

Abstract
Glucocorticoids are widely used for the treatment of hematological malignancies; however, their chronic use results in numerous metabolic side effects. Thus, the development of selective glucocorticoid receptor (GR) activators (SEGRA) with improved therapeutic index is important. GR regulates gene expression via (1) transactivation that requires GR homodimer binding to gene promoters and is linked to side effects and (2) transrepression-mediated via negative GR interaction with other transcription factors. Novel GR modulator Compound A (CpdA) prevents GR dimerization, retains glucocorticoid anti-inflammatory activity and has fewer side effects compared with glucocorticoids in vivo. Here we tested CpdA anticancer activity in human T- and B-lymphoma and multiple myeloma cells expressing GR and their counterparts with silenced GR. We found that CpdA in GR-dependent manner strongly inhibited growth and viability of human T-, B-lymphoma and multiple myeloma cells. Furthermore, primary leukemia cell cultures from T-ALL patients appeared to be equally sensitive to glucocorticoid dexamethasone and CpdA. It is known that GR expression is controlled by proteasome. We showed that pretreatment of lymphoma CEM and NCEB cells with proteasome-inhibitor Bortezomib resulted in GR accumulation and enhanced ligand properties of CpdA, shifting GR activity toward transrepression evaluated by inhibition of NFкB and AP-1 transcription factors. We also revealed remarkable GR-dependent cooperation between CpdA and Bortezomib in suppressing growth and survival of T- and B-lymphoma and multiple myeloma MM.1S cells. Overall, our data provide the rationale for novel GR-based therapy for hematological malignancies based on combination of SEGRA with proteasome inhibitors.
Keywords: glucocorticoid receptor, selective GR activator (SEGRA), lymphoma, leukemia, multiple myeloma, proteasome inhibitor
Introduction
Glucocorticoid hormones are essential physiological and pharmacological regulators of proliferation, differentiation and apoptosis of lymphoid cells and are widely used for the treatment of patients with different T- and B-cell lymphomas and multiple myeloma.1,2 Though the therapy of hematological malignancies has evolved over the past decade, the glucocorticoids still remain central for their treatment, and are often used for combination chemotherapies with traditional and newer agents such as proteasome-inhibitor Bortezomib (BZ).3,4 Unfortunately, patients chronically treated with glucocorticoids develop resistance to steroids along with numerous metabolic side effects.5,6
The biological response to glucocorticoids is mediated by glucocorticoid receptor (GR), a well-characterized transcription factor.7-9 GR controls gene expression via (1) transactivation, which requires binding of GR homodimers to palindromic glucocorticoid-responsive elements (GRE) in gene promoters and enhancers, and (2) dimerization-independent transrepression, which is chiefly mediated via negative interaction between GR and other transcription factors, including major pro-inflammatory and pro-proliferative factors NFkB and AP-1.7,9,10
In our recent work, we discovered that GR transrepression plays an important role in the tumor-suppressor effect of GR in different models.6,11,12 This makes anticancer and anti-inflammatory mechanisms of the GR signaling similar, as it is well-understood that major anti-inflammatory effects of glucocorticoids are mediated via GR transrepression.13,14 In contrast to therapeutic effects, many adverse metabolic effects of glucocorticoids are mediated by GR transactivation.13,14 Thus, selective GR activators (SEGRA, also called “dissociated” GR ligands) that preferentially activate GR transrepression could be a better option for the treatment of patients with hematological malignancies.
Recently, several non-steroidal SEGRA have been developed and entered clinical trials as anti-inflammatory GR ligands with reduced side effects.14,15 One of the novel GR modulators is a small molecule 2-(4-acetoxyphenyl)-2-chloro-N-methylethylammonium-chloride also called Compound A (CpdA). CpdA is a synthetic analog of hydroxyphenyl aziridine precursor isolated from the Namibian shrub Salsola tuberculatiformis Botschantzev.16 Others and we showed that CpdA acts as dissociated GR ligand: it strongly competes with glucocorticoids for GR binding, does not induce GR-mediated gene activation well, but efficiently induces GR transrepression.17-20 Importantly, in vivo CpdA is as effective as glucocorticoids in counteracting inflammation in different animal models.17,19,21,22 Coincidently, in contrast to glucocorticoids, it has fewer side effects related to maintenance of hypothalamic-pituitary-adrenal (HPA) axis, and bone metabolism.14,17,19,21,23
We reported recently that CpdA has anticancer potential, and inhibits both growth and survival of highly malignant prostate cancer cells in GR-dependent fashion.20 Even though anticancer potential of GR modulators is mostly pertinent to hematological malignancies, the effects of CpdA, as well as other SEGRA on T- and B-lymphoma and multiple myeloma cell growth and apoptosis, have not been studied.
Sensitivity to therapeutic effects of glucocorticoids, including apoptosis induced in lymphoid cancer cells, directly depends on the amount of functional GR.24 The 26S proteasome controls GR protein stability in untreated and hormone-treated cells and is responsible for cell desensitization to glucocorticoids via accelerated hormone-induced GR degradation.25,26 Consequently, the use of proteasome inhibitors represents a feasible pharmacological approach to elevate the level of GR in cells.27,28
Currently, Bortezomib is the only clinically used proteasome inhibitor. It was approved by the FDA first for the treatment of patients with multiple myeloma and mantle cell lymphoma.3,4 Since proteasome inhibitors stabilize GR, we hypothesized that BZ augments CpdA effects as a selective GR modulator and enhances its chemotherapeutic activity. Thus, the major goals of this study were to evaluate the anti-lymphoma potential of novel GR modulator CpdA, and to test whether BZ enhances CpdA ligand profile and increases its therapeutic potential. Using representative human T- (CEM) and B- (NCEB) lymphoma and multiple myeloma (MM.1S) cell lines expressing endogenous functional GR, and their counterparts with silenced GR expression, we showed that CpdA indeed acted as dissociated GR ligand and inhibited growth and survival of these lymphoma cells via GR. As expected, we revealed strong GR-dependent cooperation between CpdA and BZ in suppressing growth and survival of lymphoma and multiple myeloma cells.
Results
Structural and functional characteristics of GR in lymphoma cell lines
Despite the extensive use of glucocorticoids for the treatment of patients with hematological malignancies, GR status in lymphoma patient cells and in lymphoid cancer cell lines has not been well-investigated. There are several GR isoforms that arise due to the alternative splicing. The major, fully functional GR isoform is GRalpha.29 Our work is focused on this major GRalpha isoform, and we use the abbreviation “GR” throughout the text to refer to GRalpha.
To choose the most suitable cell model for our studies, we characterized GR expression and function in several T- (CEM and K562) and B-lymphoma (NCEB, Granta and Jeko) cell lines that are widely used for the testing of novel chemotherapeutical drugs. First, we analyzed whether these cells harbor any GR mutations, as there are more than 40 mutation hot spots in GR exons that could modify response to glucocorticoids and contribute to glucocorticoid resistance.30-32 Direct sequencing did not reveal any genetic abnormalities in the GR coding region.
Next, we assessed GR protein expression and nuclear translocation in response to glucocorticoid dexamethasone (Dex), widely used for blood cancer treatment (Fig. 1A and B). In untreated cells, GR was preferentially localized in the cytoplasm, and Dex induced GR nuclear translocation in ALL studied cell lines. However, the basal level of GR expression and its nuclear accumulation were significantly higher in CEM and NCEB cells and correlated well with their higher sensitivity to Dex growth-inhibitory effect (Fig. 1A–C). Indeed, after 48 h, incubation with 1 µM Dex, the number of living CEM and NCEB cells was decreased by ~40% and 70%, respectively, while in K562, Granta and Jeko growth inhibition was insignificant, in the 10–20% range (Fig. 1C).

Figure 1. Expression and function of GR in leukemia and lymphoma cell lines. (A) Expression of GR in leukemia and lymphoma cells was determined in untreated cells by western blot analysis of whole-cell lysates. Membranes were probed with anti-actin antibodies to verify equal protein loading and transfer. Image digitizing and quantitative analysis of GR: actin normalized expression was performed by the Odyssey v 1.2 software. (B) Cells were treated with solvent (Control) or Dex for 24 h, and GR nuclear translocation was determined by western blot analysis of nuclear proteins. Membranes were probed with anti-HDAC-1 antibodies to verify equal protein loading and transfer. (C) Cells were treated with solvent (Control) or Dex for 48 h, and effect on cell growth was estimated by cell counting. * Statistically significant differences (p < 0.05) between Dex- and solvent-treated cells. (D) Generation of cells with GR knockout. Cells were infected with shGR- or empty vector-expressing lentiviruses. Expression of GR in CEM-shEV, CEM-shGR, NCEB-shEV and NCEB-shGR cells was determined by western blot analysis of whole-cell lysates. Membranes were probed with anti-actin antibodies to verify equal protein loading.
Based on these experiments, we have chosen CEM and NCEB cells to study ligand properties and anticancer effects of CpdA. To prove that CpdA effects are GR-dependent, we generated CEM and NCEB with blocked GR expression using stable infection with shGR lentivirus (Fig. 1D).
Cytotoxic effect of CpdA in transformed lymphoid cells depends on GR
To evaluate CpdA anti-lymphoma potential, we first assessed CpdA and Dex growth-inhibitory effects at concentration range of 10 nM–10 µM in CEM and NCEB cells (Fig. S2). We found that both GR ligands exerted significant cytostatic effects at similar 1–3 µM concentrations (Fig. 2A; Fig. S2).

Figure 2. Cytotoxic effect of CpdA in transformed lymphoid cell lines and primary leukemia patient cells. CEM-shEV, CEM-shGR, NCEB-shEV and NCEB-shGR were treated with solvent (control), Dex or CpdA for 48 h, and the effect on cell growth (A) was estimated by cell counting. The effect on apoptosis (B) was determined by flow cytometry using propidium iodide staining (the amount of cells in sub-G1-phase was calculated as percentage to all cells in sample) and by western blot analysis of cleaved PARP level. Membranes were probed with anti-HDAC-1 antibodies to verify the equal protein loading. Image digitizing and quantitative analysis of GR:HDAC-1 normalized expression were performed by the Odyssey v1.2 software. (C) Primary leukemia cells from four different T-ALL patients at acute stage of disease were treated with solvent (Control), Dex and CpdA for 48 h, and the growth was estimated by cell counting. (A–C) *Statistically significant differences (p < 0.05)—between Dex (or CpdA)—and solvent-treated cells. Note: (1) Effect of both GR ligands CpdA and Dex on transformed lymphoma cells growth depends on GR; (2) CpdA and Dex induced comparable cytoxic effect in primary T-ALL patient cells.
We also assessed CpdA cytotoxic effect in primary cell cultures obtained from the patients with acute lymphoblasic leukemia at the recurrence stage of disease. Remarkably, CpdA and Dex induced significant cell growth inhibition of primary cells at equimolar concentrations (Fig. 2C)
Next, we examined CpdA and Dex effects on cell survival, and found that both GR ligands induced apoptosis in NCEB and CEM cells. Indeed, flow cytometry analysis revealed that the number of apoptotic cells (sub-G1-phase cell population) reached 30–40% after 48 h incubation with CpdA or with Dex compared with 4–6% in control (Fig. 2B). These results were confirmed by western blot analysis of PARP cleavage, which revealed similar (in CEM cells), and stronger (in NCEB cells), CpdA cytotoxic effect compared with Dex (Fig. 2B).
Importantly, GR blockage by shGR-expressing lentivirus (Fig. 1D) resulted in a drastic loss of sensitivity to CpdA and Dex in lymphoma cells (Fig. 2A). These data provide the direct proof that CpdA anti-lymphoma effects depend on GR.
CpdA induces GR transrepression in lymphoma cells
CpdA properties as GR ligand were characterized in non-lymphoid cells.17-19,33 As gene expression regulation by GR is cell context-dependent and varies significantly in different cells and tissues, we assessed CpdA effect on GR transactivation and transrepression in comparison to Dex by Luciferase assay in CEM leukemia and NCEB lymphoma cells. To overcome notorious resistance of lymphoid cells to transient transfections, we generated cells stably infected with lentiviruses expressing GRE, NFkB and AP1 Luciferase reporters.
We found that Dex induced GR activity (evaluated in cells infected with GRE.Luc reporter) by 2–3 folds in CEM and ~20 folds in NCEB cells (Fig. 3A). At the same time, CpdA did not increase or slightly inhibited basal GR activity in both cell lines. Weak antagonistic effect of CpdA was most noticeable in NCEB cells, and was GR-dependent, as shown by lack of CpdA effect on GRE.Luciferase in cells infected with shGR-lentivirus (Fig. 3A).
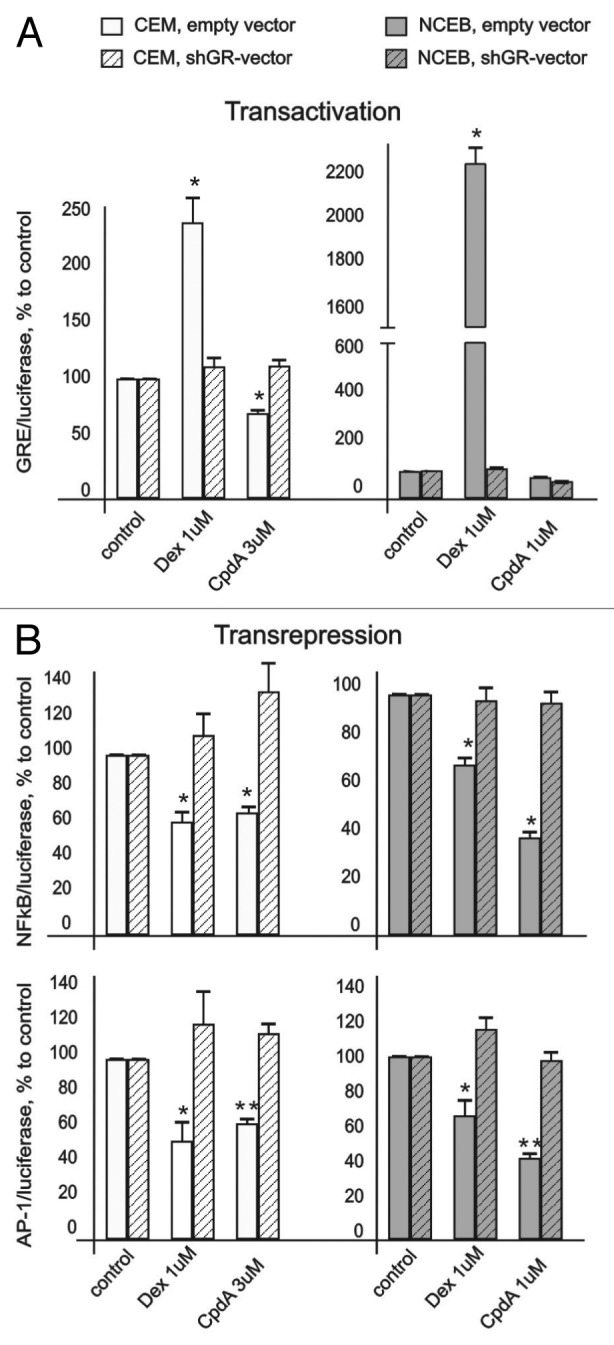
Figure 3. CpdA does not affect GR transactivation but induces GR transrepression in transformed lymphoid cells. CEM and NCEB cells stably infected with lentiviruses expressing luciferase reporters GRE.Luc (A), NFkB.Luc or AP1.Luc cells (B) were incubated for 8 h with solvent (Control), Dex or CpdA. Luciferase activity was determined as described in “Materials and Methods.” Statistically significant difference (*p < 0.05; ** p < 0.01) between Dex (or CpdA)- and solvent-treated cells.
It is well-accepted that GR transrepression is mostly mediated via negative interaction between GR and other transcription factors, including NFkB and AP-1.6 We revealed similar negative effects of both GR ligands on NFkB and AP-1 in lymphoid cells in luciferase assay. Basal activity of these transcription factors was inhibited by CpdA and Dex (Fig. 3B). Importantly, we proved that CpdA’s inhibitory effect on NFkB and AP-1 is GR-dependent. Indeed, CpdA, similar to Dex, was not able to decrease NFkB and AP-1 activity in NCEB and CEM cells with blocked GR expression (Fig. 3B). Overall, our observations are in line with previous reports suggesting that CpdA is a unique dissociated GR ligand.
CpdA does not induce cell desensitization
Chronic treatment with glucocorticoids frequently results in the development of resistance to steroids (tachyphylaxis).34 One of the important mechanisms of cell desensitization to glucocorticoids involves hormone-induced GR degradation.35 We compared effects of both ligands on GR stability in CEM and NCEB cells. Remarkably, in contrast to Dex, which induced significant GR degradation in both cell types, CpdA did not affect GR level within 24 h of treatment (Fig. 4A and B).
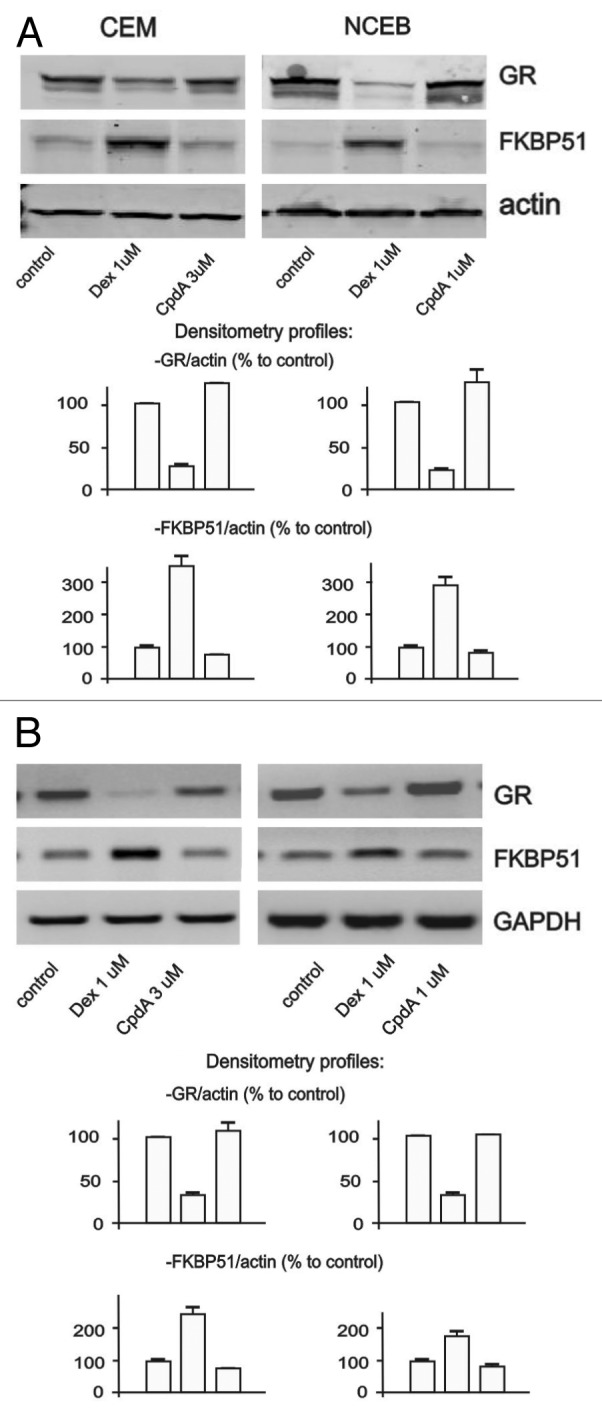
Figure 4. CpdA does not induce cell desensitization. Cells were treated with solvent (control), Dex and CpdA for 24 h. The expression of GR and FKBP51 was analyzed by western blotting of whole cell protein extracts (A), and by SQ-RT-PCR (B). To verify equal loading and adequate transfer in western blot analysis, the membranes were probed with anti-actin antibodies. Amounts of PCR-products were normalized to the amount of GAPDH PCR-product.
Another common mechanism of cell desensitization to glucocorticoids is mediated by a negative feedback loop via GR molecular chaperone FKBP51 (also called FK506-binding protein 536). FKBP51 sequesters GR in cytoplasm and prevents its nuclear translocation.37,38 We found that in contrast to Dex, CpdA did not activate FKBP51 gene/protein expression (Fig. 4A and B). Overall, our experiments showed that CpdA failed to induce two important mechanisms underlying development of cell resistance to activated GR signaling.
Bortezomib enhanced CpdA properties as “dissociated” GR ligand
It is known that proteasome inhibitors can stabilize GR both in steroid-treated and -untreated cells.26,35 Thus, we expected that BZ will accumulate GR protein and, consequently, enhance CpdA ligand properties, further shifting GR activity toward transrepression.
Indeed, we found that after a relatively short-time BZ treatment, the amount of GR protein was significantly increased in both CEM and NCEB cells (Fig. 5C). We next evaluated the effect of BZ on GR function modulation induced by CpdA in lymphoid cells. To reduce the cytotoxic effects on CpdA and BZ on Luciferase expression, we treated cells with BZ and CpdA at marginally toxic concentrations (as shown in Figs. S2 and 3) for a relatively short time (8 h).
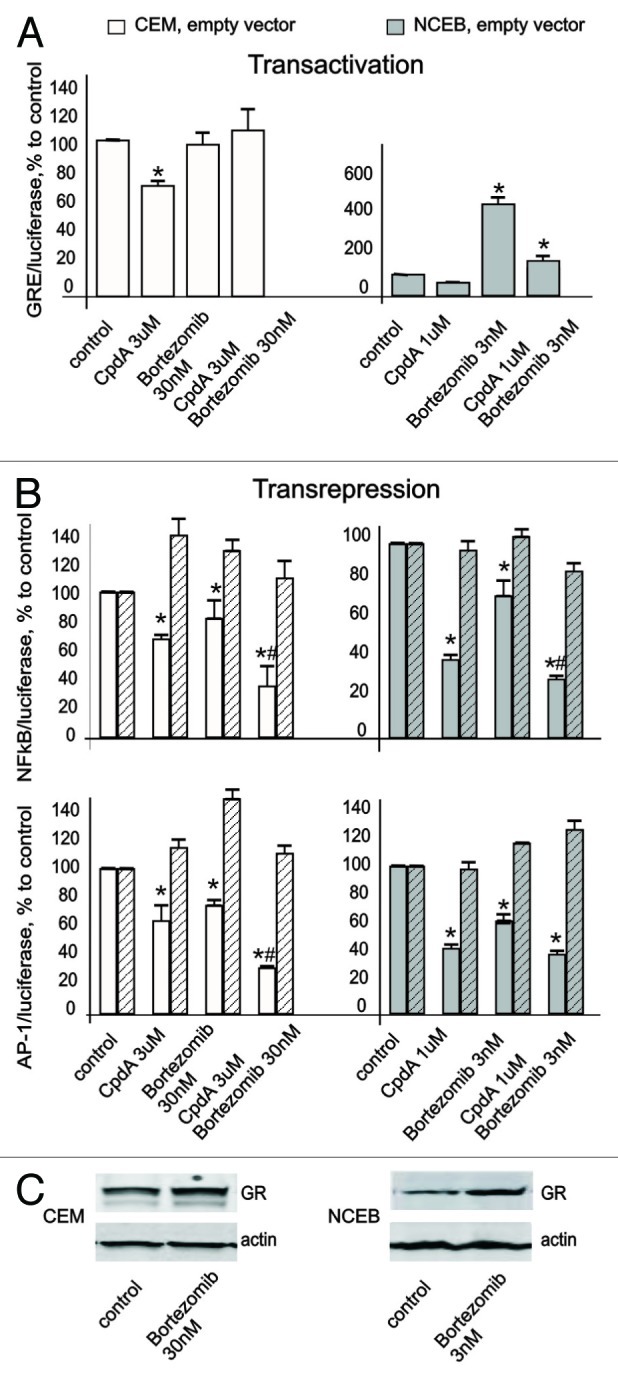
Figure 5. Bortezomib induced GR accumulation, and enhanced CpdA properties as “dissociated” GR ligand. CEM and NCEB cells stably infected with lentiviruses expressing luciferase reporters: GRE.Luc (A), NFkB.Luc or AP1.Luc cells (B) were incubated for 8 h with solvent (control), Bortezomib (BZ), CpdA or BZ + CpdA. Luciferase activity was determined as described in “Materials and Methods.” Statistically significant difference (*p < 0.05; ** p < 0.01) between treated- and control cells; # statistically significant difference (p < 0.05) between CpdA and BZ+CpdA. (C). Cells were incubated for 8 h with BZ or solvent (Control), and GR expression was determined by western blot analysis of whole CEM and NCEB cell extracts. To verify equal loading and adequate transfer, the membranes were probed with anti-actin antibodies.
It was shown previously that in some cells, BZ alone could induce GR nuclear translocation and activate GR. Using GRE.Luc reporter assay, we found that BZ either did not affect (in CEM) or induced (in NCEB) GR activity in lymphoid cells. However, GR activation by BZ in NCEB cells was 4–5 fold lower compared with activation induced by Dex (4–5-fold induction by CpdA vs. 20-fold induction by Dex, Figs. 3A and 5A). This is an important finding, suggesting that a large portion of GR in BZ-treated cells remained inactive in the absence of ligand. Thus, we assumed that the functional profile of accumulated GR will be defined by ligand. Indeed, the exposure of NCEB cells to CpdA+BZ resulted in decreased GR transactivation potential, compared with BZ only (Fig. 5A). In CEM cells, luciferase activity after BZ+CpdA treatment was similar to basal control level.
The results of NFkB and AP-1 luciferase assays showed that both CpdA and BZ in single treatments inhibited activity of those transcription factors (Fig. 5B). The effects of BZ were relatively weak, as we used BZ at low doses. The combined treatment with CpdA+BZ further inhibited NFkB and AP-1. This was especially pronounced in CEM cells, where CpdA and BZ strongly cooperated to block NFkB and AP-1 activity (Fig. 5B). The same trend was observed in NCEB cells, even though the difference between CpdA+BZ vs. CpdA only was not statistically significant (Fig. 5B). Importantly, the parallel experiments with shGR-expressing CEM and NCEB cells revealed that inhibitory effect of CpdA+BZ on NFkB and AP-1 activity significantly depended on GR.
Overall, these findings suggest that BZ exerts potentiating effects on CpdA-mediated GR transrepression, and that BZ+CpdA does not efficiently induce GR transactivation.
Cooperative anticancer effect of proteasome-inhibitor Bortezomib and CpdA in transformed lymphoid cells
To test our major hypothesis that BZ increases anticancer activity of CpdA via GR accumulation, we treated CEM and NCEB cells with CpdA and BZ at low, minimally toxic concentrations (1–3 μM as shown in Figs. S2 and 3) for 24–48 h. We observed remarkable cooperation between CpdA and BZ in induction of anticancer effects (Fig. 6). In both NCEB and CEM cells, growth inhibition by CpdA+BZ combination far exceeded that caused by CpdA or BZ in single treatments (Fig. 6A), and led to 80–90% inhibition of NCEB and CEM cell growth. The cytotoxic effect of all treatments, CpdA, BZ and CpdA+BZ, strongly depended on GR (Fig. 6A).

Figure 6. Cooperative anticancer effect of proteasome inhibitor Bortezomib and CpdA in transformed lymphoid cells. CEM-shEV, CEM-shGR, NCEB-shEV and NCEB-shGR cells were incubated with solvent, CpdA, BZ or CpdA/BZ for 48 h. (A) Cell growth was evaluated by cell counting. (B and C) Apoptosis in CEM and NCEB cells was analyzed by western blot analysis of PARP cleavage (B) and by flow cytometry using propidium iodide staining (C). The number of apoptotic cells was calculated as percentage to all cells in sample (C andD). Number of cells in S-phase was determined by flow cytometry using propidium iodide staining. Statistically significant difference (*p < 0.05; ** p < 0.01) between treated and control cells; #, statistically significant difference (p < 0.05) between CpdA and BZ+CpdA.
Using FACS analysis, we confirmed cooperation between CpdA and BZ in CEM and NCEB cells as assessed by (1) inhibition of proliferation defined by the number of cells in S phase, and (2) apoptosis evaluated as the increase in sub-G1-phase cell population (Fig. 6C and D). For example, CpdA+BZ treatment resulted in a 42% increase of apoptotic cell numbers in CEM cells, while individual application of these compounds gave rise to 4% and 27% cells in sub-G1-phase, accordingly (Fig. 6C). These findings were further supported by the results of PARP cleavage evaluated by western blotting (Fig. 6B).
As multiple myeloma (MM) represents a terminal stage of B cell differentiation, we investigated anti-lymphoma effect of CpdA in combination with BZ in multiple myeloma MM.1S (GR-positive, sensitive to glucocorticoids) and MM.1R (GR negative, resistant to glucocorticoids) cell lines. These MM.1 cell lines were established from myeloma patients who failed chemotherapy.2 MM.1 cell lines were extensively characterized and are widely used for the pre-clinical studies.2 We established MM.1R-GR cell line stably infected with GR-expressing lentivirus to further address GR-dependence of CpdA anticancer effects (Fig. 7).

Figure 7. Cooperative GR-dependent anticancer effect of proteasome-inhibitor Bortezomib and CpdA in multiple myeloma cells. (A) Expression of GR in MM cells. MM1.S, MM1.R and MM.1R-GR cells stably infected with GR-expressing lentivirus was determined by western blot analysis of whole-cell lysates. (B and C) GR-dependent cytotoxic effect of GR ligands FA and CpdA in MM cells. MM1.S and MM1.R, were treated with solvent (control), FA or CpdA for 72 h, and the effect on apoptosis (B) was determined by western blot analysis of PARP cleavage after 36 h of treatment. The effect on cell growth (B) was estimated by cell counting. (D) MM.1S cells are more sensitive to cytotoxic effect of BZ than MM.1R cells. MM cells with different GR status were treated with BZ for 72 h, and the number of cells/well was counted. (E) Cooperative anti-MM effect of CpdA and BZ depends on GR. MM.1R and MM.1S cells were incubated with solvent, CpdA, BZ or CpdA/BZ for 72 h. Cell growth was evaluated by cell counting. Apoptosis in MM cells was analyzed by western blot analysis of PARP cleavage. Note: MM.1R cells are resistant and MM.1S and MM.1R-GR cells are sensitive to cytotoxic and cytostatic effect of CpdA and FA.
As MM cells are very sensitive to glucocorticoids and BZ, we modified the treatment protocol and used lower doses of both compounds (up to 10−10 M). As shown in Figure 1, MM.1R cells were resistant to both glucocorticoid fluocinolone acetonide (FA) and GR modulator CpdA tested at a wide range of concentrations in growth inhibition and apoptosis assays. After we expressed exogenous GR in MM.1R cells, they gained sensitivity to apoptosis (Fig. 7) and growth inhibition (data not shown) induced by both GR ligands. Importantly, via comparison of sensitivity of multiple myeloma cells with different GR status to BZ cytotoxic effects, we confirmed one of our important observations that BZ’s anticancer effect depends on GR, which suggests that GR is a novel molecular target for proteasome inhibitors in cancer cells.
Finally, using MM.1S and MM.1R cells, we confirmed GR-dependent cooperation between anticancer effects of BZ and CpdA (Fig. 7). Overall, our observations made in multiple myeloma cell models strongly support findings in lymphoma cells.
Discussion
There is a continuous interest in the development of new selective GR activators, SEGRA, that preserve therapeutic potential of classical steroids yet have reduced adverse effects. From the molecular point of view, “dissociated” GR modulators that do not support GR dimerization, but activate GR transrepression via tethering of monomeric GR to other transcription factors and should have improved effect/side effect ratio (therapeutic index).39 This molecular concept is strongly supported by the results obtained in GRdim-knocking mice with normal GR transrepression, but severely impaired GR dimerization and transactivation. These animals expressing the GR dimerization mutant appeared to be very sensitive to anti-inflammatory, but not to the side effects of glucocorticoids.19
CpdA represents a remarkable example of such “dissociated” GR ligand. The unique ligand properties of CpdA are largely attributed to its inability to induce GR dimerization.20 Other mechanisms that add to CpdA “dissociated” profile include recruitment of cofactors such as NCoR or SMRT that inhibit GR-dependent transcription, and extensive export of GR bound to CpdA from the nucleus, resulting in the decreased amount of nuclear GR necessary for transactivation.18,20,40 These changes in GR functions elicited by CpdA probably reflect the conformational changes in GR molecule compared with GR activated by steroids; yet the structure of GR ligand-binding domain bound to CpdA remains to be resolved.
Traditionally, the focus in the search for novel dissociated GR ligands has been on compounds for the treatment of chronic inflammatory diseases.15,41 The anticancer effect of SEGRA has not been well-investigated. In this work, we (1) tested the anti-lymphoma potential of CpdA, and (2) corroborated our hypothesis that proteasome inhibitors enhance CpdA ligand properties and anticancer activity.
Overall, our results obtained in lymphoid malignant cells further confirmed the unique CpdA capability to shift GR activity toward transrepression (Figs. 3 and 4). We also found that CpdA strongly inhibited both growth and viability of CEM, NCEB and MM1.S transformed cells in a GR-dependent manner. Importantly, lymphoma cell lines and primary ALL cell cultures appeared equally sensitive to anticancer effects of CpdA and glucocorticoids Dex and FA (Figs. 2 and 7).
This is an unexpected finding, as CpdA usually exerts biological effects at higher doses than glucocorticoids both in vitro and in vivo.17,18,42 Moreover, the similar potential of CpdA and Dex to inhibit growth and apoptosis highlights the important role of GR transrepression in anti-lymphoma effects of GR ligands.
As discussed above, CpdA has fewer side effects in vivo compared with glucocorticoids.19,21-23 CpdA is also less likely to induce therapy resistance, as cell exposure to CpdA does not lead to homologous GR downregulation (Fig. 4). It is possible that the altered pattern of GR phosphorylation induced by CpdA compared with classical glucocorticoids does not initiate ubiquitination and typical proteasome degradation of receptor. CpdA also does not activate FKBP51 (Fig. 4), the direct GR target gene43 involved in feedback control of GR signaling via retention of GR in the cytoplasm. Interestingly, high level of FKBP51 is associated with glucocorticoid resistance in patients with brain and prostate cancer, melanomas and lymphomas.44,45
We hypothesized that proteasome inhibitor BZ induces GR accumulation, augments CpdA effects as a selective GR modulator and consequently maximizes CpdA anticancer activity. This hypothesis is based on the previous findings that 26S proteasome inhibitors stabilize GR in the presence and in the absence of glucocorticoids.26,37,46,47
Definitely, BZ treatment increased GR protein levels in T- and B-lymphoid cells; however, the significant part of accumulated GR after BZ treatment remained inactive (Fig. 5). We assumed that in such a case, the ligand would ultimately define GR functional profile. Indeed, when combined with BZ, CpdA only weakly induced GR transactivation, but strongly augmented transrepression assessed by inhibition of NFkB and AP-1(Fig. 5A,B). Interestingly, even though there are other known molecular mechanisms of NFkB blockage by proteasome inhibitors, including stabilization of its inhibitor IkBalpha,4 the effect of BZ+/− CpdA on NFkB activity in lymphoid cells was at least partially GR-dependent (Fig. 5). It was reported previously that in some cells, proteasome inhibitors could activate AP-1 via decreased turnover of its subunits, the proteins from Jun/Fos families.50 In our experiments, BZ at low sub-toxic doses inhibited activity of AP-1 (Fig. 5). We observed strong GR-dependent cooperation between CpdA and BZ in inhibition of AP-1 and NFkB (Fig. 5). Overall, our results strongly, even though indirectly, suggest that BZ may induce accumulation of significant amounts of monomeric GR whose function is defined by the ligand.
Although the ability of proteasome inhibitors to modulate GR activity is known, the role of GR in the proteasome-induced apoptosis has never been considered. The comparison of BZ cytotoxic effects in syngenic lymphoid cells, with and without GR, revealed that GR is critically important for BZ anticancer activity. These results are in agreement with our recent findings in prostate carcinoma cells25 and allow us to establish GR as a novel molecular target of proteasome inhibitors in cancer cells.
In this work, we also demonstrated the remarkable cooperation between CpdA and BZ in their anticancer effects assessed by different growth and apoptosis tests (Figs. 6 and 7). These data are especially compelling in the case of multiple myeloma, suggesting the potential for both CpdA and BZ dose reduction when they are used in combination. The latter is important, because in vivo, as stand-alone agents, both are effective at high, maximally tolerated doses (MTD), and in the clinic BZ doses also approach MTD, with multiple adverse effects.3,21,23
The endoplasmic reticulum (ER) stress as a part of more general proteotoxic stress is induced in cells by accumulation of un- or misfolded proteins.49,50 Protracted lethal ER stress is one of the major molecular mechanisms underlying the anticancer effects of proteasome inhibitors, and there is an extensive search for the approaches/compounds to further sensitize cancer cells to ER stress induced by proteasome inhibitors.51,52 In our recent work, we showed that CpdA enhanced ER stress induced by BZ.25 The involvement of ER stress in cooperation between BZ and CpdA in lymphoma cells remains to be investigated.
In conclusion, CpdA represents first GR modulator, which abrogates GR dimerization. Results presented here along with literature data indicate that CpdA has a unique and very beneficial pharmacological profile: it retains the therapeutic anti-inflammatory and anticancer potential of glucocorticoids, but induces fewer side effects and will less likely induce patient resistance to the treatments. This makes CpdA a very attractive candidate for further investigations of future therapeutic applications in hematological oncology. We also demonstrated here that pretreatment with proteasome inhibitors, followed by selective GR modulators like CpdA, could release the anticancer GR signaling at its maximal potential. This approach establishes a novel strategy for GR-targeted chemotherapy of hematologic malignancies.
Materials and Methods
Reagents
2-(4-acetoxyphenyl)-2-chloro-N-methyl-ethylammonium chloride (CpdA) was synthesized from (±) Synephrine and acetyl chloride in glacial acetic acid, as described before.53 Synthetic glucocorticoids dexamethasone (Dex) and fluocinolone acetonide (FA) were obtained from Sigma-Aldric, Bortezomib (BZ) was from Millenium Pharmaceuticals.
Cell cultures and treatments
CEM T-cell acute lymphoblastic leukemia (kind gift from Dr. Posypanova, Moscow Research Institute of Medical Ecology), K562 chronic myeloblastic leukemia cells and NCEB mantle cell lymphoma cells (ATCC), Granta and Jeko mantle cell lymphoma cells (kind gift from Dr. S. Bernstein, University of Rochester) and multiple myeloma MM.1R (glucocorticoid resistant) and MM.1S (glucocorticoid sensitive) cells (established by S. Rosen2) were cultured in RPMI-1640 (Life Technologies) with 10% FBS (HyClone), sodium piruvate (10 mM), HEPES (10 mM) and antibiotics (Gibco BRL Life Technologies). Peripheral blood samples were collected from 1–10-y-old pediatric patients with newly diagnosed T-ALL in acute phase of disease at Blokhin Cancer Research Center. The ethical Committee of the Blokhin Cancer Research Center approved this study, and parents or guardians of the patients provided written informed consent. Leukemic cells were isolated by 1.077 g/ml Ficoll-Isopaque density-gradient centrifugation and cultured in the complete medium.
Lentiviral technology
Lentiviral stocks were generated using lentiviral expression vectors as described previously11,54: pGIPZ-encoding shRNA against GR or empty vector as control (Open Biosystems); AP1-Luc, NFkB.Luc, GRE.Luc-encoding Firefly Luciferase reporters or Firefly Luciferase under minimal CMV promoter as control (System Biosciences). PL6-V5/TOPO-encoding GR (Invitrogen) was generated as described previously.11 Lymphoma cell lines stably infected with lentiviruses were selected using 6 ug/ul blasticidin or 5 ug/ul puromicin.
Luciferase assay
Cells stably expressing Firefly Luciferase under NFkB, AP-1 or GRE promoters, were treated for 8 h with CpdA, Dex, Bortezomib or appropriate vehicle (0.01% DMSO and/or 0.01% ethanol). Luciferase activity was measured using commercial Luciferase Assay (Promega, Corp.) and Luminometer TD 20/20 (Turner Design Instruments). Cells stably expressing Firefly Luciferase under minimal CMV promoter were used as a control to adjust for non-specific toxicity.
Sequencing
DNA fragments obtained after PCR amplification of GR exons 2–9 of CEM, K562, NCEB, Granta and Jeko cells were sequenced on automatic sequencer ABI PRISM 3730 (Applied Biosystems) using Big Dye Sequencing Kit (Perkin-Elmer) according to manufacturer protocol. Samples were analyzed by ABI-377 DNA Sequencer Software.
Western blot analysis
Western blot analysis of whole-cell extracts, cytoplasmic and nuclear fractions was performed as described previously.11 Proteins were resolved by SDS-PAGE. The following antibodies were used: anti-GR, (Santa Cruz Biotechnology), anti-poly(ADP-ribose) polymerase (PARP; BD PharMingen) and anti- FK506 binding protein 5 (FKBP5; Santa Cruz Biotechnology). Membranes were blocked with 5% Blotto in TBS and incubated with primary antibody overnight at 4°C, followed by IRDye 800CW and IRDye 680CW-conjugated anti-rabbit/anti-mouse IgG secondary antibodies (LI-COR). To verify equal protein loading and adequate transfer, the membranes were probed with anti-actin or histone deacetylase-1 (HDAC-1) antibodies (Santa Cruz Biotechnology). Protein bands were visualized by Odyssey IR Scanner (LI-COR) with 680-nm and 800-nm scanner channel. Quantitative analysis was performed by Odyssey v1.2 software (LI-COR).
PCR
PCR of GR exons 2–9 was performed using Taq DNA Polymerase, PCR-Supermix (Invitrogen Corp.), appropriate PCR primers (Fig. S1) and genomic DNA isolated by TRIzol Reagent (Invitrogen Corp.) according to the manufacturer protocol. The PCR primers were designed using the Primer-Bank database (www.pga.mgh.harvard.edu/primerbank/) and Oligo 6 software.
Semiquantitative RT-PCR (SQ-RT-PCR)
SQ-RT-PCR was performed using reverse MLV transcriptase with random primers and PCR-Supermix (Invitrogen Corp.) with appropriate PCR primers (Fig. S1). Total RNA was isolated with RNAeasy kit (Qiagen). PCR products were run on 1.5% agarose gels, the actual amount of PCR products was measured by Quantity One 1-D Analysis Software and normalized to the amount of glyceraldehydes-3-phosphatase dehydrogenase (GAPDH) PCR products.
Proliferation assay
The proliferation was measured by direct cell counting using Celigo Cytometer (Cyntellect Inc.). Cells were plated at 104 cells/well onto 24-well plates and cultured in complete medium in the presence of CpdA, Dex, Bortezomib or vehicle (0.1% ethanol or DMSO).
Apoptosis detection
To evaluate apoptosis, we used western blot analysis of PARP cleavage and PI staining. Cells were cultured on 10-cm dishes and treated with CpdA, Dex, Bortezomib or vehicle for 16–48 h. PARP cleavage was evaluated by western blot analysis with anti-PARP antibody using nuclear protein extracts. For PI staining, cells were resuspended in 70% ethanol, fixed for 2 h at -20°C, placed in PBS containing 5 μL PI, 0.1% sodium citrate and 0.3% NP-40 and incubated for 30 min at room temperature. Analysis by FACScan flow cytometer (Becton Dickinson) was performed to discriminate between live and apoptotic cells.
Statistical analysis
All experiments were repeated at least three times. Mean ± SE values were calculated using Microsoft Excel software and compared using Student’s t-test. P value of 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We are grateful to Dr. O. Volpert (Northwestern University) for fruitful discussion of our work. We thank SDRC DNA/RNA delivery Core and RHLCCC Flow Cytometry Core Facility for technical support. Work is supported by grants: RO1CA118890, SPORE in Prostate Cancer, P30 CA090386, Northwestern University RHLCCC (to I.B.), ACS IL #160185 (to A.Y.), RFBR # 10–04–00979 (to M.Y.), UICC ICRETT-09–137 (to E.L.), EACR Travel Fellowship Award (to E.L.), RFBR grant 12–04–31148 (to E.L.).
Glossary
Abbreviations:
- AP-1
activator protein 1
- BZ
Bortezomib
- CpdA
Compound A
- Dex
Dexamethason
- FKBP5
FK506 binding protein 5
- FA
fluocinolone acetonide
- GR
glucocorticoid receptor
- GRE
glucocorticoid responsive element
- HDAC1
histone deacetylase-1
- NFκB
nuclear factor kappa-B
- PARP
poly (ADP-ribose) polymerase
- SEGRA
selective glucocorticoid receptor activator
- SQ-RT-PCR
semi-quantitative reverse transcription polymerase chain reaction
- TF
transcription factor
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Notes
E.L. designed and performed research, analyzed and interpreted data, prepared figures; A.Y. designed and performed research, analyzed and interpreted data, prepared figures; K.K. performed research, analyzed and interpreted data; A.P. collected data, analyzed and interpreted data; G.B. analyzed and interpreted data; M.Y. designed research, analyzed and interpreted data; L.G. analyzed and interpreted data; S.R. analyzed and interpreted data; I.B. designed research, analyzed and interpreted data, wrote the manuscript.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/23048
References
- 1.Rosenthal MC, Saunders RH, Schwartz LI, Zannos L, Perez Santiago E, Dameshek W. The use of adrenocorticotropic hormone and cortisone in the treatment of leukemia and leukosarcoma. Blood. 1951;6:804–23. [PubMed] [Google Scholar]
- 2.Greenstein S, Krett NL, Kurosawa Y, Ma C, Chauhan D, Hideshima T, et al. Characterization of the MM.1 human multiple myeloma (MM) cell lines: a model system to elucidate the characteristics, behavior, and signaling of steroid-sensitive and -resistant MM cells. Exp Hematol. 2003;31:271–82. doi: 10.1016/S0301-472X(03)00023-7. [DOI] [PubMed] [Google Scholar]
- 3.Moreau P, Avet-Loiseau H, Facon T, Attal M, Tiab M, Hulin C, et al. Bortezomib plus dexamethasone versus reduced-dose bortezomib, thalidomide plus dexamethasone as induction treatment before autologous stem cell transplantation in newly diagnosed multiple myeloma. Blood. 2011;118:5752–8, quiz 5982. doi: 10.1182/blood-2011-05-355081. [DOI] [PubMed] [Google Scholar]
- 4.McConkey DJ, Zhu K. Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist Updat. 2008;11:164–79. doi: 10.1016/j.drup.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 5.Schlossmacher G, Stevens A, White A. Glucocorticoid receptor-mediated apoptosis: mechanisms of resistance in cancer cells. J Endocrinol. 2011;211:17–25. doi: 10.1530/JOE-11-0135. [DOI] [PubMed] [Google Scholar]
- 6.Chebotaev D, Yemelyanov A, Budunova I. The mechanisms of tumor suppressor effect of glucocorticoid receptor in skin. Mol Carcinog. 2007;46:732–40. doi: 10.1002/mc.20349. [DOI] [PubMed] [Google Scholar]
- 7.Adcock IM. Glucocorticoid-regulated transcription factors. Pulm Pharmacol Ther. 2001;14:211–9. doi: 10.1006/pupt.2001.0283. [DOI] [PubMed] [Google Scholar]
- 8.Barnes PJ. Glucocorticosteroids: current and future directions. Br J Pharmacol. 2011;163:29–43. doi: 10.1111/j.1476-5381.2010.01199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Necela BM, Cidlowski JA. Mechanisms of glucocorticoid receptor action in noninflammatory and inflammatory cells. Proc Am Thorac Soc. 2004;1:239–46. doi: 10.1513/pats.200402-005MS. [DOI] [PubMed] [Google Scholar]
- 10.De Bosscher K, Vanden Berghe W, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev. 2003;24:488–522. doi: 10.1210/er.2002-0006. [DOI] [PubMed] [Google Scholar]
- 11.Yemelyanov A, Czwornog J, Chebotaev D, Karseladze A, Kulevitch E, Yang X, et al. Tumor suppressor activity of glucocorticoid receptor in the prostate. Oncogene. 2007;26:1885–96. doi: 10.1038/sj.onc.1209991. [DOI] [PubMed] [Google Scholar]
- 12.Chebotaev D, Yemelyanov A, Zhu L, Lavker RM, Budunova I. The tumor suppressor effect of the glucocorticoid receptor in skin is mediated via its effect on follicular epithelial stem cells. Oncogene. 2007;26:3060–8. doi: 10.1038/sj.onc.1210108. [DOI] [PubMed] [Google Scholar]
- 13.Schäcke H, Schottelius A, Döcke WD, Strehlke P, Jaroch S, Schmees N, et al. Dissociation of transactivation from transrepression by a selective glucocorticoid receptor agonist leads to separation of therapeutic effects from side effects. Proc Natl Acad Sci USA. 2004;101:227–32. doi: 10.1073/pnas.0300372101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schäcke H, Zollner TM, Döcke WD, Rehwinkel H, Jaroch S, Skuballa W, et al. Characterization of ZK 245186, a novel, selective glucocorticoid receptor agonist for the topical treatment of inflammatory skin diseases. Br J Pharmacol. 2009;158:1088–103. doi: 10.1111/j.1476-5381.2009.00238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu X, Du S, Tunca C, Braden T, Long KR, Lee J, et al. The antagonists but not partial agonists of glucocorticoid receptor ligands show substantial side effect dissociation. Endocrinology. 2011;152:3123–34. doi: 10.1210/en.2010-1447. [DOI] [PubMed] [Google Scholar]
- 16.Swart P, Swart AC, Louw A, van der Merwe KJ. Biological activities of the shrub Salsola tuberculatiformis Botsch.: contraceptive or stress alleviator? Bioessays. 2003;25:612–9. doi: 10.1002/bies.10285. [DOI] [PubMed] [Google Scholar]
- 17.De Bosscher K, Vanden Berghe W, Beck IM, Van Molle W, Hennuyer N, Hapgood J, et al. A fully dissociated compound of plant origin for inflammatory gene repression. Proc Natl Acad Sci USA. 2005;102:15827–32. doi: 10.1073/pnas.0505554102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yemelyanov A, Czwornog J, Gera L, Joshi S, Chatterton RT, Jr., Budunova I. Novel steroid receptor phyto-modulator compound a inhibits growth and survival of prostate cancer cells. Cancer Res. 2008;68:4763–73. doi: 10.1158/0008-5472.CAN-07-6104. [DOI] [PubMed] [Google Scholar]
- 19.Dewint P, Gossye V, De Bosscher K, Vanden Berghe W, Van Beneden K, Deforce D, et al. A plant-derived ligand favoring monomeric glucocorticoid receptor conformation with impaired transactivation potential attenuates collagen-induced arthritis. J Immunol. 2008;180:2608–15. doi: 10.4049/jimmunol.180.4.2608. [DOI] [PubMed] [Google Scholar]
- 20.Robertson S, Allie-Reid F, Vanden Berghe W, Visser K, Binder A, Africander D, et al. Abrogation of glucocorticoid receptor dimerization correlates with dissociated glucocorticoid behavior of compound a. J Biol Chem. 2010;285:8061–75. doi: 10.1074/jbc.M109.087866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Loo G, Sze M, Bougarne N, Praet J, Mc Guire C, Ullrich A, et al. Antiinflammatory properties of a plant-derived nonsteroidal, dissociated glucocorticoid receptor modulator in experimental autoimmune encephalomyelitis. Mol Endocrinol. 2010;24:310–22. doi: 10.1210/me.2009-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wüst S, Tischner D, John M, Tuckermann JP, Menzfeld C, Hanisch UK, et al. Therapeutic and adverse effects of a non-steroidal glucocorticoid receptor ligand in a mouse model of multiple sclerosis. PLoS One. 2009;4:e8202. doi: 10.1371/journal.pone.0008202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rauner M, Goettsch C, Stein N, Thiele S, Bornhaeuser M, De Bosscher K, et al. Dissociation of osteogenic and immunological effects by the selective glucocorticoid receptor agonist, compound A, in human bone marrow stromal cells. Endocrinology. 2011;152:103–12. doi: 10.1210/en.2010-0456. [DOI] [PubMed] [Google Scholar]
- 24.Geley S, Hartmann BL, Hala M, Strasser-Wozak EM, Kapelari K, Kofler R. Resistance to glucocorticoid-induced apoptosis in human T-cell acute lymphoblastic leukemia CEM-C1 cells is due to insufficient glucocorticoid receptor expression. Cancer Res. 1996;56:5033–8. [PubMed] [Google Scholar]
- 25.Yemelyanov A, Bhalla P, Yang X, Ugolkov A, Iwadate K, Karseladze A, et al. Differential targeting of androgen and glucocorticoid receptors induces ER stress and apoptosis in prostate cancer cells: a novel therapeutic modality. Cell Cycle. 2012;11:395–406. doi: 10.4161/cc.11.2.18945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wallace AD, Cao Y, Chandramouleeswaran S, Cidlowski JA. Lysine 419 targets human glucocorticoid receptor for proteasomal degradation. Steroids. 2010;75:1016–23. doi: 10.1016/j.steroids.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kofler R, Schmidt S, Kofler A, Ausserlechner MJ. Resistance to glucocorticoid-induced apoptosis in lymphoblastic leukemia. J Endocrinol. 2003;178:19–27. doi: 10.1677/joe.0.1780019. [DOI] [PubMed] [Google Scholar]
- 28.Gross KL, Oakley RH, Scoltock AB, Jewell CM, Cidlowski JA. Glucocorticoid receptor alpha isoform-selective regulation of antiapoptotic genes in osteosarcoma cells: a new mechanism for glucocorticoid resistance. Mol Endocrinol. 2011;25:1087–99. doi: 10.1210/me.2010-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gross KL, Lu NZ, Cidlowski JA. Molecular mechanisms regulating glucocorticoid sensitivity and resistance. Mol Cell Endocrinol. 2009;300:7–16. doi: 10.1016/j.mce.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruiz M, Lind U, Gåfvels M, Eggertsen G, Carlstedt-Duke J, Nilsson L, et al. Characterization of two novel mutations in the glucocorticoid receptor gene in patients with primary cortisol resistance. Clin Endocrinol (Oxf) 2001;55:363–71. doi: 10.1046/j.1365-2265.2001.01323.x. [DOI] [PubMed] [Google Scholar]
- 31.Beesley AH, Weller RE, Senanayake S, Welch M, Kees UR. Receptor mutation is not a common mechanism of naturally occurring glucocorticoid resistance in leukaemia cell lines. Leuk Res. 2009;33:321–5. doi: 10.1016/j.leukres.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 32.Irving JA, Minto L, Bailey S, Hall AG. Loss of heterozygosity and somatic mutations of the glucocorticoid receptor gene are rarely found at relapse in pediatric acute lymphoblastic leukemia but may occur in a subpopulation early in the disease course. Cancer Res. 2005;65:9712–8. doi: 10.1158/0008-5472.CAN-05-1227. [DOI] [PubMed] [Google Scholar]
- 33.De Bosscher K, Vanden Berghe W, Haegeman G. Cross-talk between nuclear receptors and nuclear factor kappaB. Oncogene. 2006;25:6868–86. doi: 10.1038/sj.onc.1209935. [DOI] [PubMed] [Google Scholar]
- 34.van Rossum EF, van den Akker EL. Glucocorticoid resistance. Endocr Dev. 2011;20:127–36. doi: 10.1159/000321234. [DOI] [PubMed] [Google Scholar]
- 35.Wallace AD, Cidlowski JA. Proteasome-mediated glucocorticoid receptor degradation restricts transcriptional signaling by glucocorticoids. J Biol Chem. 2001;276:42714–21. doi: 10.1074/jbc.M106033200. [DOI] [PubMed] [Google Scholar]
- 36.Wiederrecht G, Hung S, Chan HK, Marcy A, Martin M, Calaycay J, et al. Characterization of high molecular weight FK-506 binding activities reveals a novel FK-506-binding protein as well as a protein complex. J Biol Chem. 1992;267:21753–60. [PubMed] [Google Scholar]
- 37.Stechschulte LA, Sanchez ER. FKBP51-a selective modulator of glucocorticoid and androgen sensitivity. Curr Opin Pharmacol. 2011;11:332–7. doi: 10.1016/j.coph.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chun E, Lee HS, Bang BR, Kim TW, Lee SH, Kim JH, et al. Dexamethasone-induced FKBP51 expression in peripheral blood mononuclear cells could play a role in predicting the response of asthmatics to treatment with corticosteroids. J Clin Immunol. 2011;31:122–7. doi: 10.1007/s10875-010-9463-9. [DOI] [PubMed] [Google Scholar]
- 39.Schäcke H, Berger M, Rehwinkel H, Asadullah K. Selective glucocorticoid receptor agonists (SEGRAs): novel ligands with an improved therapeutic index. Mol Cell Endocrinol. 2007;275:109–17. doi: 10.1016/j.mce.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 40.Tanner T, Claessens F, Haelens A. The hinge region of the androgen receptor plays a role in proteasome-mediated transcriptional activation. Ann N Y Acad Sci. 2004;1030:587–92. doi: 10.1196/annals.1329.068. [DOI] [PubMed] [Google Scholar]
- 41.López FJ, Ardecky RJ, Bebo B, Benbatoul K, De Grandpre L, Liu S, et al. LGD-5552, an antiinflammatory glucocorticoid receptor ligand with reduced side effects, in vivo. Endocrinology. 2008;149:2080–9. doi: 10.1210/en.2007-1353. [DOI] [PubMed] [Google Scholar]
- 42.Gossye V, Elewaut D, Bougarne N, Bracke D, Van Calenbergh S, Haegeman G, et al. Differential mechanism of NF-kappaB inhibition by two glucocorticoid receptor modulators in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2009;60:3241–50. doi: 10.1002/art.24963. [DOI] [PubMed] [Google Scholar]
- 43.U M, Shen L, Oshida T, Miyauchi J, Yamada M, Miyashita T, U M Identification of novel direct transcriptional targets of glucocorticoid receptor. Leukemia. 2004;18:1850–6. doi: 10.1038/sj.leu.2403516. [DOI] [PubMed] [Google Scholar]
- 44.Li L, Lou Z, Wang L. The role of FKBP5 in cancer aetiology and chemoresistance. Br J Cancer. 2011;104:19–23. doi: 10.1038/sj.bjc.6606014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W, et al. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell. 2009;16:259–66. doi: 10.1016/j.ccr.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kinyamu HK, Jefferson WN, Archer TK. Intersection of nuclear receptors and the proteasome on the epigenetic landscape. Environ Mol Mutagen. 2008;49:83–95. doi: 10.1002/em.20360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, DeFranco DB. Alternative effects of the ubiquitin-proteasome pathway on glucocorticoid receptor down-regulation and transactivation are mediated by CHIP, an E3 ligase. Mol Endocrinol. 2005;19:1474–82. doi: 10.1210/me.2004-0383. [DOI] [PubMed] [Google Scholar]
- 48.Adler J, Reuven N, Kahana C, Shaul Y. c-Fos proteasomal degradation is activated by a default mechanism, and its regulation by NAD(P)H:quinone oxidoreductase 1 determines c-Fos serum response kinetics. Mol Cell Biol. 2010;30:3767–78. doi: 10.1128/MCB.00899-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Neznanov N, Komarov AP, Neznanova L, Stanhope-Baker P, Gudkov AV. Proteotoxic stress targeted therapy (PSTT): induction of protein misfolding enhances the antitumor effect of the proteasome inhibitor bortezomib. Oncotarget. 2011;2:209–21. doi: 10.18632/oncotarget.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grant S. Enhancing proteotoxic stress as an anticancer strategy. Oncotarget. 2011;2:284–6. doi: 10.18632/oncotarget.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fulda S. Novel insights into the synergistic interaction of Bortezomib and TRAIL: tBid provides the link. Oncotarget. 2011;2:418–21. doi: 10.18632/oncotarget.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pandit B, Gartel AL. FoxM1 knockdown sensitizes human cancer cells to proteasome inhibitor-induced apoptosis but not to autophagy. Cell Cycle. 2011;10:3269–73. doi: 10.4161/cc.10.19.17735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bretschneider H, Biemann K, Sachsenmaier W, Phenylalkanolamines V. Chlorophenethylamine derivatives and their conversion into halogen-free compounds. Monatsh Chem. 1948;78:82–116. doi: 10.1007/BF00942491. [DOI] [Google Scholar]
- 54.Zufferey R, Dull T, Mandel RJ, Bukovsky A, Quiroz D, Naldini L, et al. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol. 1998;72:9873–80. doi: 10.1128/jvi.72.12.9873-9880.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.