophage PVs acts as a trigger for differentiation of Leishmania into the mammalian-infective amastigote forms.
We found that growth in iron-poor medium results in up-regulation of FeSOD activity in L. amazonensis promastigotes, with a kinetics that coincides with the first appearance of amastigotes in the cultures. This finding suggests a role for the parasite’s intracellular redox balance in determining cell fate, a process which is becoming increasingly evident in eukaryotes (Owusu-Ansah and Banerjee, 2009; Theopold, 2009; Hamanaka and Chandel, 2010). In higher eukaryotes, mitochondrial and cytosolic SODs contain different metal cofactors, which allows for these activities to be distinguished biochemically. In contrast, all Leishmania SODs (FeSODA and FeSODB1/B2) exclusively use iron as a cofactor essential for functionality (Taylor and Kelly, 2010). This points to a mechanism by which iron availability in the environment can strongly and directly impact ROS homeostasis in the parasites. Consistent with this view, FeSOD plays an important role in the virulence and intracellular survival of L. donovani, L. tropica, and L. chagasi (Ghosh et al., 2003; Plewes et al., 2003). FeSODA corresponds to the mitochondrial isoform (Getachew and Gedamu, 2007), whereas FeSODB1/B2, which is equivalent to the cytosolic enzyme from higher eukaryotes, appears to be localized in glycosomes (Plewes et al., 2003). FeSODB is apparently essential for promastigote survival, as indicated by several failed attempts to generate double knockouts (Ghosh et al., 2003; Plewes et al., 2003). Based on our results, we hypothesize that the expression of LIT1 induced in an iron-poor environment stimulates iron uptake and stimulates FeSOD activity, which in turn controls the parasite’s redox balance and regulates signaling pathways leading to differentiation.
Intracellular iron deficiency has been associated with increased ROS generation and a highly oxidative cellular state (Nagababu et al., 2008), suggesting that the same may be occurring within iron-deprived L. amazonensis before LIT1 expression. Although dedicated cellular ROS producers such as NADPH oxidases also participate in signaling pathways, the tightly regulated ROS production that occurs in mitochondria plays an important role in the maintenance of cellular oxidative homeostasis (Hamanaka and Chandel, 2009, 2010). Inefficiencies in the electron transport chain are thought to be the main source of mitochondrial ROS production in a process that can be triggered by stress conditions such as increased respiratory rate, changes in mitochondrial inner membrane potential, or damage to respiratory chain components. A damaged electron transport chain “leaks” electrons, which generate superoxide in the presence of molecular oxygen. In this context, the markedly elevated levels of superoxide radical that we observed in Δlit1 promastigotes are likely to result from at least two factors related to intracellular iron deprivation: reduced levels of FeSOD activity and stress-related enhancement of superoxide generation in mitochondria. Conversely, the ability to import iron and to assemble active FeSOD correlates well with the low superoxide levels observed in WT parasites and their ability to survive and differentiate in response to iron deprivation. The sudden death of Δlit1 L. amazonensis promastigotes in iron-poor medium is likely to result from a combination of superoxide radical accumulation and a dysfunctional respiratory chain because intracellular iron deprivation leads to loss of Fe/S cluster proteins essential for electron transport (Levi and Rovida, 2009).
ROS are largely viewed as deleterious for eukaryotic cells (Balaban et al., 2005; Wallace, 2005). However, recent studies revealed that superoxide and H2O2 are also important regulators of cell proliferation and differentiation in higher eukaryotes (Dröge, 2002; Finkel, 2003; Owusu-Ansah and Banerjee, 2009; Hamanaka and Chandel, 2010). It is becoming increasingly clear that the relative concentrations of O2•− and H2O2 can determine cell fate by regulating the switch between cell proliferation and differentiation (Buetler et al., 2004; Rigoulet et al., 2011). Our study shows that this mechanism can also determine cell fate in protozoan parasites. Importantly, recent results implicated H2O2 in signaling leading to macroautophagy (Azad et al., 2009; Wang et al., 2010), a process which our current study and others (Besteiro et al., 2006; Williams et al., 2006, 2009, 2012) have linked to the promastigote to amastigote transition. We detected up-regulation of the autophagy-related genes ATG8, ATG4.1, and CPB soon after shifting L. amazonensis promastigotes to iron-deficient medium, indicating that up-regulation of the autophagy machinery is an early step in the cellular remodeling process that generates amastigotes in response to iron deprivation. Also in agreement with our results, higher levels of intracellular H2O2 and increased virulence were observed in L. major promastigotes lacking the APX LmAPX (Pal et al., 2010).
A reduced replication rate of L. chagasi promastigotes in low iron media was reported in a previous study, which explored different iron sources for their ability to support parasite growth. Similar to what we observed with L. amazonensis, the reduced growth rate of L. chagasi could be restored to normal levels by complementing the culture medium with Fe-NTA (Wilson et al., 1994). However, these authors did not report any morphological changes during culture in iron-depleted medium or after iron add-back, so it remains to be determined whether L. amazonensis and L. chagasi, which are New World agents of cutaneous/mucocutaneous or visceral leishmaniasis, respectively, show similar responses to iron availability. Interestingly, L. chagasi promastigotes treated with sublethal doses of the superoxide-generating drug menadione also became more virulent (Wilson et al., 1994), although that study did not link this observation to iron homeostasis or to amastigote differentiation.
The iron-dependent amastigote differentiation process that we described here occurs while parasites are maintained at the low temperature (26°C) and neutral pH characteristic of the insect vector midgut. This demonstrates that iron availability is a strong signal that overrides the classical elevated temperature/low pH triggers for differentiation in Leishmania (Zilberstein and Shapira, 1994). Intriguingly, there is evidence that elevated temperature and low pH can also trigger ROS generation in Leishmania (Wilson et al., 1994; Alzate et al., 2006, 2007; Riemann et al., 2011). The extensive information available on transcriptome and proteome changes during promastigote to amastigote differentiation in L. donovani (Saxena et al., 2007; Rosenzweig et al., 2008a,b; Lahav et al., 2011) should facilitate future studies to determine whether common pathways are triggered downstream of the iron and temperature/pH stimuli. Preliminary comparison of SL RNA-seq data from both systems already points to many similarities, and a few differences, between the pathways.
In the vast majority of earlier studies, the enhanced antioxidant pathways observed in infective stages of Leishmania have been interpreted as a defense mechanism against the oxidative burst produced by host macrophages (Ghosh et al., 2003; Plewes et al., 2003; Getachew and Gedamu, 2007). However, SOD-generated H2O2 is also known to enhance protein tyrosine phosphorylation by inactivating tyrosine phosphatases that are particularly susceptible to oxidation as the result of low pKa cysteines in their active sites (Rhee, 2006; Brandes et al., 2009). In this context, it is noteworthy that elFeα dephosphorylation delays promastigote to amastigote differentiation in L. infantum (Chow et al., 2011). By revealing a functional link between iron uptake, increased FeSOD activity and redox balance in Leishmania parasites, our work provides insight into the mechanism by which ROS can act as signaling molecules responsible for the generation of virulent life cycle stages.
MATERIALS AND METHODS
Parasites.
L. amazonensis IFLA/BR/67/PH8 WT or LIT1-null mutant (Δlit1::NEO/Δlit1::HYG; Huynh et al., 2006) promastigotes were maintained in vitro at 26°C in M199 growth media containing 40 mM Hepes, pH 7.4, and supplemented with 10% heat-inactivated FBS, 0.1% hemin (25 mg/ml in 50% triethanolamine; Frontier Scientific), 0.1 mM adenine, pH 7.5, 0.0001% vol/vol biotin, 5 mM l-glutamine, and 100 U/ml penicillin/0.1 mg/ml streptomycin. Axenic amastigotes were generated by mixing late-log phase promastigote cultures (∼2 × 106/ml) with equal volumes of acidic amastigote media (M199 supplemented with 0.25% glucose, 0.5% trypticase, and 40 mM Na succinate, pH 4.5) and elevating the temperature to 32°C and maintained in amastigote media at 32°C.
Iron depletion and supplementation.
Iron-depleted FBS was prepared by treating 100 ml FBS with 10 mM ascorbic acid for 6–7 h at 37°C, until the OD 405 was reduced from 1.2–1.4 to 0.6–0.8. 5 g/100 ml Chelex was then added, followed by stirring at 50 rpm for 3–4 h, filtration through Whatman paper to remove the Chelex, and dialysis (cut-off 2,000 D) against 4 liters of cold sterile PBS with three buffer changes every 6 h. The iron-depleted FBS was then filtered (0.22-µm filter pore size), aliquoted, and kept at −20°C until use.
Iron-depleted media was prepared similarly to regular M199 growth media except that hemin was omitted and iron-depleted FBS replaced the regular FBS. The media was stirred with 5 g/100 ml Chelex for 1 h at room temperature (RT) and filtered through a 0.22-µm filter. Chelex-treated and regular media samples were analyzed for Li, B, Na, Mg, P, K, Ca, Mn, Fe, Co, Ni, Cu, Zn, As, Se, Mo, and Cd ion content using an Elan DRCe ICP-MS instrument (PerkinElmer). The metal ions Fe, Ca, Cu, Mn, Mg, and Zn were present at 11.3, 2,140, 1,120, 0.08, 0.98, and 7.42 µM, respectively, in the regular media before treatment and at 3.4, 1,930, 690, 0.03, 0.87, and 1.04 µM, respectively, after Chelex treatment. The other elements measured by ICP-MS showed no significant changes after the Chelex treatment. Using these values, all metal ions except iron were added back at the original concentrations to generate the iron-depleted media.
Iron supplementation was performed by adding Fe-NTA at 8 µM. Fe-NTA was prepared by dissolving 1.64 g NaHCO3 in 80 ml of water, followed by additions of 2.56 g trisodium NTA and 2.7 g FeCl3.6H2O (Liu et al., 1991). The solution was brought up to 100 ml final volume and purged with nitrogen, filter sterilized, and stored in sterile anaerobic bottles.
Parasite culture in iron-depleted media.
L. amazonensis promastigotes grown to mid-log phase (1–2 × 106/ml) in regular growth media were harvested by centrifugation (930 g for 5 min at RT) and resuspended in iron-depleted media at a final concentration of 106 or 4 × 106/ml, as indicated in the experiments. Live and dead parasites were distinguished by incubating culture samples for 5 min at RT with 100 mM fluorescein diacetate (FDA) to detect living parasites and 150 µM propidium iodide to detect dead parasites. Parasites were transferred to a Bright-Line hemocytometer and viewed on an Eclipse E200 microscope with a 40× NA 0.75 objective (Nikon). Green fluorescent parasites after FDA treatment were scored as “live,” whereas red fluorescent parasites after propidium iodide were scored as “dead.” Parasite growth was determined by counting the FDA-stained cells in culture samples at 24-h intervals.
To determine the percentage of flagellated versus nonflagellated parasites in culture samples, FDA-stained parasites were visualized via phase contrast. At least 200 viable cells per sample were scored based on the criteria of possessing or not long flagella.
Intracellular iron content determination.
Total cellular iron was determined using a colorimetric ferrozine-based assay (Riemer et al., 2004). 108 L. amazonensis promastigotes were collected at various times after culture in iron-depleted medium and washed three times with 1× PBS (pretreated with Chelex). The cells were lysed with 100 µl of 50 mM NaOH followed by addition of 100 µl of 10 mM HCl. 100 µl of iron-releasing solution prepared by mixing equal volumes of 1.4 M HCl and 4.5% potassium permanganate was added to the lysates followed by incubation at 60°C for 2 h and addition of 30 µl of iron detection reagent containing 6.5 mM ferrozine, 6.5 mM neocuproine, 2.5 M ammonium acetate, and 1 M ascorbic acid dissolved in water. After 30 min of incubation at RT, 280 µl of each sample was transferred to a 96-well plate, and absorbance at 550-nm wavelength was measured. Iron contents were determined from standard curves generated using ferric chloride solutions of known molarity (0–75 µM).
SL RNA-seq library preparation.
SL RNA-seq libraries were prepared using a protocol adapted from Armour et al. (2009). First strand cDNA was synthesized from 1 µg of DNase-treated total RNA using Superscript III (Invitrogen) and primer 1 (5′-TCCGATCTCTNNNNNNN-3′). The reaction was performed by incubating for 90 min at 40°C followed by 15 min at 70°C. The samples were then treated with 2U RNase H (Invitrogen; 37°C for 20 min) to remove the RNA component of the RNA/DNA heteroduplex, and the resulting first strand DNA was purified on a QIAquick PCR purification column (QIAGEN). Second strand synthesis reaction was performed using the first strand DNA as template in a reaction mix containing a primer (5′-TCAGTTTCTGTA-3′) that matches the Leishmania SL sequence and 3′-5′ exo- Klenow Fragment (New England Biolabs, Inc.) according to the manufacturer’s protocol (incubation at 37°C for 30 min). The resulting double-stranded DNA was purified using the QIAquick PCR purification kit. Enrichment of DNA fragments with adapters was performed by PCR amplification of the purified double-stranded DNA samples using the ExpandPlus High Fidelity PCR System (Roche) and primers (5′-CAAGCAGAAGACGGCATACGAGCTCTTCCGATCTCT-3′ and 5′-AATGATACGGCGACCACCGACACTCTTTCCCTACATCAGTTTCTGTACTTTA-3′). A 2-min denaturation step at 94°C followed by two cycles (94°C for 10 s, 40°C for 2 min, 72°C for 1 min) and an additional 13–16 cycles (94°C for 10 s, 60°C for 30 s, and 72°C for 1 min) with a final 5-min, 72°C elongation step were performed. The resulting enriched fragments were purified on a QIAquick PCR purification column and represented the SL RNA library. Libraries were sequenced using the Genome Analyzer IIx (Illumina) at the High Throughput Genomics Unit at the University of Washington to generate 36-nt long single-end reads.
Mapping of high-throughput sequencing reads.
Reads containing a TTG tag at the 5′ end, indicating the presence of authentic SL sequences, were separated from non–TTG-containing reads. The first three bases (TTG) of the reads were trimmed, and the remaining sequence was aligned against the L. mexicana genome (TriTrypDB version 4.0; Aslett et al., 2010) using bowtie (Langmead et al., 2009) configured to allow roughly one to three mismatches depending on quality of base where the mismatch occurred. Alignments were processed using SAMtools (Li et al., 2009), and internally developed Perl scripts were used to calculate the number of reads aligned against each gene (including the CDS and the 5′ upstream intergenic region). edgeR software (Robinson et al., 2010) was used for differential expression (DE) analysis of count data from all four libraries. Genes that did not have one read per million (cpm, counts per million) aligned reads in all four samples were excluded from further DE analysis. The biological coefficient of variation within the biological replicates was estimated using edgeR and used in DE calculations. Based on a negative binomial error model, edgeR was used to fit the count data to a generalized linear model and to calculate DE for each gene. P-values were adjusted for multiple comparisons as described in Klipper-Aurbach et al. (1995). The SL RNA-seq data have been submitted to the GEO database under accession no. GSE41641.
Determination of gene expression by qPCR.
Approximately 108 L. amazonensis cells were harvested, washed with PBS (930 g for 5 min at RT), and used to isolate total RNA using the RNeasy kit (QIAGEN) as per the manufacturer’s protocol. An on-column DNase digestion step was performed using an RNase-Free DNase Set (QIAGEN) to ensure removal of any contaminating DNA. Synthesis of cDNA was performed using 1 µg total RNA and qScript cDNA supermix (Quanta Biosciences). To quantify levels of specific mRNA transcripts in individual samples, 2 µl of each cDNA sample was amplified with gene-specific primers (Table S1) in PerfeCTa SYBR green FastMix (Quanta Biosciences) according to the manufacturer’s protocol, using a C1000 thermocycler fitted with a CFX96 real-time system (Bio-Rad Laboratories). Three technical and three biological replicates of each reaction were performed, amplification efficiencies were validated, and expression for every test gene was normalized to the mRNAs encoding ubiquitin hydrolase, which is known to be expressed constitutively (Rochette et al., 2008; Depledge et al., 2009).
Determination of SOD activity and superoxide concentration.
L. amazonensis promastigotes (∼2 × 108) were harvested by centrifugation (930 g for 5 min at RT), washed twice with PBS, and resuspended in hypotonic buffer (5 mM Tris–HCl, pH 7.5, 0.1 mM EDTA, 5 mM phenylmethylsulfonyl fluoride, and 1× Complete Mini EDTA-free protease inhibitor cocktail [Roche]) at a final concentration of ∼5 × 108 cells/ml. The cells were flash-frozen in liquid nitrogen and stored at −80°C in a freezer until use. Frozen cells were ruptured by three freeze–thaw cycles alternating liquid nitrogen and a 37°C water bath; cell lysis was monitored microscopically. Whole cell extract supernatants were obtained after centrifugation of the lysates at 12,000 g for 30 min at 4°C, and protein contents were determined using a BCA protein assay kit (Thermo Fisher Scientific). SOD activity in whole cell extracts was measured using SOD Assay kit-WST (Dojindo Molecular Technologies, Inc.) according to the manufacturer’s protocol. The assay system utilizes the enzyme xanthine oxidase, which oxidizes its substrate xanthine to produce superoxide anions, making the water-soluble tetrazolium salt (WST), normally colorless, to turn yellow after reduction to formazan dye. Reduction of WST to formazan is inhibited by the presence of SOD activity. Inhibition of yellow formazan dye formation, after the addition of whole cell extracts to the SOD assay mix, was determined as a measure of SOD activity in cell extracts. Standard inhibition curves were generated using known concentrations of horseradish SOD (Sigma-Aldrich).
The intracellular superoxide anion concentration was determined using LumiMax Superoxide Anion Detection kit (Agilent Technologies). 108 cells from individual cultures growing in iron-depleted media were harvested, washed with PBS, and assayed per the manufacturer’s protocol.
Immunofluorescence microscopy.
Detection of LIT1 protein expression was performed on live parasites to detect surface-expressed molecules. In brief, L. amazonensis promastigotes growing in regular growth media or iron-depleted growth media were washed three times with ice-cold PBS. The cells were then incubated in blocking buffer (10% goat serum and 50 mM ammonium chloride in PBS) for 1 h on ice, followed by 1-h incubation with anti-LIT1 rabbit polyclonal antibody generated against the C-terminal peptide fragment of LIT1 (Huynh et al., 2006) in blocking buffer at 4°C. The parasites were then washed five times with PBS, incubated with Alexa Fluor 488–conjugated goat anti–rabbit antibodies (Invitrogen) for 1 h, and washed again five times with PBS before imaging.
To detect expression of the amastigote marker P4 endonuclease (Kar et al., 2000), 108 cells were harvested, washed three times with ice-cold PBS, and fixed in 2% paraformaldehyde (PFA) for 5 min on ice. Fixed parasites were permeabilized with 0.05% Triton X-100, washed with PBS, blocked with 5% goat serum in PBS for 1 h, and incubated with mouse monoclonal antibodies against P4 (courtesy of D. McMahon-Pratt, Yale University, New Haven, CT) for 1 h at RT. The cells were washed three times with PBS and incubated with Alexa Fluor 488–conjugated goat anti–mouse antibodies (Invitrogen) for 1 h at RT. The cells were then washed three times with PBS, incubated with 10 µg/ml DAPI, washed with PBS, and mounted in ProLong mounting media (Invitrogen) before imaging.
Images were acquired on an Axiovert 200 fluorescence microscope (Carl Zeiss) equipped with a charge-coupled device camera (CoolSNAP HQ; Photometrics) controlled by MetaMorph software (Molecular Devices). Image analysis was performed with the Volocity Software Suite (PerkinElmer).
Immunoblot analysis.
L. amazonensis whole cell extracts were subjected to 10% SDS-PAGE electrophoresis, followed by transfer into nitrocellulose membranes (Bio-Rad Laboratories). Immunoblots were probed with specific primary antibodies against Leishmania P4, PFR, and CPB (provided by D. McMahon-Pratt) followed by incubation with HRP-conjugated goat anti–mouse secondary antibodies (Invitrogen). Bands corresponding to parasite proteins were detected by developing blots with Immun-star WesternC kit (Bio-Rad Laboratories) and chemiluminescence scanning using a LAS-300 imaging system and LAS-300 imaging software (Fujifilm).
Treatment of L. amazonensis promastigotes with ROS-generating agents.
Mid-log phase promastigotes grown in regular media were resuspended at 4 × 106 cells/ml in regular growth media supplemented with the ROS-generating reagents, menadione (Sigma-Aldrich) or H2O2 (J.T. Baker) and incubated overnight at 26°C. Cell viability was confirmed by staining with FDA. To determine parasite length after treatment with ROS-generating agents, parasites were washed, resuspended in PBS, and imaged by differential interference contrast microscopy on an Eclipse Ti inverted microscope with a 100× NA 1.4 objective (Nikon) equipped with a C9100-50 camera (Hamamatsu Photonics). Acquired images were analyzed with the Volocity Software Suite. Cell lengths were measured along the longest body axis in a minimum of 300 cells representative of each population, and body length distributions were plotted to estimate morphological changes (Williams et al., 2006; Flannery et al., 2011).
Quantification of Leishmania intracellular growth in macrophages.
A total of 105 BMDMs prepared as previously described (Becker et al., 2009) were plated on glass coverslips in 3-cm dishes 24 h before experiments. Promastigotes grown in regular or iron-depleted media for 4 d were harvested, washed, and resuspended in PBS, and the numbers of flagellated and nonflagellated forms were microscopically estimated. Parasites from regular or iron-depleted cultures and axenically transformed amastigotes following the classical temperature/pH shift protocol (Bates et al., 1992) were then incubated with the adherent macrophages at an MOI of 2 in RPMI 10% FBS for 1 h at 34°C. After invasion, cells were washed three times in PBS and incubated for the indicated times at 34°C. Coverslips were fixed in 4% PFA after 1 (baseline infection) 24, 48, and 72 h of incubation, permeabilized with 0.1% Triton X-100 for 10 min, and stained with 10 µg/ml DAPI for 1 h. The number of intracellular parasites was quantified by scoring the total number of macrophages and the total number of intracellular parasites per microscopic field (100× NA 1.3 oil immersion objective, E200 epifluorescence microscope; Nikon), and the results were expressed as intracellular parasites per 100 macrophages. At least 300 host cells, in triplicate, were analyzed for each time point.
Infected macrophages were processed for immunofluorescence as described previously (Cortez et al., 2011). PFA-fixed cells were washed with PBS, quenched with 15 mM NH4 Cl for 15 min, and permeabilized with saponin/0.1% PBS. PV membranes were stained with rat anti–mouse Lamp1 mAb (Developmental Studies Hybridoma Bank) for 1 h, followed by 1-h incubation with anti–rabbit IgG Alexa Fluor 488. For parasite staining, coverslips were permeabilized with 0.1% Triton X-100 for 3 min and incubated with mouse polyclonal antibodies against axenic amastigotes of L. amazonensis, followed by anti–mouse IgG–Texas red. All samples were incubated with 10 µg/ml DAPI for nuclei staining.
In vivo virulence and persistence assays.
Female BALB/c mice were injected in the left hind footpad with 5.0 × 106 of L. amazonensis (Frame et al., 2000) parasites grown for 4 d in either regular or iron-depleted media. The effect of ROS on virulence was determined by treating promastigotes from regular cultures (day 4) with 5 µM menadione for 16 h before inoculation into mice. Axenic amastigotes generated through the classical temperature/pH shift protocol (Bates et al., 1992) were injected as positive controls at 2.5 × 106 per mice. Lesion progression was followed by blindly measuring left and right hind footpads every week with a caliper. The parasite tissue loads in infected footpads were determined after 8 wk using a limiting dilution assay (Tabbara et al., 2005).
Online supplemental material.
Table S1 lists primers used for qPCR reactions. Table S2 shows SL RNA-seq analysis of L. amazonensis promastigotes cultured with or without iron. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20121368/DC1.
Supplementary Material
Acknowledgments
We thank Dr. D. McMahon-Pratt for the generous gift of antibodies, D. Zilberstein, D. Sacks, D.C. Miguel, and A. Flannery for critical reading of the manuscript, and N. Beck for excellent technical assistance. We are particularly grateful to A. Flannery for advice with iron content determinations.
This work was supported by National Institutes of Health grant RO1AI067979 to N.W. Andrews.
The authors have no conflicting financial interests.
Footnotes
Abbreviations used:
- APX
- ascorbate peroxidase
- CPB
- cysteine protease B
- DE
- differential expression
- FDA
- fluorescein diacetate
- Fe-NTA
- ferric nitrolotriacetate
- FeSOD
- iron SOD
- PFA
- paraformaldehyde
- PV
- parasitophorous vacuole
- qPCR
- quantitative real-time PCR
- ROS
- reactive oxygen species
- RT
- room temperature
- SL
- splice leader
- SOD
- superoxide dismutase
References
- Alzate J.F., Alvarez-Barrientos A., González V.M., Jiménez-Ruiz A. 2006. Heat-induced programmed cell death in Leishmania infantum is reverted by Bcl-X(L) expression. Apoptosis. 11:161–171 10.1007/s10495-006-4570-z [DOI] [PubMed] [Google Scholar]
- Alzate J.F., Arias A.A., Moreno-Mateos D., Alvarez-Barrientos A., Jiménez-Ruiz A. 2007. Mitochondrial superoxide mediates heat-induced apoptotic-like death in Leishmania infantum. Mol. Biochem. Parasitol. 152:192–202 10.1016/j.molbiopara.2007.01.006 [DOI] [PubMed] [Google Scholar]
- Antoine J.C., Prina E., Lang T., Courret N. 1998. The biogenesis and properties of the parasitophorous vacuoles that harbour Leishmania in murine macrophages. Trends Microbiol. 6:392–401 10.1016/S0966-842X(98)01324-9 [DOI] [PubMed] [Google Scholar]
- Armour C.D., Castle J.C., Chen R., Babak T., Loerch P., Jackson S., Shah J.K., Dey J., Rohl C.A., Johnson J.M., Raymond C.K. 2009. Digital transcriptome profiling using selective hexamer priming for cDNA synthesis. Nat. Methods. 6:647–649 10.1038/nmeth.1360 [DOI] [PubMed] [Google Scholar]
- Aslett M., Aurrecoechea C., Berriman M., Brestelli J., Brunk B.P., Carrington M., Depledge D.P., Fischer S., Gajria B., Gao X., et al. 2010. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 38:D457–D462 10.1093/nar/gkp851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad M.B., Chen Y., Gibson S.B. 2009. Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxid. Redox Signal. 11:777–790 10.1089/ars.2008.2270 [DOI] [PubMed] [Google Scholar]
- Balaban R.S., Nemoto S., Finkel T. 2005. Mitochondria, oxidants, and aging. Cell. 120:483–495 10.1016/j.cell.2005.02.001 [DOI] [PubMed] [Google Scholar]
- Barak E., Amin-Spector S., Gerliak E., Goyard S., Holland N., Zilberstein D. 2005. Differentiation of Leishmania donovani in host-free system: analysis of signal perception and response. Mol. Biochem. Parasitol. 141:99–108 10.1016/j.molbiopara.2005.02.004 [DOI] [PubMed] [Google Scholar]
- Bates P.A. 2006. Housekeeping by Leishmania. Trends Parasitol. 22:447–448 10.1016/j.pt.2006.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates P.A., Robertson C.D., Tetley L., Coombs G.H. 1992. Axenic cultivation and characterization of Leishmania mexicana amastigote-like forms. Parasitology. 105:193–202 10.1017/S0031182000074102 [DOI] [PubMed] [Google Scholar]
- Becker S.M., Delamarre L., Mellman I., Andrews N.W. 2009. Differential role of the Ca(2+) sensor synaptotagmin VII in macrophages and dendritic cells. Immunobiology. 214:495–505 10.1016/j.imbio.2008.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besteiro S., Williams R.A., Morrison L.S., Coombs G.H., Mottram J.C. 2006. Endosome sorting and autophagy are essential for differentiation and virulence of Leishmania major. J. Biol. Chem. 281:11384–11396 10.1074/jbc.M512307200 [DOI] [PubMed] [Google Scholar]
- Blackwell J.M., Goswami T., Evans C.A., Sibthorpe D., Papo N., White J.K., Searle S., Miller E.N., Peacock C.S., Mohammed H., Ibrahim M. 2001. SLC11A1 (formerly NRAMP1) and disease resistance. Cell. Microbiol. 3:773–784 10.1046/j.1462-5822.2001.00150.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes N., Schmitt S., Jakob U. 2009. Thiol-based redox switches in eukaryotic proteins. Antioxid. Redox Signal. 11:997–1014 10.1089/ars.2008.2285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buetler T.M., Krauskopf A., Ruegg U.T. 2004. Role of superoxide as a signaling molecule. News Physiol. Sci. 19:120–123 [DOI] [PubMed] [Google Scholar]
- Chow C., Cloutier S., Dumas C., Chou M.N., Papadopoulou B. 2011. Promastigote to amastigote differentiation of Leishmania is markedly delayed in the absence of PERK eIF2alpha kinase-dependent eIF2alpha phosphorylation. Cell. Microbiol. 13:1059–1077 10.1111/j.1462-5822.2011.01602.x [DOI] [PubMed] [Google Scholar]
- Cortez M., Huynh C., Fernandes M.C., Kennedy K.A., Aderem A., Andrews N.W. 2011. Leishmania promotes its own virulence by inducing expression of the host immune inhibitory ligand CD200. Cell Host Microbe. 9:463–471 10.1016/j.chom.2011.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debrabant A., Joshi M.B., Pimenta P.F., Dwyer D.M. 2004. Generation of Leishmania donovani axenic amastigotes: their growth and biological characteristics. Int. J. Parasitol. 34:205–217 10.1016/j.ijpara.2003.10.011 [DOI] [PubMed] [Google Scholar]
- Depledge D.P., Evans K.J., Ivens A.C., Aziz N., Maroof A., Kaye P.M., Smith D.F. 2009. Comparative expression profiling of Leishmania: modulation in gene expression between species and in different host genetic backgrounds. PLoS Negl. Trop. Dis. 3:e476 10.1371/journal.pntd.0000476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey R., Meneses C., Salotra P., Kamhawi S., Nakhasi H.L., Duncan R. 2010. Characterization of a Leishmania stage-specific mitochondrial membrane protein that enhances the activity of cytochrome c oxidase and its role in virulence. Mol. Microbiol. 77:399–414 10.1111/j.1365-2958.2010.07214.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dröge W. 2002. Free radicals in the physiological control of cell function. Physiol. Rev. 82:47–95 [DOI] [PubMed] [Google Scholar]
- Duboise S.M., Vannier-Santos M.A., Costa-Pinto D., Rivas L., Pan A.A., Traub-Cseko Y., De Souza W., McMahon-Pratt D. 1994. The biosynthesis, processing, and immunolocalization of Leishmania pifanoi amastigote cysteine proteinases. Mol. Biochem. Parasitol. 68:119–132 10.1016/0166-6851(94)00157-X [DOI] [PubMed] [Google Scholar]
- Finkel T. 2003. Oxidant signals and oxidative stress. Curr. Opin. Cell Biol. 15:247–254 10.1016/S0955-0674(03)00002-4 [DOI] [PubMed] [Google Scholar]
- Flannery A.R., Huynh C., Mittra B., Mortara R.A., Andrews N.W. 2011. LFR1 ferric iron reductase of Leishmania amazonensis is essential for the generation of infective parasite forms. J. Biol. Chem. 286:23266–23279 10.1074/jbc.M111.229674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame M.J., Mottram J.C., Coombs G.H. 2000. Analysis of the roles of cysteine proteinases of Leishmania mexicana in the host-parasite interaction. Parasitology. 121:367–377 10.1017/S0031182099006435 [DOI] [PubMed] [Google Scholar]
- Getachew F., Gedamu L. 2007. Leishmania donovani iron superoxide dismutase A is targeted to the mitochondria by its N-terminal positively charged amino acids. Mol. Biochem. Parasitol. 154:62–69 10.1016/j.molbiopara.2007.04.007 [DOI] [PubMed] [Google Scholar]
- Ghosh S., Goswami S., Adhya S. 2003. Role of superoxide dismutase in survival of Leishmania within the macrophage. Biochem. J. 369:447–452 10.1042/BJ20021684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossage S.M., Rogers M.E., Bates P.A. 2003. Two separate growth phases during the development of Leishmania in sand flies: implications for understanding the life cycle. Int. J. Parasitol. 33:1027–1034 10.1016/S0020-7519(03)00142-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta N., Goyal N., Rastogi A.K. 2001. In vitro cultivation and characterization of axenic amastigotes of Leishmania. Trends Parasitol. 17:150–153 10.1016/S1471-4922(00)01811-0 [DOI] [PubMed] [Google Scholar]
- Hamanaka R.B., Chandel N.S. 2009. Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr. Opin. Cell Biol. 21:894–899 10.1016/j.ceb.2009.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanaka R.B., Chandel N.S. 2010. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 35:505–513 10.1016/j.tibs.2010.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer T.R., McMaster W.R., Forney J.D. 2006. Expression profiling by whole-genome interspecies microarray hybridization reveals differential gene expression in procyclic promastigotes, lesion-derived amastigotes, and axenic amastigotes in Leishmania mexicana. Mol. Biochem. Parasitol. 146:198–218 10.1016/j.molbiopara.2005.12.009 [DOI] [PubMed] [Google Scholar]
- Huynh C., Sacks D.L., Andrews N.W. 2006. A Leishmania amazonensis ZIP family iron transporter is essential for parasite replication within macrophage phagolysosomes. J. Exp. Med. 203:2363–2375 10.1084/jem.20060559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar S., Soong L., Colmenares M., Goldsmith-Pestana K., McMahon-Pratt D. 2000. The immunologically protective P-4 antigen of Leishmania amastigotes. A developmentally regulated single strand-specific nuclease associated with the endoplasmic reticulum. J. Biol. Chem. 275:37789–37797 10.1074/jbc.M002149200 [DOI] [PubMed] [Google Scholar]
- Kinnula V.L., Crapo J.D. 2004. Superoxide dismutases in malignant cells and human tumors. Free Radic. Biol. Med. 36:718–744 10.1016/j.freeradbiomed.2003.12.010 [DOI] [PubMed] [Google Scholar]
- Klipper-Aurbach Y., Wasserman M., Braunspiegel-Weintrob N., Borstein D., Peleg S., Assa S., Karp M., Benjamini Y., Hochberg Y., Laron Z. 1995. Mathematical formulae for the prediction of the residual beta cell function during the first two years of disease in children and adolescents with insulin-dependent diabetes mellitus. Med. Hypotheses. 45:486–490 10.1016/0306-9877(95)90228-7 [DOI] [PubMed] [Google Scholar]
- Lahav T., Sivam D., Volpin H., Ronen M., Tsigankov P., Green A., Holland N., Kuzyk M., Borchers C., Zilberstein D., Myler P.J. 2011. Multiple levels of gene regulation mediate differentiation of the intracellular pathogen Leishmania. FASEB J. 25:515–525 10.1096/fj.10-157529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Trapnell C., Pop M., Salzberg S.L. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi S., Rovida E. 2009. The role of iron in mitochondrial function. Biochim. Biophys. Acta. 1790:629–636 10.1016/j.bbagen.2008.09.008 [DOI] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25:2078–2079 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M., Okada S., Kawabata T. 1991. Radical-promoting “free” iron level in the serum of rats treated with ferric nitrilotriacetate: comparison with other iron chelate complexes. Acta Med. Okayama. 45:401–408 [DOI] [PubMed] [Google Scholar]
- Marquis J.F., Gros P. 2007. Intracellular Leishmania: your iron or mine? Trends Microbiol. 15:93–95 10.1016/j.tim.2007.01.001 [DOI] [PubMed] [Google Scholar]
- Nagababu E., Gulyani S., Earley C.J., Cutler R.G., Mattson M.P., Rifkind J.M. 2008. Iron-deficiency anaemia enhances red blood cell oxidative stress. Free Radic. Res. 42:824–829 10.1080/10715760802459879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owusu-Ansah E., Banerjee U. 2009. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature. 461:537–541 10.1038/nature08313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S., Dolai S., Yadav R.K., Adak S. 2010. Ascorbate peroxidase from Leishmania major controls the virulence of infective stage of promastigotes by regulating oxidative stress. PLoS ONE. 5:e11271 10.1371/journal.pone.0011271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plewes K.A., Barr S.D., Gedamu L. 2003. Iron superoxide dismutases targeted to the glycosomes of Leishmania chagasi are important for survival. Infect. Immun. 71:5910–5920 10.1128/IAI.71.10.5910-5920.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee S.G. 2006. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 312:1882–1883 10.1126/science.1130481 [DOI] [PubMed] [Google Scholar]
- Riemann A., Schneider B., Ihling A., Nowak M., Sauvant C., Thews O., Gekle M. 2011. Acidic environment leads to ROS-induced MAPK signaling in cancer cells. PLoS ONE. 6:e22445 10.1371/journal.pone.0022445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemer J., Hoepken H.H., Czerwinska H., Robinson S.R., Dringen R. 2004. Colorimetric ferrozine-based assay for the quantitation of iron in cultured cells. Anal. Biochem. 331:370–375 10.1016/j.ab.2004.03.049 [DOI] [PubMed] [Google Scholar]
- Rigoulet M., Yoboue E.D., Devin A. 2011. Mitochondrial ROS generation and its regulation: mechanisms involved in H(2)O(2) signaling. Antioxid. Redox Signal. 14:459–468 10.1089/ars.2010.3363 [DOI] [PubMed] [Google Scholar]
- Robinson M.D., McCarthy D.J., Smyth G.K. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 26:139–140 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochette A., Raymond F., Ubeda J.M., Smith M., Messier N., Boisvert S., Rigault P., Corbeil J., Ouellette M., Papadopoulou B. 2008. Genome-wide gene expression profiling analysis of Leishmania major and Leishmania infantum developmental stages reveals substantial differences between the two species. BMC Genomics. 9:255 10.1186/1471-2164-9-255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig D., Smith D., Myler P.J., Olafson R.W., Zilberstein D. 2008a. Post-translational modification of cellular proteins during Leishmania donovani differentiation. Proteomics. 8:1843–1850 10.1002/pmic.200701043 [DOI] [PubMed] [Google Scholar]
- Rosenzweig D., Smith D., Opperdoes F., Stern S., Olafson R.W., Zilberstein D. 2008b. Retooling Leishmania metabolism: from sand fly gut to human macrophage. FASEB J. 22:590–602 10.1096/fj.07-9254com [DOI] [PubMed] [Google Scholar]
- Saar Y., Ransford A., Waldman E., Mazareb S., Amin-Spector S., Plumblee J., Turco S.J., Zilberstein D. 1998. Characterization of developmentally-regulated activities in axenic amastigotes of Leishmania donovani. Mol. Biochem. Parasitol. 95:9–20 10.1016/S0166-6851(98)00062-0 [DOI] [PubMed] [Google Scholar]
- Sacks D., Kamhawi S. 2001. Molecular aspects of parasite-vector and vector-host interactions in leishmaniasis. Annu. Rev. Microbiol. 55:453–483 10.1146/annurev.micro.55.1.453 [DOI] [PubMed] [Google Scholar]
- Sacks D.L., Melby P.C. 2001. Animal models for the analysis of immune responses to leishmaniasis. Curr. Protoc. Immunol. Chapter 19:19–: 2. 10.1002/0471142735.im1902s28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacks D.L., Perkins P.V. 1984. Identification of an infective stage of Leishmania promastigotes. Science. 223:1417–1419 10.1126/science.6701528 [DOI] [PubMed] [Google Scholar]
- Sarsour E.H., Kumar M.G., Chaudhuri L., Kalen A.L., Goswami P.C. 2009. Redox control of the cell cycle in health and disease. Antioxid. Redox Signal. 11:2985–3011 10.1089/ars.2009.2513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena A., Lahav T., Holland N., Aggarwal G., Anupama A., Huang Y., Volpin H., Myler P.J., Zilberstein D. 2007. Analysis of the Leishmania donovani transcriptome reveals an ordered progression of transient and permanent changes in gene expression during differentiation. Mol. Biochem. Parasitol. 152:53–65 10.1016/j.molbiopara.2006.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabbara K.S., Peters N.C., Afrin F., Mendez S., Bertholet S., Belkaid Y., Sacks D.L. 2005. Conditions influencing the efficacy of vaccination with live organisms against Leishmania major infection. Infect. Immun. 73:4714–4722 10.1128/IAI.73.8.4714-4722.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor M.C., Kelly J.M. 2010. Iron metabolism in trypanosomatids, and its crucial role in infection. Parasitology. 137:899–917 10.1017/S0031182009991880 [DOI] [PubMed] [Google Scholar]
- Theopold U. 2009. Developmental biology: A bad boy comes good. Nature. 461:486–487 10.1038/461486a [DOI] [PubMed] [Google Scholar]
- Tsigankov P., Gherardini P.F., Helmer-Citterich M., Zilberstein D. 2012. What has proteomics taught us about Leishmania development? Parasitology. 139:1146–1157 10.1017/S0031182012000157 [DOI] [PubMed] [Google Scholar]
- Tsukagoshi H., Busch W., Benfey P.N. 2010. Transcriptional regulation of ROS controls transition from proliferation to differentiation in the root. Cell. 143:606–616 10.1016/j.cell.2010.10.020 [DOI] [PubMed] [Google Scholar]
- Wallace D.C. 2005. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet. 39:359–407 10.1146/annurev.genet.39.110304.095751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Singh R., Xiang Y., Czaja M.J. 2010. Macroautophagy and chaperone-mediated autophagy are required for hepatocyte resistance to oxidant stress. Hepatology. 52:266–277 10.1002/hep.23645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R.A., Tetley L., Mottram J.C., Coombs G.H. 2006. Cysteine peptidases CPA and CPB are vital for autophagy and differentiation in Leishmania mexicana. Mol. Microbiol. 61:655–674 10.1111/j.1365-2958.2006.05274.x [DOI] [PubMed] [Google Scholar]
- Williams R.A., Woods K.L., Juliano L., Mottram J.C., Coombs G.H. 2009. Characterization of unusual families of ATG8-like proteins and ATG12 in the protozoan parasite Leishmania major. Autophagy. 5:159–172 10.4161/auto.5.2.7328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R.A., Smith T.K., Cull B., Mottram J.C., Coombs G.H. 2012. ATG5 is essential for ATG8-dependent autophagy and mitochondrial homeostasis in Leishmania major. PLoS Pathog. 8:e1002695 10.1371/journal.ppat.1002695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson M.E., Andersen K.A., Britigan B.E. 1994. Response of Leishmania chagasi promastigotes to oxidant stress. Infect. Immun. 62:5133–5141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., El Fakhry Y., Sereno D., Tamar S., Papadopoulou B. 2000. A new developmentally regulated gene family in Leishmania amastigotes encoding a homolog of amastin surface proteins. Mol. Biochem. Parasitol. 110:345–357 10.1016/S0166-6851(00)00290-5 [DOI] [PubMed] [Google Scholar]
- Zilberstein D., Shapira M. 1994. The role of pH and temperature in the development of Leishmania parasites. Annu. Rev. Microbiol. 48:449–470 10.1146/annurev.mi.48.100194.002313 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.