

Abstract
Optic neuritis (ON), which is an acute inflammatory autoimmune demyelinating disease of the central nervous system (CNS), often occurs in multiple sclerosis (MS). ON is an early diagnostic sign in most MS patients caused by damage to the optic nerve leading to visual dysfunction. Various features of both MS and ON can be studied following induction of experimental autoimmune encephalomyelitis (EAE), an animal model of MS, in Lewis rats. Inflammation and cell death in the optic nerve, with subsequent damage to the retinal ganglion cells in the retina, are thought to correlate with visual dysfunction. Thus, characterizing the pathophysiological changes that lead to visual dysfunction in EAE animals may help develop novel targets for therapeutic intervention. We treated EAE animals with and without the calpain inhibitor calpeptin (CP). Our studies demonstrated that the Ca2+-activated neutral protease calpain was upregulated in the optic nerve following induction of EAE at the onset of clinical signs (OCS) of the disease and these changes were attenuated following treatment with CP. These reductions correlated with decreases in inflammation (cytokines, iNOS, COX-2, NF-κB), and microgliosis (i.e. activated microglia). We observed that calpain inhibition reduced astrogliosis (reactive astroglia) and expression of aquaporin 4 (AQP4). The balance of Th1/Th2 cytokine production and also expression of the Th1-related CCR5 and CXCR3 chemokine receptors influence many pathological processes and play both causative and protective roles in neuron damage. Our data indicated that CP suppressed cytokine imbalances. Also, Bax:Bcl-2 ratio, production of tBid, PARP-1, expression and activities of calpain and caspases, and internucleosomal DNA fragmentation were attenuated after treatment with CP. Our results demonstrated that CP decreased demyelination [loss of myelin basic protein (MBP)] and axonal damage [increase in dephosphorylated neurofilament protein (de-NFP), and also promoted intracellular neuroprotective pathways in optic nerve in EAE rats. Thus, these data suggest that calpain is involved in inflammatory as well as in neurodegenerative aspects of the disease and may be a promising target for treating ON in EAE and MS.
Keywords: apoptosis, calpain, chemokines, cytokines, EAE, inflammation, optic neuritis
Introduction
Multiple sclerosis (MS) and its animal model experimental autoimmune encephalomyelitis (EAE) are neurodegenerative diseases with characteristic inflammation and demyelination in the central nervous system (CNS) areas, including the optic nerve (Potter and Bigazzi 1992; Guyton et al. 2005a). Neuronal and axonal damage is considered to be the main cause of long-term disability in patients with MS. Optic neuritis (ON), inflammation of the optic nerve, is the first diagnosed sign in approximately 20% of patients with MS and as many as 50% of patients with ON eventually develop the disease. Electroretinogram (ERG) studies, which assess retinal function and visual-evoked potential (VEP) data that assess optic nerve abnormalities, have demonstrated that visual function is impaired in MS patients and in animals with EAE (Kornek et al. 2000; Meyer et al. 2001; Fisher et al. 2006). MS and EAE are caused by an immune attack on the CNS by auto-reactive T cells and activated macrophages. As a result, there is a significant increase in pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF-α) and interferon gamma (IFN-γ), as well as other mediators (Williams et al. 1994). However, current research suggests that MS and EAE are also neurodegenerative diseases, leading to axonal degeneration and neuronal death (Trapp et al. 1999; Guyton et al. 2009; 2010). Since pathophysiological changes that occur in EAE spinal cord also occur in EAE optic nerve; the EAE animal model is ideal for studying ON (Davie et al. 1995; Shields et al. 1998a and 1998b). In the CNS, aquaporin 4 (AQP4) expression promotes inflammation and causes demyelinating lesions. This idea is a subject of intense speculation. Recently, increased AQP4 expression was found in brain and spinal cord in EAE, providing further support for the possible involvement of AQP4 in CNS inflammation (Misu et al. 2007; Miyamoto et al. 2009). No data were available about modifications in the expression of both AQP4 and glial fibrillary acidic protein (GFAP) in the reported EAE models in the optic nerve. A recent study in a myelin oligodendrocyte glycoprotein (MOG)-induced EAE model showed the attenuation of disease progression in AQP4 knockout mice (Misu et al. 2007; Li et al. 2009). Other experimental data were obtained from the MOG-induced EAE models in nonobese diabetic (NOD)/Lt and C57BL/6 mice, known to develop a diffuse inflammatory process not limited to the opticospinal location. Furthermore, complete Freund’s adjuvant (CFA) and pertussis toxin, which are known to alter the blood-brain-barrier permeability, are largely used (Lu et al. 2008; Namer et al. 1994).
Neuropathological studies of GFAP, a specific marker of reactive astrocytes, and AQP4, particularly abundant in the optic nerves, spinal cord, and periependymal regions (Nagelhus et al. 1998; Wujek et al. 2002; Trip et al. 2005; Vitellaro-Zuccarello et al. 2005; Pittock et al. 2006; Tsoi et al. 2006; Takano et al. 2008), have demonstrated decreases in expression of GFAP and AQP4 in the optic nerve and spinal cord of neuromyelitis optica (NMO) patients prior to demyelination (Misu et al. 2007; Roemer et al. 2007). This pattern of reactivity seems different from that encountered in MS where loss of AQP4 has been found mainly in strongly demyelinated regions in intensive and acute MS lesions, whereas overexpression of AQP4 has mostly been observed in other chronic demyelinated tissue due to astrogliosis. The inflammatory nature of ON implicates the participation of immunoregulatory cytokines, including the T-helper type 1 (Th1) cell-associated IFN-γ, the Th2 cell-related interleukin-4 (IL-4), and the immune response-downregulating cytokine transforming growth factor-beta (TGF-β), but proof for their involvement in ON has been lacking (Guan et al. 2006). Moreover, patients with ON had elevated percentages of CXCR3 and CCR5 expressing T cells compared with patients with other non-inflammatory neurological diseases (OND). Greater chemokine receptor expression may be one prerequisite for Th1 cells to migrate to the CNS.
Studies in our laboratory have demonstrated that calpain expression is upregulated in splenic cells isolated from EAE animals before onset of clinical signs of disease (Schaecher et al. 2002; Guyton et al. 2005b; Das et al. 2008a). Calpain degrades myelin proteins into immunogenic fragments that can potentially activate myelin-specific autoreactive T cells. Calpain also degrades many cytoskeletal substrates, including myelin basic protein (MBP), neurofilament protein (NFP), and spectrin, which can lead to cellular damage. In addition, calpain cleaves many signaling proteins that are involved in the regulation of apoptosis (Ray and Banik 2003). Also, studies in our laboratory have demonstrated increased calpain activity in immune cells and astrocytes in the optic nerves of animals with EAE (Guyton et al. 2005b; Shindler et al. 2006; Shields et al. 1998b); however, we have not previously examined if calpain is involved in cell death in EAE optic nerve before the onset of clinical signs. Since calpain is involved in myelin degradation in MS and since optic nerve damage arises before appearance of EAE signs in animal models of EAE, we have hypothesized that increase in calpain activity, as a result of increased Ca2+ influx, may correlate with cell death in EAE optic nerve, thus resulting in impaired visual function. Our studies demonstrated that calpain expression and cell death were increased in the optic nerve at the onset of clinical symptoms of disease and persisted during the acute EAE attack. These findings suggest that early treatment with calpain inhibitors may be of therapeutic value in preserving retinal function in EAE. ON manifests as an acute, self-limited episode of optic nerve inflammation with decrease in vision that recovers over several weeks in the majority of patients (Beck et al. 1992; Berkelaar et al. 1994; Bettelli et al. 2003; Shao et al. 2004; Arnold et al. 2005). However, some level of permanent vision loss occurred in approximately 40% of patients in the ON Treatment Trial (Beck et al. 1992), and subsequent studies have shown that retinal nerve fiber layer thinning, which is used as a surrogate marker for retinal ganglion cell (RGC) axonal loss, correlates with vision loss after an episode of ON. Inhibition of chemokine receptor expression may constitute a potentially important therapeutic effect.
In the current study, we examined the timing and extent of several parameters (e.g., cell death, inflammation, anti-apoptotic factors, cytokines, chemokines) in optic nerve damage during acute ON in EAE rats, and we also examined whether cell death occurred by an apoptotic mechanism. Our current results suggest that inflammatory cells infiltrating into the optic nerve mediate the mechanisms of apoptotic cell death and CP attenuates these deleterious effects.
Materials and methods
Induction of EAE and isolation of optic nerve
Adult male Lewis rats (180 to 200g) were purchased from Charles River Breeding Laboratories (Wilmington, MA), provided water and food ad libitum, and maintained and used in the proposed experiments in accordance with the Laboratory Welfare Act and the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (Bethesda, MD). To induce EAE, rats were immunized subcutaneously with a 0.2 ml emulsion (1:1) of CFA containing Mycobacterium tuberculosis H37Ra (10 mg/ml; Difco) and phosphate-buffered saline (PBS) containing guinea pig spinal cord (GPSC) homogenate (200 mg/ml) and MBP (280 μg/ml). Control animals received PBS/CFA without GPSC/MBP. Two hours later all rats received an intraperitoneal (i.p.) injection of pertussis toxin (2 μg/rat). After induction of EAE, the rats were monitored daily for weight loss and clinical signs of paralysis. The clinical scores for severity of EAE symptoms were based on the following grades: 0-no change, 1-limp tail, 2-hind-limb weakness with difficulty righting, 3-one-limb plegic, 4-two limb plegic, 5-quadriplegic or moribund. Animals were sacrificed under anesthesia (95 mg/kg ketamine, 5 mg/kg xylazine) on day 10-11 post-EAE inductions. Optic nerves were removed and cut into two sections for time course studies (onset EAE, EAE Grade 2 and EAE Grade 3-4). One segment was snap frozen for ELISpot and DNA fragmentation studies, and the other segment was snap frozen for Western blotting. In subsequent studies, animals were treated twice daily with the calpain inhibitor (calpeptin: CP) and clinical scores were monitored or were sacrificed on day 10 post-EAE induction, and optic nerve tissues were collected. The effects of calpain inhibition on clinical signs of disease were assessed in an acute EAE model by treating control (CON) and EAE animals with i.p. injections of either vehicle (1.0% DMSO in saline) or CP (50–250 μg/kg) twice daily during days 1–9 post-EAE induction (Guyton et al. 2010; Smith et al. 2011). These doses of CP were chosen based on efficacious effects achieved at this dose range in other neurological disorders (Guyton et al. 2010).
Analysis of DNA fragmentation
Genomic DNA fragmentation was analyzed by agarose gel electrophoresis of genomic DNA isolated from optic nerve tissues, as we reported previously (Das et al. 2008b). Briefly, optic nerve segments were homogenized in a buffer (10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 50 mM EDTA) using a battery-operated homogenizer (Kontes Instruments, Vineland, NJ). The homogenates were digested in a solution (10 mM Tris-HCl, pH 8.0, 50 mM NaCl, 10 mM EDTA, 0.5% SDS, 250 ng/ml proteinase K) at 37°C for 24 hr. The digests were extracted twice with a 1:1 (v/v) mixture of phenol and chloroform and once with chloroform alone. Total genomic DNA was precipitated, dried in air, and dissolved in TE (10 mM Tris-HCl, pH 8.0, 1 mM EDTA) buffer containing RNase A (50 ng/ml) for 1 hr at 37°C. Equal amounts of the DNA samples were loaded onto 1.6% agarose gels and electrophoresed in a TAE (40 mM Tris-acetate, pH 8.3, 1 mM EDTA) buffer. Gels were stained with ethidium bromide (1 μg/ml), destained the background in water, and photographed on a UV (303 nm) transilluminator using Alpha Innotech (San Leandro, CA).
Antibodies
Monoclonal antibody against β-actin (Sigma) was used to standardize cytosolic protein loading on SDS-PAGE gels. All primary IgG antibodies were purchased from Santa Cruz Biotech (Santa Cruz, CA) or Calbiochem (Gibbstown, NJ). Secondary antibodies were horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (ICN Biomedicals, Aurora, OH,) and HRP-conjugated goat anti-rabbit IgG (ICN Biomedicals, Solon, OH, USA).
Detection of Ca2+ influx
Optic nerve tissues were dissected and set in agarose, as we previously described (Wingrave et al. 2003). The tissue block was then oriented in a container with rat serum buffer (RSB) and sectioned (200 μm) with a vibratome section slicer (EMS 4000). Slices were stained in RSB containing 1 μM calcium green-2AM (Molecular Probes, Eugene, OR) for 10 min. Sections were washed twice for 10 min each with RSB and then placed on a slide for viewing with a confocal microscope (Bio-Rad, Hercules, CA). Because calcium green-2AM will not fluoresce until it is inside the cell, observation of emission intensity allows for assessment of intracellular Ca2+ levels.
Western blotting
Western blot analysis was performed by standard procedures, as we described previously (Das et al. 2006, 2008b). The films were scanned using Photoshop software (Adobe Systems, Seattle, WA) and the optical density (OD) of each band was determined using Quantity One software (Bio-Rad, Hercules, CA).
ELISpot cytokine and chemokines array assay
Cytokines and chemokines present in the optic nerves were analyzed using an ELISpot Cytokine Array kit according to the manufacturer’s instructions (R&D Biosystems, Minneapolis, MN). Each nitrocellulose membrane provided in the kit contained a marker for 29 different cytokines and chemokines. The membranes were first blocked for 1 hr at room temperature with 2 ml of blocking buffer. Then, the homogenate (0.1 ml) was mixed with 0.5 ml of the provided array buffer and 15 μl of detection antibody cocktail and left to incubate at room temperature for another 1 hr. The cytokine-chemokines/detection antibody cocktail was then poured over another nitrocellulose membrane and allowed to incubate on a rocking platform overnight at 4°C. Each membrane was then washed three times for 10 min with 20 ml of wash buffer. About 1.5 ml of streptavidin-HRP was then added to each membrane and allowed to incubate for 30 min at room temperature. The membranes were again washed three times for 10 min each with 20 ml of wash buffer. Membranes were then drained and exposed to 2 ml each of Chemiglow West Substrate (Alpha Innotech, San Leandro, CA) for 3 min. The membranes were scanned using Photoshop software (Adobe Systems, Seattle, WA).
Statistical analysis
Data from Western blot analysis for detection of significant differences between two groups were analyzed by one-way analysis of variance (ANOVA) followed by the Fisher’s protected least significant difference (PLSD) post-hoc tests using Statview software (Abacus Concepts, Berkeley, CA). Results were expressed as mean ± standard error of mean (SEM) of independent experiments (n ≥ 3). The null hypothesis was rejected at p < 0.05.
Results
CP therapy attenuated calpain expression and activity in optic nerve in EAE rats
The production of pro-inflammatory mediators by the infiltrating immune cells and resident glial cells can lead to increases in intracellular free Ca2+ levels that cause calpain activation. Here, we demonstrated that Ca2+ levels were increased after onset of EAE, when control and EAE optic nerve sections were subjected to calcium green-2AM staining, which would fluoresce only in the presence of intracellular free Ca2+. The Ca2+ levels were greatly increased in cells and processes in EAE sections compared with controls (Figure 1A). These findings suggested that Ca2+ influx during acute EAE in optic nerve could be responsible for calpain activation. Our previous results showed that CP reduced clinical signs of the disease in acute EAE animals (Guyton et al. 2010). Lewis rats with acute EAE were i.p. injected twice daily with CP (50-250 μg/kg) or vehicle at the onset of EAE, and during progression of EAE (Grade 2 and Grade 3-4) and monitored for clinical signs of disease until day of sacrifice (n = 8 to 12 rats per group). Clinical scores for the EAE-vehicle group significantly increased (p = 0.05) over EAE-CP before onset and CP at onset. Both treatment groups showed significantly improved clinical scores compared with vehicle-treated EAE rats, and the difference between CP-treated groups was minimal. Since calpain expression and activity are upregulated in optic nerve from EAE-induced ON Lewis rats (Shields et al. 1998b), in the present study, we have also examined the time-dependent increase in calpain expression and activity to determine if this upregulation coincides with pathophysiological changes in the optic nerve. Optic nerve tissues were collected for biochemical analysis from animals at the onset of EAE, and during progression of EAE (Grade 2 and Grade 3-4). Western blot analysis was performed using antibody against intact m-calpain and 76 kD active calpain to determine calpain content and activitation (Figure 1B). Anti-calpastatin and anti-spectrin antibodies, which recognized the 110 kD calpastatin and 145 kD calpain-cleaved spectrin degradation product (SBDP), respectively, were used to determine their levels. Our data indicated that calpain expression was increased in optic nerve tissue that developed clinical signs at the disease onset, as measured in terms of m-calpain/active calpain/145 kD SBDP with respect to its endogenous inhibitor calpastatin (Figure 1C). CP treatments significantly attenuated both calpain expression and activity. Calpastatin was degraded and thus decreased in EAE and its level was recovered following treatment with CP.
Figure 1.
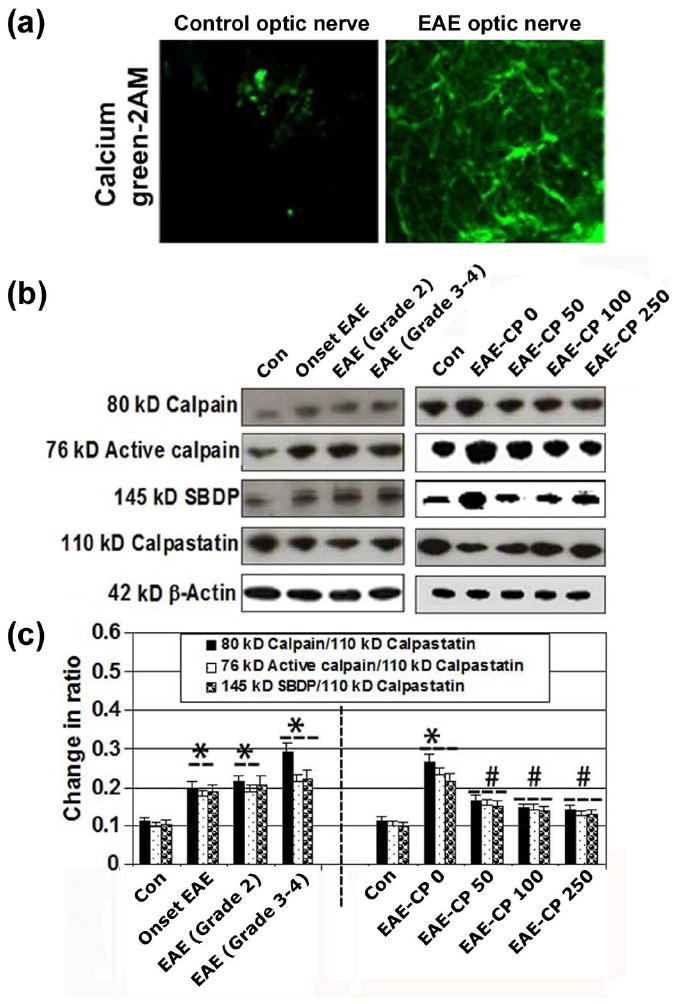
CP therapy attenuated calpain expression and activity in optic nerve in EAE rats. (A) Increase in intracellular Ca2+ level was examined in acute EAE optic nerve (at onset of EAE) in Lewis rats by staining the slices with Calcium green-2AM. Slices were examined under the laser scanning confocal microscopy at 400x magnification. (B) Western blotting for examination of levels of expression of 80 kD and 76 kD calpain, 145 kD SBDP, and 110 kD calpastatin in optic nerve of EAE rats. (C) Determination of calpain:calpastatin, active calpain:calpastatin and SBDP:calpastatin ratios (n ≥ 3 per group, *p ≤ 0.05 compared with untreated control (Con); #p ≤ 0.05 compared with EAE-CP0).
CP therapy attenuated inflammation and microgliosis in optic nerve in EAE rats
Studies have demonstrated that the optic nerve is attacked before the onset of clinical signs in EAE animals, indicating that pathophysiological changes begin early in optic nerve in the EAE rats. Calpain expression and activity in spinal cord tissues increase with onset of clinical signs of EAE (Schaecher, et al. 2002; Guyton, et al. 2005b). Control animals that received either vehicle or CP (250 μg/kg) therapy did not show clinical signs of EAE, but EAE rats that did not receive CP (EAE-0) showed classic signs of EAE, including tail limpness and hindlimb paralysis, with a cumulative clinical score of 4.5 (Guyton et al. 2010). Nuclear factor-kappa B (NF-κB) is a transcription factor that plays an important role in the inflammatory process and it has become a candidate target for new anti-inflammatory therapy. Thus, we have investigated the changes in levels of NF-κB and the expression of inflammatory molecules such as cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) in EAE rats, and we also examined the effect of calpain inhibitor on these parameters (Figure 2). Our Western blot analysis demonstrated that NF-κB was significantly upregulated in the optic nerve of EAE tissues before onset, at onset, and during the progression of EAE at the onset of EAE, and during progression of EAE (Grade 2 and Grade 3-4) (Figure 2A). The effects of CP on expression of signaling proteins involved in pro-inflammatory events were also examined. CP resulted in a significant decrease in the expression of NF-κB from 91.7 ± 12.0% in EAE CP-0 group of rats to around 30% in EAE CP 50-250 group of rats (Figure 2B). The levels of COX-2 expression were decreased by 50% in EAE rats that received CP. Further, the levels of expression of iNOS, the enzyme involved in nitric oxide production, were also significantly decreased to 60% in optic nerve tissues as a result of CP treatment. In order to examine the effects of CP therapy on microgliosis (i.e. microglial activation) during acute EAE, optic nerve sections were stained with antibody against CD11b, which identified microgliosis (Figure 2C). Treatment of EAE animals with vehicle alone exhibited notable increases in microgliosis, compared with control animals treated with either vehicle or CP (50-250 μg/kg). In contrast, intensty of staining for CD11b in optic nerve tissues from Lewis rats with acute EAE were reduced in the EAE-250 group.
Figure 2.
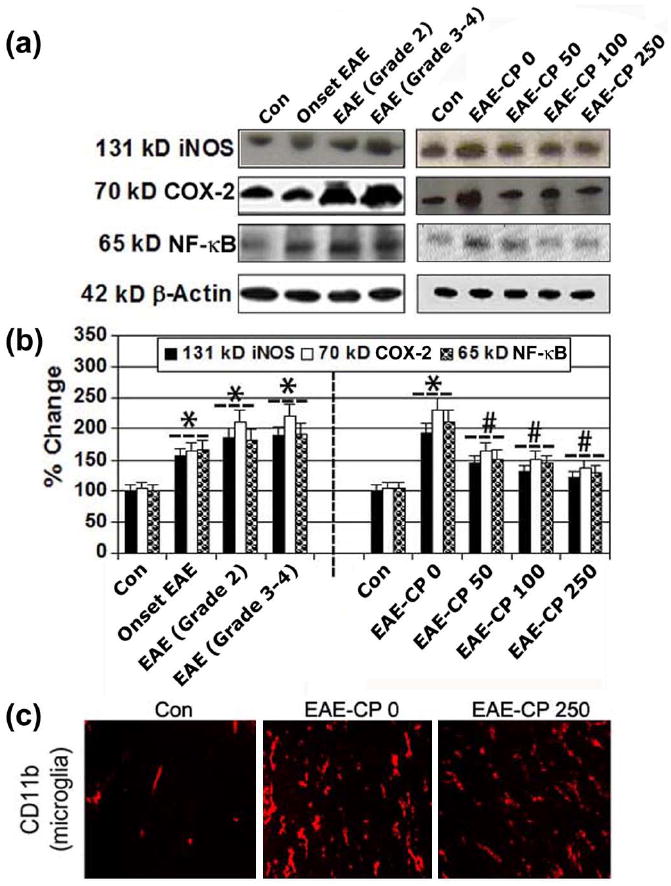
CP therapy attenuated inflammation and microgliosis in optic nerve in EAE rats. Representative (A) Western blots and corresponding (B) bar diagrams (densitometry) showing 131 kD iNOS, 70 kD COX-2, and 65 kD NF-kB in optic nerve of EAE rats (n = 3 per group, *p ≤ 0.05 compared with untreated control; #p ≤ 0.05 compared with EAE-CP0). (C) Microgliosis was examined in EAE-CP and compared with control (Con) EAE optic nerve sections via immunohistochemical staining with CD11b (red) antibody (n ≥ 3, 200x magnification).
CP therapy attenuated expression of GFAP and AQP4 in optic nerve in EAE rats
Increasing attention has recently been focused on the astrocyte markers, GFAP and AQP4, in ON. Because increased levels of expression GFAP and AQP4 show the early involvement of astrocytes before demyelination in the optic nerve. The overexpression of AQP4 was particularly pronounced in the optic nerve and was concomitant with demyelination, astrocyte activation, and apoptosis. We have previously demonstrated reactive gliosis in the optic nerves in EAE animals (Sheild et al. 1998b). In the present study, we examined levels of expression of GFAP and AQP4 in the animals after the treatments (Figure 3). Reactive Astroglia was confirmed by determining the levels of GFAP, which was increased in the EAE optic nerve compared with control animals (Figure 3A, 3B). Reactive astroglia was significantly attenuated following treatment with CP. We investigated the involvement of AQP4 in disease severity in EAE animals (Figure 3A, 3B). In the optic nerve, levels of expression GFAP and AQP4 were increased at the onset of EAE, and during progression of EAE (Grade 2 and Grade 3-4) with visible inflammation (Figure 3B). During the disease stage, AQP4 levels continued to increase significantly with GFAP and remained high until the late stage of the disease. Additionally, CP at all three doses significantly attenuated the levels of expression of GFAP and AQP4.
Figure 3.
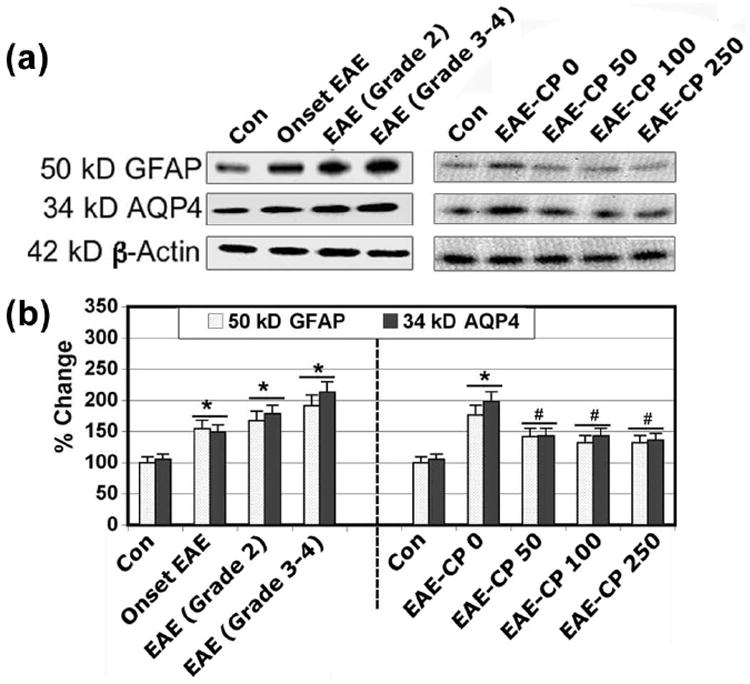
CP therapy attenuated the levels of expression of GFAP and AQP4. Representative (A) Western blots and corresponding (B) bar diagrams (densitometry) showing levels of expression of 50 kD GFAP and 34 kD AQP4 in optic nerve of EAE rats (n ≥ 3 per group, *p ≤ 0.05 compared with untreated control (Con); #p ≤ 0.05 compared with EAE-CP0).
CP modulated the production of cytokines and chemokines in optic nerve in EAE rats
Cytokines and chemokines play critical roles in the development of the inflammatory process in EAE. While Th1 cells, which produce IL-1α/β, IL-2, IL-ra, IL-3, IFN-γ, and TNF-α, have been implicated in the pathological processes, on the other hand, Th2 cells that secrete IL-4, IL-6, and IL-10 are involved in down regulation of those in diseases. However, the relative contributions of cytokines (Th1 or Th2) and chemokines to the pathogenic process remain to be elucidated. We investigated whether CP treatment affected the production of cytokines and chemokines (Figure 4). Our results from ELISpot (Figure 4A) showed the increases in expression of IL-1α/β, IL-2, IL-ra, IL-3, IFN-γ, and TNF-α (Figure 4B), decreases in expression of IL-4, IL-6, and IL-10 (Figure 4C), and increases in expression of chemokines (CXCL-10, CXCL-9, CCL-3, and CCL-5) (Figure 4D) in the optic nerve at onset of EAE. CP treatment at all three doses significantly attenuated the production of inflammatory cytokines and chemokines (Figure 4B-4D). Thus, our data demonstrated that CP treatments upregulate Th2 types of cytokines in EAE rats. Increased Th2 cytokines following CP treatment correlated well with improved clinical scores.
Figure 4.
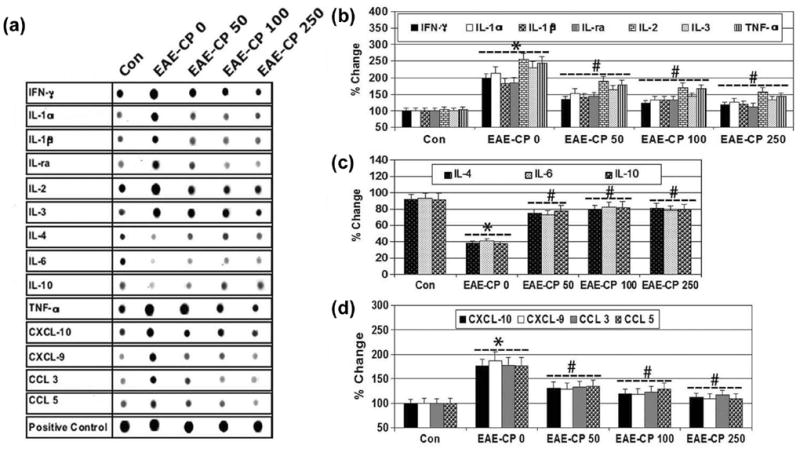
CP modulated the production of cytokines and chemokines in optic nerve in EAE rats. (A) Cytokines and chemokines in in optic nerve of EAE rats, determined using an ELISpot Cytokine Array kit. Bar diagrams for levels of the pro-iflammatory cytokines (A): IL-1a/b, IL-2, IL-ra, IL-3, IFN-γ, and TNF-a; anti-iflammatory cytokines (B): IL-4, IL-6, and IL-10; and chemokines (D): CXCL-10, CXCL-9, CCL-3, CCL-9 expression (n ≥ 3 per group, *p ≤ 0.05 compared with untreated control (Con); #p ≤ 0.05 compared with EAE-CP0).
Calpain inhibition decreased apoptosis in optic nerve in EAE rats
Understanding the timing and mechanisms of optic nerve damage may be important for developing potential neuroprotective therapies to prevent permanent vision loss from ON. Thus, we examined the timing and the effects of calpain inhibition on proteins involved in receptor mediated (e.g., caspase-8, tBid) and mitochondria mediated (e.g., increased Bax:Bcl-2 ratio) pathways of apoptosis in optic nerves in EAE animals (Figure 5). Western blot analysis revelaed a significant increase in Bax:Bcl-2 ratio, 18 kD active capase-8 and 15 kD tBid in optic nerve at the onset of EAE, and during progression of EAE (Grade 2 and Grade 3-4) when compared with control animals (Figure 5A-5C). In contrast, calpain inhibition at all three doses significantly reduced the Bax:Bcl-2 ratio, caspase-8 activation, and formation of tBid when compared with those levels in untreated EAE animals (Figure 5A-5C). The timing and effects of calpain and caspase-3 inhibition were also assessed via Western blotting. The expression of 12 kD caspase-3 active fragment was significantly decreased following treatment with 50-250 μg/kg CP (Figure 5A, 5C). Capase-3 activity, as assessed in the production of 120 kD SBDP and formation of the 85 kD PARP-1 fragments, was significantly increased in EAE-0 optic nerve compared with control animals (Figure 5C). CP at all three doses significantly attenuated the production of 120 kD SBDP in EAE animals when compared with those levels in control animals (Figure 5C). These results indicated that CP treatment significantly reduced apoptotic death in EAE optic nerve. We also correlated molecular changes due to apoptosis with internucleosomal DNA fragmentation during the development of acute EAE (Figure 5D). Unlike the control animals, substantial DNA laddering was found in EAE-ON before onset and after onset of EAE. Also, there was dramatic decrease in DNA laddering in EAE-ON from CP treated animals (Figure 5D), compared with the control animals. These studies showed a substantial increase in optic nerve death during progression of EAE. These data also indicated that cell death in ON occurred during the late phase of disease progression. Thus, our results demonstrated decrease in apoptosis due to CP treatment in EAE-ON, indicating that CP treatment protected cells in the optic nerve.
Figure 5.
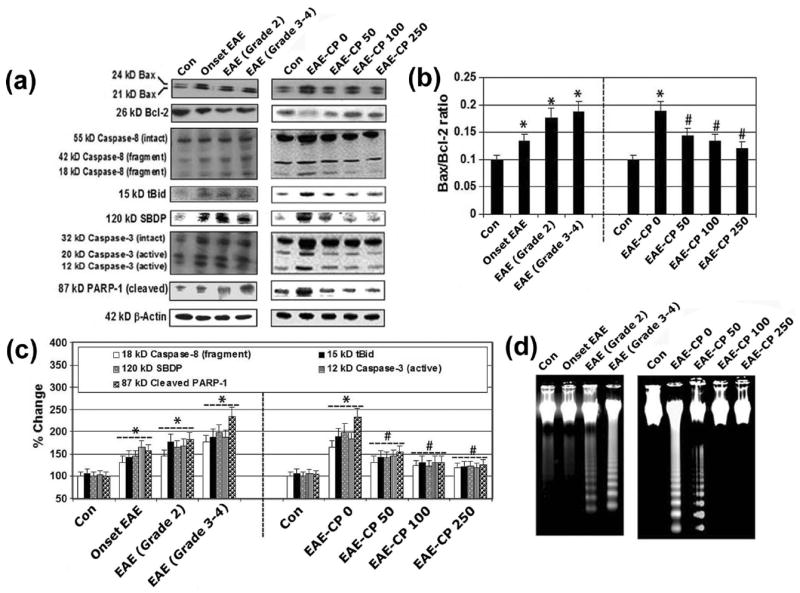
Calpain inhibition decreased apoptosis in optic nerve in EAE Rats. Representative (A) Western blots and corresponding (B) Bax:Bcl-2 ratio; (C) bar diagrams (densitometry) showing levels of expression of 24 and 21 kD Bax, 26 kD Bcl-2, 18 kD caspase-8, 15 kD tBid, 120 kD SBDP, 12 kD caspase-3, and 87 kD PARP-1 in optic nerve of EAE rats (n ≥ 3 per group, *p ≤ 0.05 compared with untreated control (Con); #p ≤ 0.05 compared with EAE-CP0). (D) Agarose gel electrophoresis of genomic DNA samples for detection of internucleosomal DNA fragmentation.
CP therapy attenuated MBP degradation and axonal damage in optic nerve in EAE rats
Demyelination and axonal degeneration are hallmarks of MS and EAE pathology and can lead to severe disability. Therefore, the effects of calpain inhibition on myelin and axonal damage were assessed in the optic nerve of EAE rats using Western blot analysis of MBP (a major component of myelin) and de-NFP (a marker of axonal degeneration) in optic nerve tissues (Figure 6). In comparison with the CON-0 group, MBP expression was decreased in EAE-0 animals (Figure 6A, 6B). MBP degradation was significantly attenuated in the EAE-CP-50 to 250 groups compared with the EAE-0 group (Figure 6B). The effect of calpain inhibition on axonal damage was assessed by decrease in de-NFP levels. The optic nerve tissue sections revealed an increase in de-NFP in the EAE-0 group compared with the control group (Figure 6B). In contrast, de-NFP in the EAE-CP-50 to 250 groups was similar to those seen in the control group.
Figure 6.
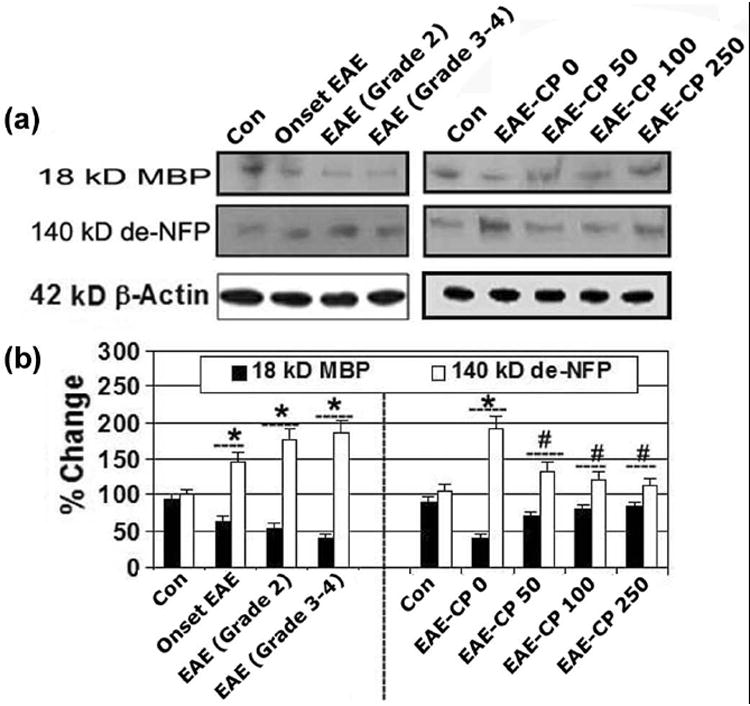
CP therapy attenuated MBP degradation and axonal damage. Representative (A) Western blots and corresponding (B) bar diagrams (densitometry) showing levels of expression of 18 kD MBP and 140 kD de-NFP in optic nerve of EAE rats (n ≥ 3 per group, *p ≤ 0.05 compared with untreated control (Con); #p ≤ 0.05 compared with EAE-CP0).
Calpain inhibition promoted intracellular neuroprotective pathways in optic nerve in EAE rats
In order to examine the involvement of intracellular signal transduction cascades in cell death mechanism, we used Western blotting to analyze phospho-Akt (p-Akt), phospho-MAPK (p-MAPK), p-Bcl-2, and p-STAT3 in CP treated and untreated EAE animals (Figure 7). Our results demonstrated a significant decrease in p-Akt, p-Bad, p44/42-MAPK, p-Bcl-2 and p-STAT3 in optic nerve tissue at the onset of EAE, and during progression of EAE (Grade 2 and Grade 3-4) when compared with control animals (Figure 7A, 7B). Additionally, CP at all three doses significantly attenuated the production of p-Akt, p-Bad, p44/42-MAPK, p-Bcl-2 and p-STAT3 in EAE animals to the levels seen in control animals (Figure 7A, 7B). Calcineurin is a Ca2+/calmodulin-dependent protein phosphatase, which is highly expressed in the CNS and is cleaved by calpain. Upon activation by calpain, calcineurin dephosphorylates and activates the pro-apoptotic protein Bad. The analysis of its expression in optic nerve of CP treated or untreated EAE demonstrated that compared with the control, a significant increase (p < 0.05) in the 60 kD calcineurin subunit and a decrease in phospho-Bad (p-Bad) in optic nerve tissue during progression of EAE (Figure 7C, 7D). Thus, this finding suggested that upregulation of calcineurin dephosphorylated Bad for apoptosis during acute EAE. CP significantly attenuated a role for calcineurin in activating Bad, thereby blocking calcineurin to prevent the apoptosis machinery in EAE.
Figure 7.
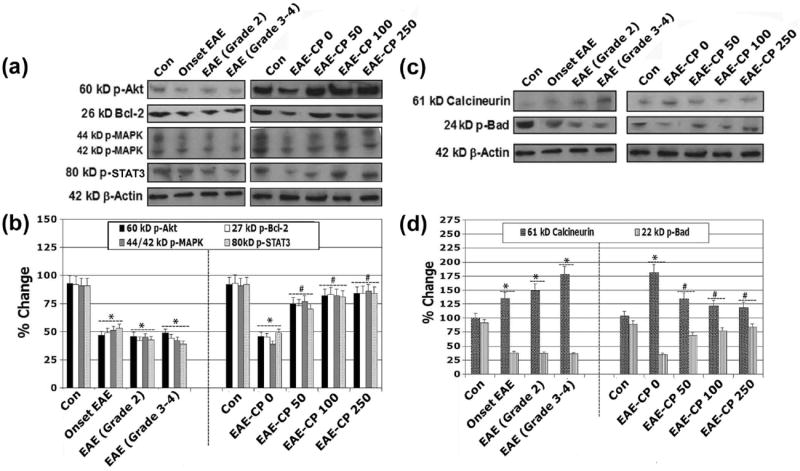
Calpain inhibition promoted intracellular neuroprotective pathways. Representative (A) Western blots and corresponding (B) bar diagrams (densitometry) showing levels of expression of 60 kD p-Akt, 26 kD Bcl-2, 44/42 kD p-MAPK, and 80 kD p-STAT-3 in optic nerve of EAE rats. Representative (C) Western blots and the corresponding (D) bar graphs (densitometry) showing levels of expression of 61 kD calcineurin and 24 kD p-Bad (n ≥ 3 per group, *p ≤ 0.05 compared with untreated control (Con); #p ≤ 0.05 compared with EAE-CP0).
Discussion
The most important findings from our current investigation indicated that calpain played an undeniable role in the pathogenesis of ON in EAE rats and CP treatment significantly attenuated both inflammatory factors and molecular alterations for prevention of apoptosis in optic nerve in EAE animals (Scheme 1). ON is one of the first presenting clinical signs in many MS patients. While not all ON patients develop MS, at least 50% of MS patients will experience visual dysfunction. Therefore, understanding the timing and mechanisms of cell death associated with the visual problem is an utmost important factor for developing potential therapies to prevent permanent vision loss from ON. Reactive oxygen species-mediated cell damage has been demonstrated in EAE optic nerves as early as 3 to 6 days after immunization (Qi et al. 2007), suggesting that the onset of ON may vary. Previous studies have demonstrated that assaults on the optic nerve occur before the appearance of clinical signs of disease in rodents with EAE (Shields et al. 1998a; Guyton et al. 2005a). The purpose of the current study was to determine if upregulation of calpain activity was a prime pathophysiological event in ON. Our study has established that calpain activity is upregulated in optic nerve during the development of acute EAE with increased axonal damage and increased infiltration of T cells and macrophages. These findings are in agreement with our previous investigations carried out in acute EAE spinal cord and retina (Schaecher et al. 2002; Guyton et al. 2005b; Smith, et al. 2011). Our previous studies prompted us to investigate now the efficacy of calpain inhibition using CP for attenuation of optic nerve damage before or after onset of clinical signs. Administration (i.p.) of CP (50-250 μg/kg) twice daily in EAE Lewis rats significantly attenuated cell death, calpain and caspase activities, inflammation, apoptotic factors, cytokines, and chemokines when compared with untreated EAE animals. Also, increases in MBP degradation and axonal damage were significantly decreased in EAE animals after CP treatment.
Scheme 1.
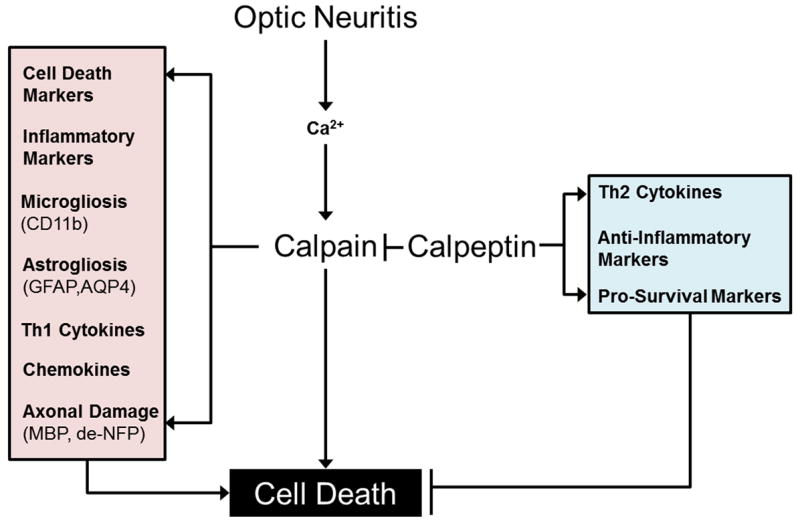
A schematic presentation of changes in factors pathways due to calpain upregulation and calpeptin (CP) treatment in EAE induced optic neuritis (ON). EAE results in increased Ca2+ in the optic nerve, which in turn triggers activation of calpain. Increased calpain is associated with elevation of cell death markers, inflammatory mediators, and Th1 cytokine and chemokine elevations, which are in turn implicated in activated microglia, reactive astroglia, axonal damage, and ultimately cell death. CP therapy attenuates these changes and results in converse increases in Th2 cytokines, anti-inflammatory mediators, and pro-survival markers that prevent cell death.
Increases in calpain expression and activity were demonstrated in splenic cells before the onset of the disease (Shields et al. 1998a and 1998b), and similar increases were also evident in infiltrating immune cells and glial cells of the spinal cord and optic nerve (Shields and Banik 1998a) even after the appearance of clinical signs. The current study confirmed our earlier findings (Guyton et al. 2005b; Schaecher et al. 2002) of increased calpain expression and activity in EAE optic nerve in a time-dependent manner. Interestingly, these increases are seen in the optic nerve of animals before they are seen in spinal cord and before apperance of clinical signs. The optic nerve degeneration and visual dysfunction occurring in EAE prior to disease onset are important determinants to MS, where the optic nerve is affected first. The increased calpain expression and activity correlated well with a decrease in expression of calpastatin, the endogenous inhibitor of calpain. This finding also suggests that calpain is involved in the neurodegenerative process in EAE and perhaps in MS.
Astrogliosis observed in this study was demonstrated by increased expression of GFAP. Also, astrocytes selectively express AQP4, a channel important for transport of both water and solutes across the plasma membrane (Das et al. 2012). Normal AQP4 activity is critical for maintenance of both cell volume and ionic gradients in the CNS. The astrocytic water channel AQP4 plays a major role in the physiopathology of ON. While animal models of EAE showing optic nerve demyelination have been developed, the involvement of astrocytes and AQP4 in these models is poorly documented. This is one of the important aspects of our current investigation in ON in EAE rats. During the disease stage, we observed that levels of expression of both GFAP and AQP4 were significantly increased in the demyelinated optic nerves. The enhanced expression of AQP4 in optic nerve astrocytes following elevation of inflammation may explain the astrocytic hypertrophy that is normally seen in MS patients. CP therapy is capable of attenuating the expression of both GFAP and AQP4 in optic nerve in EAE rats.
NF-κB is an important transcription factor that regulates both innate and adaptive immune responses. Deregulated activation of NF-κB or its regulatory kinases is associated with chronic inflammation, autoimmunity, and cancers (Bernes and Karin 1997). Our long-range goals of current investigation were to dissect the signaling pathway mediating NF-κB activation, elucidate the role of NF-κB in regulating inflammatory response, and examine expression of COX-2, iNOS, and anti-apoptotic proteins in EAE rats. Improper upregulation of COX-2 and/or iNOS has been associated with pathophysiology of inflammatory disorders (Stolina et al. 2000). Our results demonstrated a possible mechanistic target of calpain inhibition for down regulating the inflammatory responses for prevention of apoptosis in optic nerve in EAE animals. Calpain inhibition by CP is associated with the inhibition of synthesis and release of pro-inflammatory mediators and inhibition of activated immune cells and COX-2, which are responsible for the synthesis of pro-inflammatory mediators. The varying and multiple protective characteristics of CP may clinically also hold a respectable position for it as a better alternative than other anti-inflammatory drugs. These data provide evidence that CP exhibits potent anti-inflammatory activity and explain the underlying mechanism of protection of optic nerve in ON. Recent studies have indicated that inflammation plays important roles in neurodegenerative diseases (Das et al. 2008b). In EAE, factors released from damaged neurons induce reactive gliosis, especially microgliosis, which is the hallmark of inflammation (Ponomarev, et al. 2007). Reactive microgliosis may enhance the neurotoxicity by producing excessive pro-inflammatory factors, including cytotoxic superoxide, NO, and pro-inflammatory cytokines (Das et al. 2008b). Our current data demonstrated that calpain inhibition attenuated microgliosis and microglia-dependent inflammatory process that caused the progressive and self-propelling nature of EAE.
The mechanistic study of ON pathogenesis is currently seeking to understand the effects of immune responses on cell damage and protection. Cytokines are the factors that mediate most of the biological effects in both the immune and non-immune systems (Kennedy and Karpus 1999). CD4-expressing T helper (Th) cells are a major source of cytokine production and regulation. Th type 1 (Th1, inflammatory effect) cells are characterized by the production of pro-inflammatory cytokines such as IL-1α/β/ra, IL-2, IL-3, IFN-γ, and TNF-α while Th type 2 (Th2, anti-inflammatory effect) cells are characterized by the production of IL-4, IL-6, and IL-10. The balance of Th1/Th2 cytokine production influences many pathological processes and plays both destructive and protective roles in a disease process. Growing evidence indicates that imbalances of Th1/Th2 cytokine production are involved in neural damage or protection in many neurological diseases. In the current investigation, we analyzed the possible roles of Th1/Th2 cytokine production and imbalance of Th1/Th2 cytokines in optic nerve in EAE rats. Our results demonstrated a significant increase in Th1 cytokines and decrease in Th2 cytokines in acute EAE. In contrast, treatment with the calpain inhibitor CP reversed this change so that Th1 associated cytokines were decreased while Th2 associated cytokines were increased. These results suggest that calpain is involved in Th1/Th2 deregulation in EAE and CP has potential as a therapeutic in ameliorating this deregulation. Additionally, it is possible that CP therapy reduces peripheral inflammatory activation via systemic immunomodulatory effects and thus attenuation or prevents the localized inflammatory cascades mediated degenerative parameters.
A role for chemokines as mediators of Th1 cell recruitment to the CNS in MS has previously been suggested by several groups (Samson, et al. 1996; Qin et al. 1998; Quinones et al. 2008). Therefore, we studied expression of Th1 related CCR5 and CXCR3 chemokine receptors in EAE animals. The chemokine receptor CXCR3 is a Gαi protein-coupled receptor in the CXC chemokines receptor family (Clark-Lewis et al. 2003). CXCR3 binds to the CXC chemokines CXCL9 (MIG), and CXCL-10 (IP-10) and particularly the CXCR3/CXCL10 system has been implicated in inflammatory process in the CNS. This receptor is expressed on astrocytes throughout human brain and has been speculated to be upregulated in MS lesions. The CCR5 chemokine ligands bind to this receptor are RANTES (a chemotactic cytokine protein also known as CCL5) and macrophage inflammatory protein (MIP) 1α and 1β (also known as CCL3 and CCL4). CCR5 is predominantly expressed on T cells, macrophages, dendritic cells and microglia. It is likely that CCR5 plays an essential role in inflammatory responses, though its exact role in normal immune function remains unclear. We observed that optic nerve of EAE-ON rats had elevated levels of CCR5 and CXCR3 expressing cells. Such elevations disappeared in EAE rats due to treatment with the calpain inhibitor.
Since mitochondria play a key role both in maintaining cellular homeostasis and in triggering the activation of cell death pathways in EAE-ON, we evaluated the effect of timing of CP treatment in EAE rats on expression of pro-apoptotic Bax and anti-apoptotic Bcl-2. Our results showed that CP could protect cells from apoptosis by decreasing the Bax:Bcl-2 ratio. Ischemic and hypoxic injuries to the nervous system have been previously shown to involve the release of cell death-inducing cytokines and the activation of death receptors. It is likely that these events involve caspase-8 mediated cleavage of the BH3-only protein Bid to tBid. This pathway provides evidence that activation of the receptor mediated pathway of apoptosis. Once Bid is cleaved by caspase-8, tBid translocates from the cytosol to the mitochondria to cause mitochondrial damage, release of cytochrome c, increase in caspase activity, nuclear condensation, and apoptotic cell shrinkage (Das et al. 2008a and 2008b; Li et al. 2011). In addition, tBid is able to activate Bax to form Bax multimers in the mitochondria, thereby inducing the release of cytochrome c. In the present study, our results showed that caspase-8 activation correlated well with Bid cleavage to tBid in acute EAE, again demonstrating induction of cell death signals early in disease process. Our results also suggested that CP treatments (50-250 μg/kg) attenuated the processing of caspase-8 (degradation of pro-caspase-8 and appearance of active caspase-8) and Bid cleavage to tBid. Similar mechanisms of cell death may also exist in spinal cord and retina in EAE (Smith et al. 2011). Calpain interacts and modifies caspase pathways as well. Specifically, calpain has been shown to activate caspase-3 (Das et al. 2006 and 2008b). Caspase also has been shown to cleave calpastatin, indicating that caspase activity may maintain calpain as an active proteolytic enzyme, to promote apoptosis. Activation of caspase-3, the final executioner in apoptosis, which degrades the cellular target protein PARP-1, is elevated early in EAE. These results in particular indicate a pivotal co-operative role of calpain and caspase system, generating crucial cell death signals early in disease development. Calpain inhibition following treatment with CP has been found to inhibit caspase-3 and decrease apoptotic indicators, including pro-apoptotic proteins and internucleosomal DNA fragmentation. We observed increase in DNA laddering (the hallmark of apoptosis) following induction of EAE (Das et al. 2008a), indicating occurrence of apoptosis before onset (days 6 to 9) of EAE. This also suggested that the apoptotic process in optic nerve was initiated early in the inflammatory phase in EAE development, even before the appearance of clinical signs. In contrast, treatment of EAE animals with CP at all three doses that we used significantly attenuated DNA fragmentation. This observation is supported by earlier studies demonstrating the role of calpain in other neurodegenerative disorders, including ischemia, spinal cord injury, brain trauma, Alzheimer’s disease, Parkinson’s disease, and epilepsy (Ray and Banik 2003). Our results also suggest that calpain may be a potential therapeutic target for the amelioration of neurodegenerative process in MS.
Axonal damage is a key determinant in neurological disability in MS patients (Costello et al. 2006). Calpain, which is present in axon, degrades myelin and axonal cytoskeletal proteins including NFP (Li and Banik, 1995; Schaecher et al. 2002). The degeneration of axons and damage to cells were attenuated by calpain inhibitor treatment suggesting that preservation of axon-myelin structure and cell could reduce the disease severity. Calpain inhibitor treatment prevented degeneration and protected optic nerve, indicating the importance of calpain inhibition as a promising therapeutic option (Das et al. 2006 and 2008b; Guyton et al. 2009 and 2010). In the development and progression of diseases, calpain activation can induce apoptotic death through multiple mechanisms (Maier et al. 2004; Sattler et al. 2004; Das et al. 2006; Maier et al. 2007; Mckernan et al. 2007; Das et al. 2008b; Malemud and Miller 2008; Sharma et al. 2011). The finding that redistribution of Bad and suppression of apoptosis by an inhibitory mutant of calcineurin suggests that calcineurin is a significant mediator of apoptotic death signals (Li et al. 2011). Thus, the possibility that Bad activation by calcineurin activity might be involved in cell death in EAE-ON was explored. Our results suggested that calcineurin induced apoptosis by dephosphorylating Bad in EAE-ON, which was attenuated after CP treatment indicating that CP protected the optic nerve largely by interfering with the participation of calcineurin in the apoptotic process. In order to determine the intracellular signal transduction cascade, which could be involved in the optic nerve survival promoting effect of CP, we investigated whether CP treatment would promote optic nerve cell survival via intracellular signal trasduction cascades. The current study revealed the down regulations of p-Akt, p-Bcl-2, p44/42-MAPK, p-STAT-3, and p-Bad in EAE-ON. We analyzed whether CP could antagonize these pro-apoptotic mechanisms involved in optic nerve damage. Our results showed significant increases in the protein levels of p-Akt, p-Bcl-2, p44/42-MAPK, p-STAT-3, and p-Bad in EAE-ON due to CP treatment, as shown with schematic presentation also (Figure 8). The amounts of the inactive, dephosphorylated proteins were comparable in all protein lysates, indicating that not the expression level, but the degree of Akt phosphorylation could promote growth factor-mediated cell survival both directly and indirectly (data not shown). Bad is a pro-apoptotic protein of the Bcl-2 family. Akt can phosphorylate Bad on Ser136, which makes Bad dissociate it from the Bcl-2/Bcl-X complex and lose the pro-apoptotic function. STAT-3 is activated by phosphorylation at Tyr705, which induces dimerization, nuclear translocation, and DNA binding (Sharma et al. 2011). Our results demonstrated that CP protected optic nerve by potentiating the Akt/Bcl-2/MAPK/STAT-3/Bad signaling pathways (Figure 8).
In conclusion, our current study directly and significantly demonstrated that calpain inhibtion could prevent inflammation, apoptosis, and axonal degenaration in optic nerve in EAE rats (Scheme 1). Our investigation also implies that calpain inhibition may also block auto-reactive T cell-mediated inflammatory and neurodegenerative events in EAE and possibly in MS patients as well.
Acknowledgments
Completion of our current investigation was made possible by fundings in part from the National Institute of Neurological Disorders and Stroke (NINDS: NS-31622, NS-38146, NS-64556 and NS-41088), the State of South Carolina Spinal Cord Injury Research Fund, the South Carolina Clinical and Translational Research Institute, and Medical University of South Carolina’s CTSA (NIH/NCATS Grant Numbers TL1TR000061 and UL1TR000062).
Footnotes
Disclosure
None of the authors has any conflict of interest to disclose. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent.
References
- Arnold AC. Evolving management of optic neuritis and multiple sclerosis. Am J Ophthalmol. 2005;139:1101–1108. doi: 10.1016/j.ajo.2005.01.031. [DOI] [PubMed] [Google Scholar]
- Beck RW, Cleary PA, Anderson MM, Jr, Keltner JL, Shults WT, Kaufman DI, Buckley EG, Corbett JJ, Kupersmith MJ, Miller NR. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. N Engl J Med. 1992;326:581–588. doi: 10.1056/NEJM199202273260901. [DOI] [PubMed] [Google Scholar]
- Berkelaar M, Clarke DB, Wang YC, Bray GM, Aguayo AJ. Axotomy results in delayed death and apoptosis of retinal ganglion cells in adult rats. J Neurosci. 1994;14:4368–4374. doi: 10.1523/JNEUROSCI.14-07-04368.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med. 2003;197:1073–1081. doi: 10.1084/jem.20021603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- Clark-Lewis I, Mattioli I, Gong JH, Loetscher P. Structure-function relationship between the human chemokine receptor CXCR3 and its ligands. J Biol Chem. 2003;278:289–295. doi: 10.1074/jbc.M209470200. [DOI] [PubMed] [Google Scholar]
- Costello F, Coupland S, Hodge W, Lorello GR, Koroluk J, Pan YI, Freedman MS, Zackon DH, Kardon RH. Quantifying axonal loss after optic neuritis with optical coherence tomography. Ann Neurol. 2006;59:963–969. doi: 10.1002/ana.20851. [DOI] [PubMed] [Google Scholar]
- Das A, Garner DP, Del Re AM, Woodward JJ, Kumar DM, Agarwal N, Banik NL, Ray SK. Calpeptin provides functional neuroprotection to rat retinal ganglion cells following Ca2+ influx. Brain Res. 2006;1084:146–157. doi: 10.1016/j.brainres.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Das A, Wallace Iv GC, McDowell ML, Smith JA, Marshall JD, Bonilha L, Edwards JC, Glazier SS, Ray SK, Banik NL. Hippocampal tissue of patients with refractory temporal lobe epilepsy is associated with astrocyte activation, inflammation, and altered expression of channels and receptors. Neuroscience. 2012;220:237–246. doi: 10.1016/j.neuroscience.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Guyton MK, Matzelle DD, Ray SK, Banik NL. Time-dependent increases in protease activities for neuronal apoptosis in spinal cords of Lewis rats during development of acute experimental autoimmune encephalomyelitis. J Neurosci Res. 2008a;86:2992–3001. doi: 10.1002/jnr.21737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Guyton MK, Butler JT, Ray SK, Banik NL. Activation of calpain and caspase pathways in demyelination and neurodegeneration in animal model of multiple sclerosis. CNS Neurol Disord Drug Targets. 2008b;7:313–320. doi: 10.2174/187152708784936699. [DOI] [PubMed] [Google Scholar]
- Davie CA, Barker GJ, Webb S, Tofts PS, Thompson AJ, Harding AE, McDonald WI, Miller DH. Persistent functional deficit in multiple sclerosis and autosomal dominant ataxia associated with axon loss. Brain. 1995;118:1583–1592. doi: 10.1093/brain/118.6.1583. [DOI] [PubMed] [Google Scholar]
- Fisher JB, Jacobs DA, Markowitz CE, Galetta SL, Volpe NJ, Nano-Schiavi ML, Baier ML, Frohman EM, Winslow H, Frohman TC, Calabresi PA, Maguire MG, Cutter GR, Balcer LJ. Relation of visual function to retinal nerve fiber layer thickness in multiple sclerosis. Ophthalmology. 2006;113:324–332. doi: 10.1016/j.ophtha.2005.10.040. [DOI] [PubMed] [Google Scholar]
- Gran B, Zhang G-X, Yu S, Li J, Chen XH, Ventura ES, Kamoun M, Rostami A. IL-12p35-deficient mice are susceptible to experimental autoimmune encephalomyelitis: evidence for redundancy in the IL-12 system in the induction of central nervous system autoimmune demyelination. J Immunol. 2002;169:7104–7110. doi: 10.4049/jimmunol.169.12.7104. [DOI] [PubMed] [Google Scholar]
- Guan Y, Shindler KS, Tabuena P, Rostami A. Retinal ganglion cell damage induced by spontaneous autoimmune optic neuritis in MOG-specific TCR transgenic mice. J Neuroimmunol. 2006;178:40–48. doi: 10.1016/j.jneuroim.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Guyton MK, Brahmachari S, Das A, Samantaray S, Inoue J, Azuma M, Ray SK, Banik NL. Inhibition of calpain attenuates encephalitogenicity of MBP-specific T cells. J Neurochem. 2009;110:1895–1907. doi: 10.1111/j.1471-4159.2009.06287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyton MK, Das A, Samantaray S, Wallace GC, 4th, Butler JT, Ray SK, Banik NL. Calpeptin attenuated inflammation, cell death, and axonal damage in animal model of multiple sclerosis. J Neurosci Res. 2010;88:2398–2408. doi: 10.1002/jnr.22408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyton MK, Sribnick EA, Ray SK, Banik NL. A role for calpain in optic neuritis. Ann N Y Acad Sci. 2005a;1053:48–54. doi: 10.1196/annals.1344.005. [DOI] [PubMed] [Google Scholar]
- Guyton MK, Wingrave JM, Yallapragada AV, Wilford GG, Sribnick EA, Matzelle DD, Tyor WR, Ray SK, Banik NL. Upregulation of calpain correlates with increased neurodegeneration in acute experimental auto-immune encephalomyelitis. J Neurosci Res. 2005b;81:53–61. doi: 10.1002/jnr.20470. [DOI] [PubMed] [Google Scholar]
- Kennedy KJ, Karpus WJ. Role of chemokines in the regulation of Th1/Th2 and autoimmune encephalomyelitis. J Clin Immunol. 1999;19:273–279. doi: 10.1023/a:1020535423465. [DOI] [PubMed] [Google Scholar]
- Kornek B, Storch MK, Weissert R, Wallstroem E, Stefferl A, Olsson T, Linington C, Schmidbauer M, Lassmann H. Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am J Pathol. 2000;157:267–276. doi: 10.1016/S0002-9440(10)64537-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Banik NL. The localization of mcalpain in myelin: immunocytochemical evidence in different areas of rat brain and nerves. Brain Res. 1995;697:112–121. doi: 10.1016/0006-8993(95)00949-q. [DOI] [PubMed] [Google Scholar]
- Li T, Kilic A, Wei X, Wu C, Schwartzbauer G, Yankey GK, Defilippi C, Bond M, Wu ZJ, Griffith BP. Regional imbalanced activation of the calcineurin/BAD apoptotic pathway and the PI3K/Akt survival pathway after myocardial infarction. Int J Cardiol. 2011 Nov 14; doi: 10.1016/j.ijcard.2011.10.107. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Zhang H, Verkman AS. Greatly attenuated experimental autoimmune encephalomyelitis in aquaporin-4 knockout mice. BMC Neurosci. 2009;10:94. doi: 10.1186/1471-2202-10-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Pelech S, Zhang H, Bond J, Spach K, Noubade R, Blankenhorn EP, Teuscher C. Pertussis toxin induces angiogenesis in brain microvascular endothelial cells. J Neurosci Res. 2008;86:2624–2640. doi: 10.1002/jnr.21716. [DOI] [PubMed] [Google Scholar]
- Maier K, Merkler D, Gerber J, Taheri N, Kuhnert AV, Williams SK, Neusch C, Bähr M, Diem R. Multiple neuroprotective mechanisms of minocycline in autoimmune CNS inflammation. Neurobiol Dis. 2007;25:514–525. doi: 10.1016/j.nbd.2006.10.022. [DOI] [PubMed] [Google Scholar]
- Maier K, Rau CR, Storch MK, Sättler MB, Demmer I, Weissert R, Taheri N, Kuhnert AV, Bähr M, Diem R. Ciliary neurotrophic factor protects retinal ganglion cells from secondary cell death during acute autoimmune optic neuritis in rats. Brain Pathol. 2004;14:378–387. doi: 10.1111/j.1750-3639.2004.tb00081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malemud CJ, Miller AH. Pro-inflammatory cytokine-induced SAPK/MAPK and JAK/STAT in rheumatoid arthritis and the new anti-depression drugs. Expert Opin Ther Targets. 2008;12:171–183. doi: 10.1517/14728222.12.2.171. [DOI] [PubMed] [Google Scholar]
- McKernan DP, Guerin MB, O’Brien CJ, Cotter TG. A key role for calpains in retinal ganglion cell death. Invest Ophthalmol Vis Sci. 2007;48:5420–5430. doi: 10.1167/iovs.07-0287. [DOI] [PubMed] [Google Scholar]
- Meyer AL, Benson J, Song F, Javed N, Gienapp IE, Goverman J, Brabb TA, Hood L, Whitacre CC. Rapid depletion of peripheral antigen-specific T cells in TCR-transgenic mice after oral administration of myelin basic protein. J Immunol. 2001;166:5773–5781. doi: 10.4049/jimmunol.166.9.5773. [DOI] [PubMed] [Google Scholar]
- Misu T, Fujihara K, Kakita A, Konno H, Nakamura M, Watanabe S, Takahashi T, Nakashima I, Takahashi H, Itoyama Y. Loss of aquaporin 4 in lesions of neuromyelitis optica: distinction from multiple sclerosis. Brain. 2007;130(Pt 5):1224–1234. doi: 10.1093/brain/awm047. [DOI] [PubMed] [Google Scholar]
- Miyamoto K, Nagaosa N, Motoyama M, Kataoka K, Kusunoki S. Upregulation of water channel aquaporin-4 in experimental autoimmune encephalomyeritis. J Neurol Sci. 2009;276:103–107. doi: 10.1016/j.jns.2008.09.014. [DOI] [PubMed] [Google Scholar]
- Nagelhus EA, Veruki ML, Torp R, Haug FM, Laake JH, Nielsen S, Agre P, Ottersen OP. Aquaporin-4 water channel protein in the rat retina and optic nerve: polarized expression in Müller cells and fibrous astrocytes. J Neurosci. 1998;18:2506–2519. doi: 10.1523/JNEUROSCI.18-07-02506.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namer IJ, Steibel J, Poulet P, Mauss Y, Mohr M, Chambron J. The role of Mycobacterium tuberculosis in experimental allergic encephalomyelitis. Eur Neurol. 1994;34:224–227. doi: 10.1159/000117043. [DOI] [PubMed] [Google Scholar]
- Pittock SJ, Weinshenker BG, Lucchinetti CF, Wingerchuk DM, Corboy JR, Lennon VA. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol. 2006;63:964–968. doi: 10.1001/archneur.63.7.964. [DOI] [PubMed] [Google Scholar]
- Ponomarev ED, Shriver LP, Maresz K, Pedras-Vasconcelos J, Verthelyi D, Dittel BN. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J Immunol. 2007;178:39–48. doi: 10.4049/jimmunol.178.1.39. [DOI] [PubMed] [Google Scholar]
- Potter NT, Bigazzi PE. Acute optic neuritis associated with immunization with the CNS myelin proteolipid protein. Invest Ophthalmol Vis Sci. 1992;33:1717–1722. [PubMed] [Google Scholar]
- Qi X, Lewin AS, Sun L, Hauswirth WW, Guy J. Suppression of mitochondrial oxidative stress provides long-term neuroprotection in experimental optic neuritis. Invest Ophthalmol Vis Sci. 2007;48:681–691. doi: 10.1167/iovs.06-0553. [DOI] [PubMed] [Google Scholar]
- Qin S, Rottman JB, Myers P, Kassam N, Weinblatt M, Loetscher M, Koch AE, Moser B, Mackay CR. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest. 1998;101:746–754. doi: 10.1172/JCI1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinones MP, Kalkonde Y, Estrada CA, Jimenez F, Ramirez R, Mahimainathan L, Mummidi S, Choudhury GG, Martinez H, Adams L, Mack M, Reddick RL, Maffi S, Haralambous S, Probert L, Ahuja SK, Ahuja SS. Role of astrocytes and chemokine systems in acute TNFalpha induced demyelinating syndrome: CCR2-dependent signals promote astrocyte activation and survival via NF-κB and Akt. Mol Cell Neurosci. 2008;37:96–109. doi: 10.1016/j.mcn.2007.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray SK, Banik NL. Calpain and its involvement in the pathophysiology of CNS injuries and diseases: therapeutic potential of calpain inhibitors for prevention of neurodegeneration. Curr Drug Targets CNS Neurol Disord. 2003;2:173–189. doi: 10.2174/1568007033482887. [DOI] [PubMed] [Google Scholar]
- Roemer SF, Parisi JE, Lennon VA, Benarroch EE, Lassmann H, Bruck W, Mandler RN, Weinshenker BG, Pittock SJ, Wingerchuk DM, Lucchinetti CF. Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain. 2007;130:1194–1205. doi: 10.1093/brain/awl371. [DOI] [PubMed] [Google Scholar]
- Samson M, Labbe O, Mollereau C, Vassart G, Parmentier M. Molecular cloning and functional expression of a new human CC-chemokine receptor gene. Biochemistry. 1996;35:3362–3367. doi: 10.1021/bi952950g. [DOI] [PubMed] [Google Scholar]
- Sättler MB, Merkler D, Maier K, Stadelmann C, Ehrenreich H, Bähr M, Diem R. Neuroprotective effects and intracellular signaling pathways of erythropoietin in a rat model of multiple sclerosis. Cell Death Differ. 2004;(Suppl 2):S181–92. doi: 10.1038/sj.cdd.4401504. [DOI] [PubMed] [Google Scholar]
- Schaecher K, Rocchini A, Dinkins J, Matzelle DD, Banik NL. Calpain expression and infiltration of activated T cells in experimental allergic encephalomyelitis over time: increased calpain activity begins with onset of disease. J Neuroimmunol. 2002;129:1–9. doi: 10.1016/s0165-5728(02)00142-x. [DOI] [PubMed] [Google Scholar]
- Shao H, Huang Z, Sun SL, Kaplan HJ, Sun D. Myelin/oligodendrocyte glycoprotein-specific T-cells induce severe optic neuritis in the C57BL/6 mouse. Invest Ophthalmol Vis Sci. 2004;45:4060–4065. doi: 10.1167/iovs.04-0554. [DOI] [PubMed] [Google Scholar]
- Sharma S, Yang B, Xi X, Grotta JC, Aronowski J, Savitz SI. IL-10 directly protects cortical neurons by activating PI-3 kinase and STAT-3 pathways. Brain Res. 2011;1373:189–194. doi: 10.1016/j.brainres.2010.11.096. [DOI] [PubMed] [Google Scholar]
- Shields DC, Banik NL. Putative role of calpain in the pathophysiology of experimental optic neuritis. Exp Eye Res. 1998a;67:403–410. doi: 10.1006/exer.1998.0537. [DOI] [PubMed] [Google Scholar]
- Shields DC, Tyor WR, Deibler GE, Banik NL. Increased calpain expression in experimental demyelinating optic neuritis: an immunocytochemical study. Brain Res. 1998b;784:299–304. doi: 10.1016/s0006-8993(97)01381-4. [DOI] [PubMed] [Google Scholar]
- Shindler KS, Guan Y, Ventura E, Bennett J, Rostami A. Retinal ganglion cell loss induced by acute optic neuritis in a relapsing model of multiple sclerosis. Mult Scler. 2006;12:526–532. doi: 10.1177/1352458506070629. [DOI] [PubMed] [Google Scholar]
- Smith AW, Das A, Guyton MK, Ray SK, Rohrer B, Banik NL. Calpain inhibition attenuates apoptosis of retinal ganglion cells in acute optic neuritis. Invest Ophthalmol Vis Sci. 2011;52:4935–4941. doi: 10.1167/iovs.10-7027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolina M, Sharma S, Lin Y, Dohadwala M, Gardner B, Luo J, Zhu L, Kronenberg M, Miller PW, Portanova J, Lee JC, Dubinett SM. Specific inhibition of cyclooxygenase 2 restores antitumor reactivity by altering the balance of IL-10 and IL-12 synthesis. J Immunol. 2000;164:361–370. doi: 10.4049/jimmunol.164.1.361. [DOI] [PubMed] [Google Scholar]
- Takano R, Misu T, Takahashi T, Izumiyama M, Fujihara K, Itoyama Y. A prominent elevation of glial fibrillary acidic protein in the cerebrospinal fluid during relapse in neuromyelitis optica. Tohoku J Exp Med. 2008;215:55–59. doi: 10.1620/tjem.215.55. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Bö L, Mörk S, Chang A. Pathogenesis of tissue injury in MS lesions. J Neuroimmunol. 1999;98:49–56. doi: 10.1016/s0165-5728(99)00081-8. [DOI] [PubMed] [Google Scholar]
- Trip SA, Schlottmann PG, Jones SJ, Altmann DR, Garway-Heath DF, Thompson AJ, Plant GT, Miller DH. Retinal nerve fiber layer axonal loss and visual dysfunction in optic neuritis. Ann Neurol. 2005;58:383–391. doi: 10.1002/ana.20575. [DOI] [PubMed] [Google Scholar]
- Tsoi VL, Hill KE, Carlson NG, Warner JE, Rose JW. Immunohistochemical evidence of inducible nitric oxide synthase and nitrotyrosine in a case of clinically isolated optic neuritis. J Neuroophthalmol. 2006;26:87–94. doi: 10.1097/01.wno.0000223266.48447.1b. [DOI] [PubMed] [Google Scholar]
- Vitellaro-Zuccarello L, Mazzetti S, Bosisio P, Monti C, De Biasi S. Distribution of Aquaporin 4 in rodent spinal cord: relationship with astrocyte markers and chondroitin sulfate proteoglycans. Glia. 2005;51:148–159. doi: 10.1002/glia.20196. [DOI] [PubMed] [Google Scholar]
- Williams KC, Ulvestad E, Hickey WF. Immunology of multiple sclerosis. Clin Neurosci. 1994;2:229–245. [PubMed] [Google Scholar]
- Wingrave JM, Schaecher KE, Sribnick EA, Wilford GG, Ray SK, Hazen-Martin DJ, Hogan EL, Banik NL. Early induction of secondary injury factors causing activation of calpain and mitochondria-mediated neuronal apoptosis following spinal cord injury in rats. J Neurosci Res. 2003;73:95–104. doi: 10.1002/jnr.10607. [DOI] [PubMed] [Google Scholar]
- Wujek JR, Bjartmar C, Richer E, Ransohoff RM, Yu M, Tuohy VK, Trapp BD. Axon loss in the spinal cord determines permanent neurological disability in an animal model of multiple sclerosis. J Neuropathol Exp Neurol. 2002;61:23–32. doi: 10.1093/jnen/61.1.23. [DOI] [PubMed] [Google Scholar]