

Abstract
Although the interiors of membrane and water-soluble proteins are similar in their physicochemical properties, membrane proteins differ in having larger fractions of hydrophobic residues on their exteriors. Thus, it should be possible to water-solubilize membrane proteins by mutating their lipid-contacting side chains to more polar groups. Here, a computational approach was used to generate water-soluble variants of the potassium channel KcsA. As a probe of the correctness of the fold, the proteins contain an agitoxin2 binding site from a mammalian homologue of the channel. The resulting proteins express in high yield in Escherichia coli and share the intended functional and structural properties with KcsA, including secondary structure, tetrameric quaternary structure, and tight specific binding to both agitoxin2 and a small molecule channel blocker.
The 3D structures of tens of thousands of water-soluble proteins have been solved, but the structures of membrane-soluble proteins have proven much more difficult to determine. The problems associated with membrane proteins include their limited levels of expression, low stability in detergent-solubilized forms, and greater difficulty in obtaining high-resolution diffraction quality crystals. Thus, we and others have sought to develop a broadly applicable automated strategy for the preparation of water-soluble mutants of membrane proteins, which could be obtained in larger quantities for a variety of high-resolution biophysical and drug discovery studies. Initial work yielded good results for a simple transmembrane coiled-coil (1–3), but attempts to design water-soluble versions of larger proteins such as bacteriorhodopsin led to misfolded proteins with very limited solubility in water (4). Here, we demonstrate the success of a previously undescribed computational approach for the design of a water-solubilized version of a bacterial ion channel with a transplanted mammalian toxin-binding site. These findings have fundamental implications concerning the stabilization of membrane vs. water-soluble proteins (5) as well as practical implications for the design of water-soluble analogues of a variety of biologically interesting membrane proteins.
The structures of many membrane proteins are beginning to appear, allowing one to infer features common to entire families. In particular, the structures of bacterial ion channels (6–12) have provided insight into their mammalian counterparts, and the structure of rhodopsin (13) has served as a prototype for the entire family of seven-transmembrane G protein-coupled receptors. It would be advantageous to use what structural information is available for a given membrane protein to obtain water-soluble versions that retain their structure, oligomerization state, and essential ligand-binding properties. As a target for such an effort, we have chosen the bacterial KcsA potassium channel because of its available structure (6, 12), its biochemical characterization, and the interest in this family of channel proteins. Furthermore, the external vestibule of the ion-conducting pore of KcsA has been mutated to the corresponding residues in a mammalian channel (Q58A, T61S, and R64D) to allow binding of agitoxin2 (AgTx2), resulting in a protein (designated here as tKcsA) that binds AgTx2 (14). This system extends multiple clearly defined criteria for the successful design of a water-soluble version of this protein, which should: (i) be expressed at high level in a water-soluble form; (ii) show the correct helical secondary structure; (iii) associate to form tetramers; (iv) bind AgTx2 with high affinity, specificity, and in the appropriate stoichiometry; and (ν) bind small molecule channel blockers such as tetraethylammonium chloride (TEA). The designed water-soluble variants of tKcsA are referred to as WSK-1, -2, and -3.
Methods
Protein Design. A statistical entropy-based formalism has been developed for identifying amino acid probabilities from a given backbone structure (15, 16). This method takes as input a target structure, in this case a high-resolution structure of KcsA (Protein Data Bank ID code 1k4c), and energy functions that quantify sequence–structure compatibility. The output is the set of site-specific probabilities of the amino acids compatible with the structure. The site-specific probabilities of the amino acids and their discrete side chain conformational states (rotamers) are determined by maximizing an effective entropy function subject to simultaneous constraints on both the overall energy as determined by an atom-based potential and the value of an effective solvation score (“environmental energy”). In the calculation, all 20 amino acids and up to 10 of their side chain conformations [rotamer states (17)] were considered at each site where mutations were permitted. This yields a total of 129 identity-rotamer states for each variable position. C4 subunit symmetry was imposed, reducing the complexity (total number of possible sequence–rotamer combinations) from 129140 to 12935 for the first of the recursive calculations discussed below (18).
To quantify hydrophobicity and solvation effects, an environmental potential is used (16), where the local Cβ density about each residue is used to quantify its degree of exposure to solvent. The environmental energy for the entire protein (Eenv) and for the buried residue sites (Eenv_b) are constrained to the average values of water-soluble proteins in the calculation (–46.0 and –24.6). The identities of buried sites are constrained in these calculations, and the decrease in Eenv (from +20 to –46) largely results from the mutation of exposed hydrophobic residues to more hydrophilic amino acids.
Nonbonded interactions involving the side chains are calculated by using the amber 3a force field (19) with a modified hydrogen-bonding term (20) and a distance-dependent dielectric constant (ε = 4 r). To address unfolded states, a reference energy γref (α) for each amino acid is introduced into the energy Ec to represent the effects of the denatured state. The energy is calculated as a “free energy” of each amino acid in its N-acetyl-N′-methylamide derivative with averaging over multiple backbone and rotamer states. This averaging involves a sum over possible rotamers and possible backbone configurations, approximated by varying each of the backbone φ and ϕ angles in increments of 10°. This crudely approximates an average over extended unfolded states. The reference energies of each amino acid may then be estimated by using
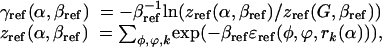 |
where γref is the conformational energy in a particular conformation of the N-acetyl-N′-methylamide derivative of the amino acid, as determined by using the molecular potential. Here βref = 1/kBT, where kB is Boltzmann's constant and T is a temperature appropriate for the conformation sampling of side chain and backbone conformations (e.g., T = 300 K). Reference energies are expressed relative to Gly (16). In the statistical formalism of Scads (statistical computationally assisted design strategy), accompanying the interatomic potential energy is a corresponding effective temperature 1/β. The probabilities used in sequence identification were determined for β = 0.5 mol/kcal; at this value, we find the sequence properties are robust with respect to slight variation in backbone structure (16).
WSK Expression and Purification. The gene encoding WSK-1 was synthesized with Pfu polymerase by using three fragments. The final WSK-1 fragment was cloned into the pET-24a(+) vector (Novagen) at the NdeI and XhoI restriction sites, expressing no tag. Mutants WSK-2 and -3 were generated by using QuikChange (Stratagene). The WSK proteins were expressed in BL21(DE3) cells (Novagen) in LB for 4 h after induction with 1 mM isopropyl-β-d-thiogalactoside. Cells were harvested by centrifugation at 4°C and 3,450 × g. Cell pellets were lysed by French press at 1,500 psi in lysis buffer containing 10 mM Tris[hydroxymethyl]aminomethane hydrochloride, pH 7.0, and 1 mM EDTA. Cell extracts were loaded onto a 40-ml Q Sepharose (Amersham Pharmacia Biosciences) column in the lysis buffer and eluted with a step gradient of the lysis buffer containing 0–500 mM KCl. Fractions containing WSK were pooled, concentrated to 50 ml, and further purified by reverse-phase HPLC on a Vydac (Hesperia, CA) C4 preparative column by using a linear gradient of water and acetonitrile containing 0.1% trifluoroacetic acid. Purity was verified by analytical reverse-phase HPLC, matrix-assisted laser desorption ionization time-of-flight mass spectrometry, and 4–12% Bis-Tris SDS/PAGE gels. The molar extinction coefficient (ε280nm = 17,781 M–1·cm–1) was determined by the difference in absorbance of a sample in 0 and 6 M guanidine·HCl, by using the calculated extinction coefficient at 280 nm as a starting point.
AgTx2 Synthesis and Purification. AgTx2 (4,097 Da) was chemically synthesized as a C-terminal carboxyamide on a 0.25 mmol scale by using an Applied Biosystems model 433A solid phase peptide synthesizer (Perkin–Elmer) with standard fluorenylmethoxycarbonyl amino acid chemistry. The tripeptide glygly-N-2,4-dinitrophenyl-Ala chromophore was coupled to the N terminus of half of the resin. AgTx2 with 2,4-dinitrophenylAla (AgTx2-DNP) has a calculated ε356nm = 11,343 M–1·cm–1, and ε280nm = 4,520 M–1·cm–1. Peptides were washed on the resin with dimethylformamide and ether, cleaved for 3.5 h with trif luoroacetic acid/water/ethanedithiol/triisopropylsilane (94.5/2.5/2.5/1) vol/vol/vol/vol, and precipitated and washed with cold ether. Purification proceeded by reverse-phase HPLC by using a preparative C4 column (Vydac) and a linear gradient of the appropriate buffers. AgTx2 required oxidative refolding to attain the proper folded state after HPLC. A series of redox buffers containing increasing proportions of oxidized (GSSG) and reduced glutathione (GSH) were made. The concentration of GSSG was determined by UV/Vis absorbance by using ε282nm = 273 M–1·cm–1, whereas GSH was monitored by titration with Elmans reagent, following absorbance at 412 nm. Optimal conditions were identical for both labeled and unlabeled peptides, and the final reaction components were determined to 125 μM toxin in the presence of air and 100% GSH (6 mM). The progress of the reaction was followed by using analytical reverse-phase HPLC and comparison of the magnitude of the properly folded peak to the misfolded peaks. Properly folded toxins were repurified by reverse-phase HPLC and tested for function.
Solution Measurements. CD spectra were collected on an Aviv 62DS (Aviv Associates, Lakewood, NJ) spectropolarimeter, by using a 1-mm path-length quartz cuvette, with protein at 44 μM, in 20 mM potassium phosphate, pH 7.0/100 mM KCl/1 mM EDTA. For ultracentrifugation, samples were centrifuged at 15,000 rpm in 20 mM potassium phosphate, pH 7.0/100 mM KCl/1 mM EDTA by using a Beckman XL-I (Beckman Coulter) analytical ultracentrifuge. Equilibrium dialysis to determine the stoichiometry of toxin binding to WSK-3 was performed by using 0.5 ml of centrifuge tubes and a dialysis membrane with a 10-kDa molecular weight cutoff. After reaching equilibrium, the concentrations of AgTx2-DNP and WSK-3 were determined by absorbance at 356 and 280 nm, respectively.
The extent of binding of AgTx2-DNP (10 μM) to WSK-3 (10 μM, tetramer) was assessed at various concentrations of TEA and tetramethylammonium ion by ultracentrifugation at 16,385 × g. Because we expected some salt dependence to the binding interaction, we also measured the extent of binding in the presence of the same concentrations of KCl. The fraction of free AgTx2-DNP was determined from the absorbance at 360 nm at the top of the cell (at this speed, the WSK-3 is essentially fully depleted, but the AgTx2-DNP has not significantly sedimented). The extent of binding, relative to the KCl control, was plotted and the IC50 determined (Fig. 5). The approximate Kdiss for binding to TEA is determined from the relationship IC50 = KTEA [fAgTx2-DNP]/KAgTx2-DNP, where fAgTx2-DNP is defined as the free AgTx2-DNP concentration, and KTEA and KAgTx2-DNP are the dissociation constants for WSK-3 with TEA and AgTx2-DNP respectively. This relationship holds approximately for [fAgTx2-DNP] ≫ KAgTx2-DNP, where [fAgTx2-DNP] = 5 μM and KAgTx2-DNP ≤ 2 μM (because tight binding is observed in the equilibrium dialysis with 10 μM total toxin concentration).
Fig. 5.

The small-molecule blocker TEA competes with AgTx2-DNP for binding to WSK-3. A competition curve for tetramethylammonium ion is also shown.
Results and Discussion
Design of WSK. The side chains of the transmembrane helices of tKcsA were stripped to the backbone and then rebuilt in accordance with the requirements for function, stability, and water solubility. We retained the pore-lining residues, the extracellular loops, the intracellular region, and the residues required for binding to AgTx2. Furthermore, we retained the buried residues, leaving 35 membrane-exposed side chains [>40% accessible to a probe with a radius of 1.4 Å (21)] per protomer as targets for design. These positions were chosen by using the computational design algorithm scads.
A key input into these calculations is an environmental energy, a database-derived quantification of solvation and hydrophobic effects (16). The value of this environmental score for WT KcsA is +20, which is well outside the range observed for soluble proteins of this size, due to the large number of exposed hydrophobic residues. Thus in calculations to determine water-soluble sequences, the environmental energy was constrained to the value expected for soluble proteins of this size, –46 (Fig. 1).
Fig. 1.

Eenv (16) vs. chain length. Eenv quantifies solvation and hydrophobic effects and has been parameterized by using a database of 500 soluble proteins (small circles) (16). For each protein structure, Eenv=∑i ε(ai, ra,i, ρCβ) where for the ith residue ai, ra,i, and ρCβ are the amino acid, side chain conformation, and local density of β carbons. Also shown is the Eenv for WT KcsA (open circle) and the value Eenv that was used as a constraint in the sequence calculations (black circle).
Scads generates profiles describing the site-dependent probabilities of the amino acids at each position allowed to mutate. A unique sequence was selected from the computed probabilities by using recursive calculations. The set of 35 exposed residues was initially targeted for variation. The identities of the remaining residues were fixed at WT, and their side chain conformations were permitted to vary to accommodate mutations (15, 16). A sequence was selected comprising the most probable amino acids at each site with the exception of several sites where alternate amino acids with appreciable probability were selected: W26E and G43A were chosen for their favorable helical propensities (22, 23) and S44, S69, and V106 were kept at WT. The WT amino acid was selected at 8 of the 35 sites. A second calculation was then performed. In the presence of the resulting designed sequence from the first iteration, an additional eight exposed hydrophobic sites were targeted for mutation. The WT was most probable at L36, V39, L40, I60, and L105; hydrophobic residues were probable at F116, and the WT was retained. The mutation V93E was most probable, and the L24D mutation was selected over the more probable W and F due to its greater polarity. At position 104, Gly and Ala had comparable probabilities, and Ala was chosen for its aqueous helix propensity. The resulting sequence was compared to 47 aligned K+ channel sequences from eight prokaryotic and eukaryotic organisms (blast, National Center for Biotechnology Information database).
The resulting sequence (WSK-1) had 29 mutations relative to tKcsA (Fig. 2). Preliminary studies discussed below indicated that this soluble protein bound toxin but formed high-order oligomers. The model of WSK-1 showed two exposed hydrophobic patches in the redesigned transmembrane helices, which might mediate aggregation. This problem was addressed in a new round of computational design varying only these exposed hydrophobic regions, resulting in WSK-2, which contained the additional mutation L116R, and WSK-3, which contained both L81R and L116R.
Fig. 2.

Depiction of KcsA and WSK-3. Only the side chains of outer helices (22–71), inner helices (89–124), and cytoplasmic and extracellular residues are shown. Side chains are colored based on frequency of occurrence in the apolar section of the lipid bilayer beginning with most probable: Ala/Ile/Leu/Val (dark green), Gly/Met/Thr (light green), and Pro/Ser/Trp/Tyr (light blue). Lys/Arg/Gln (dark blue), Asp/Glu (red). (a) KcsA. Solvent-exposed residues of KcsA allowed to vary in the design are depicted along the inner and outer helices (light green, dark green). Also shown are the cytoplasmic and extracellular residues, unchanged in our design. (b) KcsA structure with side chains of mutated residues removed. Extracellular and cytoplasmic residues held constant from KcsA to WSK are rendered. Buried residues within the interior of the structure are not shown. (c) WSK-3. Sequences of KcsA and WSK-3 are also shown, where colored residues were mutated, whereas those in black were not. Mutations to tKcsA are shown in a gray box [made by using pymol (DeLano Scientific, San Carlos, CA).
The WSK sequences result from the simultaneous imposition of effective energetic constraints on the solvation properties and the interatomic interactions of the flexible amino acid side chains. One might expect that simply mutating exposed hydrophobic residues to polar or charged amino acids might be sufficient to confer solubility, but such approaches often yield misfolded proteins (4). The calculations provide sequences with sterically consistent nontrivial patterning of amino acid identities. Among the polar mutations that were introduced, complementary charge interactions on the protein surface are apparent (see Fig. 2). Although the overall goal is to produce a water-soluble structure, it must also retain sufficient hydrophobic interactions to drive protein folding in an aqueous environment. Thus, some exposed hydrophobes are retained, and one mutation actually yields increased hydrophobic character (S102V). Others have noted the importance of careful consideration of interactions between surface residues in protein design (24).
Secondary and Quaternary Structure of WSK Variants. WSK variants were expressed in high yield (20 mg/ml) and in soluble form in Escherichia coli. The CD spectra of the purified proteins were consistent with the expected secondary structure, showing minima at 208 and 222 nm ([θ222] = –14,600 deg·cm2·dmol–1). The computed α-helical content was ≈50% (25), in good agreement with the helical content of 60% observed for KcsA (Protein Data Bank code 1k4c).
Size exclusion chromatography was used to determine the aggregation state of WSK variants. WSK-1 eluted as three peaks: one peak consistently eluted as a large aggregate in the void volume, whereas the observed molecular weights of the other two peaks were 10,600 and 50,100 Da (Fig. 3a), in good agreement with the expected masses for the monomer (11,433 Da) and tetramer (45,732 Da), respectively. Preliminary experiments (not shown) showed that both the tetramer and the higher-order aggregate bound AgTx2 in the proper stoichiometry, suggesting that the void volume peak might consist of loosely associated but otherwise properly folded tetramers. To test this possibility, the molecular weight distribution was measured in the presence of 6 M urea, a concentration lower than that required to unfold the protein (as determined from the loss of secondary structure followed by CD spectroscopy). Under this condition, only the tetrameric and monomeric peaks were observed (Fig. 3b).
Fig. 3.

Analytical gel filtration chromatography of WSK-1 (20 μM), WSK-1 (20 μM, 6 M Urea), WSK-2 (100 μM), and WSK-3 (100 μM) by using a 25-ml Superdex 200 column (Amersham Pharmacia Biosciences) in 20 mM K2PO4, pH 7.0/100 mM KCl/1 mM EDTA at 1 ml/min flow rate. The approximate volumes of elution for the monomer (Mon), tetramer (Tet), 12-mer, and high-order aggregate (Agg) are indicated. The column was calibrated by using blue dextran, BSA (66 kDa), ovalbumin (43 kDa), and carbonic anhydrase (29 kDa).
WSK-1, -2, and -3 contain progressively fewer apolar side chains in the redesigned transmembrane helices; these mutants also show a progressively smaller fraction of aggregated protein eluting in the void volume. Even in the absence of urea, WSK-3 elutes primarily as a tetramer and shows only a small peak near the position expected for a 12-mer, presumably a trimer of tetramers (Fig. 3 c and d). Thus, iterative mutagenesis guided by computation and experiment was able to minimize the nonspecific aggregates of tetramers seen in WSK-1.
AgTx2 Binding. Size exclusion chromatography was used to demonstrate that WSK-3 specifically bound to AgTx2. For these experiments, we synthesized a variant of AgTx2 with AgTx2-DNP at its N terminus, allowing the toxin to be detected from its absorbance at 356 nm (ε356nm = 11,340 M–1·cm–1). When AgTx2-DNP was chromatographed through the Superdex 200 column, it eluted as a monomer, but when incubated with WSK variants (10 μM, assuming a functional tetramer), it coeluted with the tetrameric and higher-order aggregate peaks of WSK-1, -2, and -3 (data not shown) in approximately the expected molar ratio of one AgTx2-DNP for every four molecules of WSK.
Equilibrium analytical ultracentrifugation confirmed the selective association of WSK-3 with AgTx2-DNP (Fig. 4). WSK-3 sedimented in a concentration-dependent manner, and the traces are well defined by a monomer–tetramer–12-mer equilibrium (Fig. 4a), as expected from size exclusion chromatography. Under the same conditions, AgTx2-DNP shows little sedimentation, consistent with its monomeric molecular weight (4,435 Da). When incubated with WSK-3, AgTx2-DNP cosediments with the water-solubilized channel (Fig. 4b), as determined by the DNP absorbance. At a mol ratio of one AgTx2-DNP per WSK-3 tetramer (17 μM), there was essentially no free AgTx2-DNP (26), indicating that the dissociation constant for binding was ≤2 μM (i.e., 10-fold tighter than the total toxin concentration). By contrast, when AgTx2-DNP was centrifuged in the presence of BSA (36 μM), no association was observed (Fig. 4c), demonstrating the specificity of the AgTx2–WSK-3 interaction.
Fig. 4.

Equilibrium sedimentation analytical ultracentrifugation of WSK-3 and AgTx2-DNP. (a) 17 μM WSK-3 monitored at 280 nm (o). Data were fit to a monomer–tetramer–12-mer equilibrium by using a macro in igor-pro (WaveMetrics, Lake Oswego, OR). (b) AgTx2-DNP (17 μM) in the presence (red circle) and absence (green circle) of 17 μM WSK-3 tetramer, monitored at 360 nm to detect only the AgTx2-DNP. (c) Control with BSA (36 μM) plus AgTx2-DNP (17 μM) monitored at 280 nm (blue square) to monitor the BSA, and at 360 nm (green square) to monitor AgTx2-DNP.
The stoichiometry of toxin binding was determined by equilibrium dialysis by using two starting conditions: one with equimolar AgTx2-DNP and WSK-3 tetramer (10 μM), as well as one with a 2-fold excess of AgTx2-DNP. In both experiments, the ratio of AgTx2-DNP:WSK-3 monomer was 1:4 within experimental error (1:4.4 and 1:4.1, respectively). Control experiments showed no binding of AgTx2-DNP to either BSA or carbonic anhydrase (Table 1).
Table 1. Equilibrium dialysis of WSK-3 and AgTx2-DNP.
| Experiment | [Tox] bound, μM | Stoichiometry |
|---|---|---|
| WSK-3mon (40 μM) + Tox (10 μM) | 9.0 | [WSK-3]/[Tox]bound = 4.4 |
| WSK-3mon (40 μM) + Tox (20 μM) | 9.7 | [WSK-3]/[Tox]bound = 4.1 |
| BSA (20 μM) + Tox (44 μM) | 0 | |
| CA (47 μM) + Tox (23 μM) | 0 |
Initially, WSK-3 and AgTx2-DNP (Tox) are on side a, whereas buffer is on side b. At equilibrium, the concentration of bound toxin is given by [Tox]bound = [Tox]a - [Toxfree]a = [Tox]a - [Tox]b.
TEA Competition. To determine whether small molecule channel blockers also bind specifically to WSK-3, we measured the ability of TEA to compete with AgTx2-DNP for binding to WSK-3. This small molecule blocker has been shown to compete with charybdotoxin (a homologue of AgTx2) for binding to the extracellular opening of the pore (27). TEA induces dissociation of AgTx2-DNP from WSK-3 in a concentration-dependent manner, whereas the control tetramethylammonium ion [TMA, which does not bind to K+ channels (28)] showed no significant concentration-dependent competition (Fig. 5). From the concentration of TEA required to induce 50% total toxin dissociation, we calculate a Kdiss ≤ 80 mM for TEA (assuming a Kdiss ≤ 2 μM for binding of AgTx2-DNP). This value agrees well with the reported Kdiss of 3.2–22 mM (29) for the native channel. TEA binds to the outer vestibule of the channel KcsA channel. The finding that TEA binds to WSK-3 strongly suggests that this region of the protein is properly folded. Although this finding alone does not preclude the possibility of misfolding elsewhere in the protein, the additional CD, size exclusion chromatography, and toxin-binding data together suggest that the protein has a structure similar to the WT channel.
Conclusion
WSK-3 has successfully recapitulated the target properties of tKcsA, including its ability to bind a protein toxin and a small molecule blocker. These findings should be of practical utility for the design of water-soluble analogues of membrane proteins that have proven difficult to obtain in large quantities for biophysical studies or drug screening. The method requires only a medium-resolution structure for computational modeling. Thus, it should be possible to extend this work to other ion channels and G protein-coupled receptor proteins (30) using homology models as starting points.
Acknowledgments
This work was supported by the National Institutes of Health (Grants GM 60610 and GM 61267) and the National Science Foundation (Grant CHE 99-84752). J.G.S. is a Cottrell Scholar of Research Corporation and an Arnold and Mabel Beckman Foundation Young Investigator. We thank Carol Deutsch for sharing experimental expertise and suggestions.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: AgTx2, agitoxin2; TEA, tetraethylammonium chloride; AgTx2-DNP, AgTx2 with 2,4-dinitrophenyl-Ala; Eenv, environmental energy.
References
- 1.Li, H., Cocco, M. J., Steitz, T. A. & Engelman, D. E. (2001) Biochemistry 40, 6636–6645. [DOI] [PubMed] [Google Scholar]
- 2.Slovic, A. M., Summa, C. M., Lear, J. D. & DeGrado, W. F. (2002) Protein Sci. 12, 337–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frank, S., Kammerer, R. A., Hellstern, S., Pegoraro, S., Stetefeld, J., Lustig, A., Moroder, L. & Engel, J. (2000) Biochemistry 39, 6825–6831. [DOI] [PubMed] [Google Scholar]
- 4.Mitra, K., Steitz, T. A. & Engelman, D. M. (2002) Protein Eng. 15, 485–492. [DOI] [PubMed] [Google Scholar]
- 5.Popot, J.-L. & Engelman, D. (1990) Biochemistry 29, 4031–4037. [DOI] [PubMed] [Google Scholar]
- 6.Doyle, D. A., Morais Cabral, J., Pfuetzner, R. A., Kuo, A., Gulbis, J. M., Cohen, S. L., Chait, B. T. & MacKinnon, R. (1998) Science 280, 69–77. [DOI] [PubMed] [Google Scholar]
- 7.Dutzler, R., Campbell, E. B., Cadene, M., Chait, B. T. & MacKinnon, R. (2002) Nature 415, 287–294. [DOI] [PubMed] [Google Scholar]
- 8.Bass, R. B., Strop, P., Barclay, M. & Rees, D. C. (2002) Science 298, 1582–1587. [DOI] [PubMed] [Google Scholar]
- 9.Chang, G., Spencer, R. H., Lee, A. T., Barclat, M. T. & Rees, D. C. (1998) Science 282, 2220–2226. [DOI] [PubMed] [Google Scholar]
- 10.Jiang, Y., Lee, A., Chen, J., Cadene, M., Chait, B. T. & MacKinnon, R. (2002) Nature 417, 515–522. [DOI] [PubMed] [Google Scholar]
- 11.Jiang, Y., Lee, A., Chen, J., Ruta, V., Cadene, M., Chait, B. T. & Mackinnon, R. (2003) Nature 423, 33–41. [DOI] [PubMed] [Google Scholar]
- 12.Zhou, Z., Morais-Cabral, J. H., Kauffman, A. & MacKinnon, R. (2001) Nature 414, 43–48. [DOI] [PubMed] [Google Scholar]
- 13.Palczewski, K., Kumasaka, T., Hori, T., Behnke, C. A., Motoshima, H., Fox, B. A., Le Trong, I., Teller, D. C., Okada, T., Stenkamp, R. E., et al. (2000) Science 289, 739–745. [DOI] [PubMed] [Google Scholar]
- 14.MacKinnon, R., Cohen, S. L., Kuo, A., Lee, A. & Chait, B. T. (1998) Science 280, 106–109. [DOI] [PubMed] [Google Scholar]
- 15.Zou, J. & Saven, J. G. (2000) J. Mol. Biol. 296, 281–294. [DOI] [PubMed] [Google Scholar]
- 16.Kono, H. & Saven, J. G. (2001) J. Mol. Biol. 306, 607–628. [DOI] [PubMed] [Google Scholar]
- 17.Dunbrack, R. L., Jr., & Cohen, F. E. (1997) Protein Sci. 6, 1661–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fu, X., Kono, H. & Saven, J. G. (2004) Protein Eng., in press. [DOI] [PubMed]
- 19.Weiner, S. J., Kollman, P. A., Case, D. A., Singh, U. C., Ghio, C., Alagona, G., Profeta, S. & Weiner, P. (1984) J. Am. Chem. Soc. 106, 765–784. [Google Scholar]
- 20.Kono, H. & Doi, J. (1996) J. Comput. Chem. 17, 1667–1683. [Google Scholar]
- 21.Sridharan, S., Nicholls, A. & Sharp, K. A. (1995) J. Comput. Chem. 16, 1038–1044. [Google Scholar]
- 22.Muñoz, V. & Serrano, L. (1994) Nature 1, 399–409. [DOI] [PubMed] [Google Scholar]
- 23.Muñoz, V. & Serrano, L. (1995) J. Mol. Biol. 245, 275–296. [DOI] [PubMed] [Google Scholar]
- 24.Marshall, S. A., Morgan, C. S. & Mayo, S. L. (2002) J. Mol. Biol. 316, 189–199. [DOI] [PubMed] [Google Scholar]
- 25.Chakrabartty, A., Schellman, J. A. & Baldwin, R. L. (1991) Nature 351, 586–588. [DOI] [PubMed] [Google Scholar]
- 26.Arkin, M. & Lear, J. D. (2001) Anal. Biochem. 299, 98–107. [DOI] [PubMed] [Google Scholar]
- 27.Miller, C. (1988) Neuron 1, 1003–1006. [DOI] [PubMed] [Google Scholar]
- 28.Heginbotham, L. & MacKinnon, R. (1992) Neuron 8, 483–491. [DOI] [PubMed] [Google Scholar]
- 29.Heginbotham, L., LeMasurier, M., Kolmakova-Partenky, L. & Miller, C. (1999) J. Gen. Phys. 114, 551–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Becker, O. M., Shacham, S., Marantz, Y. & Noiman, S. (2003) Curr. Opin. Drug Discov. Dev. 6, 353–361. [PubMed] [Google Scholar]