

Abstract
Purpose
Management of sepsis in critically ill patients remains difficult and requires prolonged intensive care. Genetic testing has been proposed as a strategy to identify patients at risk for adverse outcome of critical illnesses. Therefore, we wished to determine the influence of heredity on predisposition to poor outcome and on duration of ventilator support of intensive care unit (ICU) patients.
Methods
A study was conducted from July 2001 to December 2005 in heterogeneous population of patients from 12 US ICUs represented by the Genetic Predisposition to Severe Sepsis (GenPSS) archive. In 1057 Caucasian critically ill patients with SAPS II probability of survival of >0.2 in the US, six functional single nucleotide polymorphisms in relation to inflammatory cytokines and innate immunity (rs1800629, rs16944, rs1800795, rs1800871, rs2569190, and rs909253) were evaluated in terms of mortality and ventilator free days.
Results
The AA homozygote of TNF(−308) (rs1800629) was most over-represented in the deceased patient group (P = 0.015 with recessive model). The carriage of the TNF(−308)* AA genotype showed significantly higher odds ratio of 2.67(1.29–5.55) (P = 0.008) after adjustment with the covariates. However, the presence of 1, 2, or 3 acute organ dysfunctions was larger prognostic factors for the adverse outcome (OR(95%CI) = 2.98(2.00–4.45), 4.01(2.07–7.77), or 19.95(4.99–79.72), P < 0.001 for all). Kaplan–Mayer plot on ventilator duration of TNF(−308)* AA patient significantly diverged from that of TNF(−308)* (GG + GA) ((AA v GG + GA), Adjusted HR(95%CI) = 2.53(1.11–5.79) with Cox regression, P = 0.028).
Conclusions
TNF(−308)* AA is significantly associated with susceptibility to adverse outcome and to longer ventilator duration. Therefore, heredity likely affects both predisposition to ICU prognosis as well as the resource utilization.
Keywords: Genetic Predisposition to Disease, Genetic testing, Sepsis, Cytokines, Ventilators, mechanical
1. Introduction
Sepsis is life-threatening and the leading cause of multiple organ failure [1], and overcoming this formidable enemy is challenging even with the present critical care, which has been highly advanced. Sepsis is also a costly public health problem [2], therefore early prediction, detection and treatments are considered to be very important to decrease the expense of medical resources as well as the mortality of septic patients. Needless to say, various factors, including age, gender, chronic health, physiologic derangement, treatments, diagnostic strategies, and complications affect the outcome of severe sepsis. However, few studies have ever formally evaluated strengths of these determinants to predict susceptibility to and also the outcome of severe sepsis and the association among these co-factors.
On the other hand, evidence of genetic influences with respect to predisposition to various critical illnesses has started to garner attention, and susceptibility to severe sepsis has been considered to be attributed to both heritable and comorbid factors [3–5]. A number of studies have been conducted to evaluate associations between genetic variations and susceptibility to sepsis or outcome of septic patients under a similar concept to that study [3–6]. However, most of those genetic researches have been statistically underpowered, and control patients have been inappropriately selected for the purpose of determination of the genetic influence bona fide on susceptibility to diseases and on the patients’ outcome [7–9].
In an attempt to solve the problems of these population association studies, we have already performed a collaborative case-control study with respect to the susceptibility to severe sepsis in a larger, heterogeneous population of patients from 12 USA ICUs represented by the Genetic Predisposition to Severe Sepsis (GenPSS) archive [10]. In that project, we used the Project IMPACT™ (PI) critical care clinical database information management system developed by the Society of Critical Care Medicine (SCCM) [11].We have previously reported the methodology in detail, and a single nucleotide polymorphism (SNP) in the first intron of LTA at +252(LTA(+252)) was found to influence predisposition to severe sepsis, a predisposition that is modulated by gender and age [10]. However, no clinical outcome in relation to genetic polymorphisms has been shown in the article. The present work represents a continuation of this population association study to address the outcome using the six candidate SNPs which have been widely assumed to be associated with systemic inflammation [10].
Here, we performed logistic regression analyses for ranking by regression of age, chronic health, physiologic derangement, treatments, complications and the genotypes all on the outcomes of severely ill patients with the same PI database, which was utilized in our previous study [10]. Because critical illnesses often cause acute respiratory failure which requires mechanical ventilation, genetic conditions were additionally assessed with respect to the ventilator duration, i.e. the ICU resource utilization.
2. Materials and methods
2.1. Design and data sources
We conducted a retrospective analysis of outcome and ventilator duration in patients with severe sepsis in a heterogeneous, large population in the US. In the present study, we examined whether genetic conditions affect the outcome of critical illnesses and how large the effect is relative to acquired conditions. All participating clinical centers utilized the PI database information management system developed by SCCM [11] and managed by Tri-Analytics, Inc. (http://www.trianalytics.com/, accessed 11 November 2011) and Cerner Corporation (Kansas City, MO). PI is an administrative database designed for critical care units across all disciplines [11].
2.2. Patient selection and definitions of severe sepsis
The diagnosis of severe sepsis was made when a patient met the criteria proposed by the American College of Chest Physicians/SCCM Consensus Conference [10,12,13]. A previously validated approach was applied to the identification of severe sepsis involving the co-occurrence of International Classification of Diseases, Ninth Revision (ICD-9) codes for a bacterial or fungal infectious process and acute organ dysfunction [10].
During the 4 years since Jul 2001, there were more than 200,000 patients among 120 centers in the PI database. The GenPSS archive includes critically ill patients from 12 intensive care units across the US, and the archive contains a total of 854 severely septic patients with genetic material. The patients with SAPS (simplified acute physiology score) II probability of survival [14] of <0.20, i.e. poor chance of survival, are thought to be too severe to evaluate their genetic effect on their clinical outcome; therefore we excluded the severest patient population from the 1708 patients (Fig. 1). Also, the distribution of the polymorphisms assessed in this study (vide infra) was different among African Americans and Caucasians, and these polymorphisms identify different haplotypes in different ethnic groups (data not shown). Therefore, analyses were stratified on presented only for Caucasians. The study population of the critically ill patients is shown in Table 1. Even with the non-random elimination of the severe patient population, SAPS II probability of survival [14] was similar as the actual survival rate (Table 1). Selected treatments for critical illnesses were also evaluated with respect to outcome of the ICU patients in GenPSS archive. Because the patient characteristics are largely diverse, the purpose of the treatments even with the same drug, e.g. steroids, might vary in the critical care settings. And several treatments and procedures have been proven not to be associated with the clinical outcome with our preliminary analyses (data not shown). Therefore, the number of treatments as confounders was strictly limited.
Fig. 1.

Flow chart of patients’ selection in the GenPSS project (GenPSS, Genetic Predisposition to Severe Sepsis).
Table 1.
Baseline characteristics of the study population.
| Genotyped critically ill patients (n = 1057) | |
|---|---|
| Age (median, range) | 66, 18–90 |
| Male sex; n (%) | 556 (52.6) |
| Survivors; n (%) | 744 (70.4) |
| Severe sepsis; n (%) | 585 (55.3) |
| SAPS II prob. of survival (mean ± SD, range) | 70.4 ± 29.6, 20.0–100 |
| Ventilator free days (mean ± SE, range) | 17.3 ± 0.4, 0–28 |
| Pre-existing conditions; n (%) | |
| Cardiovascular diseases | 960 (90.8) |
| Diabetes mellitus | 751 (71.1) |
| COPD | 702 (66.4) |
| Malignant neoplasm | 449 (42.5) |
| Trauma | 71 (6.7) |
| Obesity | 402 (38.0) |
| Treatments/procedures; n (%) | |
| Renal replacement therapy | 86 (8.1) |
| Vasopressin | 36 (3.4) |
| Blood product administrations | 151 (14.3) |
| Enteral feeding | 392 (37.1) |
| Acute organ dysfunctions and sequelae; n (%) | |
| Respiratory | 523 (49.5) |
| Cardiovascular | 93 (8.8) |
| Renal | 154 (14.6) |
| Hematologic | 40 (3.8) |
| Neurologic | 36 (3.4) |
| Hepatic | 14 (1.3) |
| DIC | 32 (3.0) |
| DVT | 68 (6.4) |
| VAP | 55 (5.2) |
| GI bleeding | 11 (1.0) |
| Number of acute organ dysfunctions (mean ± SD) | 0.81 ± 0.76 |
SAPS, simplified acute physiology score; COPD, chronic obstructive pulmonary diseases; DIC, disseminated intravascular coagulation; DVT, deep venous thrombosis; VAP, ventilator associated pneumonia; GI, gastrointestinal, SE, standard error; SD, standard deviation.
2.3. Definitions of ventilator free days
Ventilator free days (VFDs) were defined as the number of days between successful weaning from mechanical ventilation and 28 day after study enrollment by a formula by Schoenfeld and Bernard [15], The formulae are as follows:
VFDs = 0: If the patient dies before 28 days.
VFDs = (28 − x): If the patient is successfully weaned from mechanical ventilation within 28 days, where x is the number of days spent receiving mechanical ventilation.
VFDs = 0: If the patient requires mechanical ventilation for 28 days or more.
2.4. ICD-9. Codes used to identify pre-existing conditions, acute organ dysfunction, and sequelae
Preexisting conditions (cardiovascular diseases, diabetes mellitus, chronic obstructive pulmonary diseases (COPD), malignant neoplasm, trauma, and obesity) and comorbidities during ICU stay (disseminated intravascular coagulation (DIC), deep venous thrombosis (DVT)) were defined in accordance with the ICD-9 codes as follows. 393–429, Pre-existing cardiovascular diseases; 250, Diabetes mellitus; 490–495, COPD; 140–159, 161–165, 170–208, 230– 234, Malignant neoplasm; 800–848, 850–854, 860–879, 881–887, 890–897, 900–904, 925, 928, 929, 950–959, Trauma; 278.00, 278.01, Obesity; 286.6, 286.7, 286.9, DIC; 451–454, DVT. We adapted ICD-9 based classification of acute organ dysfunction from a stuff by Weycker et al. [16].
2.5. Genotyping
Detailed information about sample collection, patient confidentiality, and genotyping methods has been described previously [10]. Genomic DNA for genotyping was isolated from these blood stains. First, we amplified the target region of DNA by polymerase chain reaction (PCR) with primers specific for the sequences of each of our six selected markers. The selected markers include SNPs present before the first codon of TNF at −308 (rs1800629), IL1B at −511 (rs16944), IL6 at −174 (rs1800795), IL10 at −819 (rs1800871), and CD14 at −159 (rs2569190) (TNF(−308), IL1B(−511), IL6 (−174), IL10 (−819), and CD14 (−159)), and in the first intron of lymphotoxin-α (formerly known as TNF-β) at +252 (rs909253) (LTA(+252)). These six polymorphisms were selected from 22 candidate markers related to systemic inflammation to infection, i.e. sepsis, based on our preliminary analysis of a separate pilot patient cohort [10]. We focused on genes whose products had a known functional role in the inflammatory response and selected the same six candidate SNPs which were evaluated at the former GenPSS study [10].
We introduced a single nucleotide extension strategy for genotyping using dideoxy terminator nucleotides, or, the template-directed, dye-terminator incorporation assay with fluorescence polarization detection (TDI-FP) methods [10]. The PCR primers and the conditions for the six SNPs are shown in Appendix 1. Also, the TDI-FP oligonucleotide probes, dye-terminator combinations, and reaction conditions are shown in Appendix 2.
2.6. Statistical analysis
We performed a multivariable logistic regression analysis applying a forced entry method with SPSS software (SPSS Inc, Chicago, IL) in order to assess risk factors, including heritable and acquired conditions, which are considered to be associated with outcome of critical illnesses. We additionally evaluated treatments’ effects against several critical illnesses by the logistic regression. Usages of mechanical ventilation, acute renal replacement therapy, vasopressin, blood products administration, and enteral feeding for the patients were queried by Microsoft Access database. Ventilator durations in the ICU patients with regard to the common allele presence of a selected SNP were also assessed by Cox regression analysis using the statistical software program SPSS (SPSS Inc., Chicago, IL).
The Hardy–Weinberg equilibrium for the population distribution of the variant alleles was determined according to the approach described by Guo and Thompson [17]. Allelic chi-squares were examined for each SNP. We considered the differences significant with respect to mortality and VFD at a full scan permutation of the correlation/trend test P-value of ≤0.05. The multiple logistic regression model was established to evaluate the independent relationship between a SNP variant and the presence of the potential confounders in relation to the outcome of critical illnesses. Odds ratios (ORs) were calculated in 95% confidence interval (CI) and all P values were two-tailed, with statistical significance defined by P ≤ 0.05. Hazard ratios (HRs) for Cox regression analysis of ventilator durations were also calculated and all P values of these multivariable regression analyses were with statistical significance defined by P ≤ 0.05. Adjustment for multiple testing was not performed due to the exploratory hypothesis generating nature of the study. All of both heritable and acquired covariates included in the models were indicated in Table 1.
The protocol, which includes waiver of informed consent, was approved by the Human Studies Committee at the Washington University School of Medicine. The GenPSS project design meets the confidentiality and specimen access controls required of a typical NIGMS Human Genetic Cell Repository (http://ccr.coriell.org/Sections/Collections/NIGMS/?SsId=8, accessed 11 November 2011).
3. Results
3.1. Population characteristics
To investigate the association between heritable and acquired factors of the critically ill in the heterogeneous US population, we analyzed a total of 1057 Caucasian ICU patients from the Gen- PSS archive and genotyped for the six SNPs. The baseline characteristics of the genotyped patients are presented in Table 1. Patients with severe sepsis were 55.3% of total population and male gender was 52.6%. Median age (range) was 66.0 (18–90) years old and their ventilator free days was 17.3 (0.4) (median, (SE)). SAPS II probability of survival (mean (SD)) [14] was 70.4 (29.6) and the actual in-hospital survival rate was also 70.4%. Cardiovascular disease was the most prominent pre-existing conditions appearing in the patient cohort according to the ICD-9 entry at admission. Enteral feeding was performed for 37.1% of the patients. Mechanical ventilation was introduced for 49.5% of the patients and the number of acute organ dysfunction (mean (SD)) was 0.81 (0.76).
3.2. Genotypic distributions of the six inflammation gene polymorphisms and the outcome of the critically ill in the genotype categories
Genotype call rate of the six SNPs was 91.5–99.0%, although the genotypic distribution in LTA(+252) diverged from Hardy–Weinberg equilibrium(HWE) in the studied subjects (P < 0.001; Table 2). The TNF(−308)* AA homozygote showed the highest in-hospital mortality (48.7%) among the 18 genotypes evaluated and recessive model of TNF(−308) and IL10(−819) showed statistically significant association with outcome of the critically ill (P = 0.015 and P = 0.040, respectively with correlation/trend test). As expected, given their collocation on chromosome six, TNF(−308) and LTA(+252) were in linkage disequilibrium (LD correlation R = 0.502, D′= 0.808, P < 0.001).
Table 2.
Genotypic distributions of the six SNPs and mortality for the group of the critically ill patients in each genotype category.
| SNP locus | Genotype; n (genotypic distribution (%)) | P values of additive model on mortality | P values of recessive model on mortality | P values of dominant model on mortality | Genotype call rate | Hardy–Weinberg equilibrium test P value | ||
|---|---|---|---|---|---|---|---|---|
| TNF(−308) | GG | GA | AA | |||||
| n = 967 | 661(68.4%) | 269(27.8%) | 37(3.83%) | 0.091 | 0.015a | 0.318 | 0.915 | 0.147 |
| Mortality (%) | 29.5 | 30.5 | 48.7a | |||||
| LTA(+252) | AA | AG | GG | |||||
| n = 1046 | 451 (43.1%) | 433 (41.4%) | 162 (15.5%) | 0.824 | 0.573 | 0.928 | 0.990 | <0.001 |
| Mortality (%) | 29.5 | 30.5 | 27.8 | |||||
| IL1B(−511) | CC | CT | TT | |||||
| n = 983 | 464(47.2%) | 408 (41.5%) | 111 (11.3%) | 0.655 | 0.748 | 0.419 | 0.930 | 0.141 |
| Mortality (%) | 31.5 | 28.4 | 31.5 | |||||
| IL6(−174) | GG | GC | CC | |||||
| n = 1041 | 358 (34.4%) | 512 (49.2%) | 171 (16.4%) | 0.251 | 0.400 | 0.312 | 0.985 | 0.595 |
| Mortality (%) | 31.6 | 29.1 | 26.9 | |||||
| IL10(−819) | CC | CT | TT | |||||
| n = 991 | 587 (59.2%) | 355 (35.8%) | 49 (4.94%) | 0.231 | 0.040a | 0.597 | 0.938 | 0.618 |
| Mortality (%) | 29.1 | 29.0 | 42.9a | |||||
| CD14(−159) | CC | CT | TT | |||||
| n = 978 | 269(27.5%) | 485(49.6%) | 224(22.9%) | 0.729 | 0.882 | 0.491 | 0.925 | 0.849 |
| Mortality (%) | 31.6 | 28.9 | 30.4 | |||||
SNP, single nucleotide polymorphism; TNF(−308), SNP at position −308 nucleotides 5′ of the first exon of tumor necrosis factor-α; LTA(+252), SNP at position 252 site of lymphotoxin-α; IL1B(−511), SNP at position −511 nucleotides 5′of the first exon of interleukin-1β; IL6(−174), SNP at position −174 nucleotides before the first exon of IL-6; IL10(−819), SNP at position −819 nucleotides before the first exon of IL-10; CD14(−159), SNP at position −159 nucleotides before the first exon of CD14.
TNF(−308)*AA and IL10(−819)*TT genotypes were observed at a frequency higher in non-survivors than in survivors (P = 0.015 and 0.040, respectively with correlation/trend test).
3.3. Ranking by the regression of the prognostic factors on critical illnesses
We performed the ranking of heritable and acquired risk factors for severe sepsis (Fig. 2). The more organ dysfunctions the septic patients obtained, the higher mortality they had, hence presence of three organ dysfunctions was the highest prognostic factor for the adverse outcome (OR(95%CI) = 19.96(4.99–79.72), P < 0.001). Gastrointestinal bleeding was also the significant risk factors for deterioration (OR(95% CI) = 7.64(1.76–33.12), P = 0.007).
Fig. 2.

Multivariable logistic regression model for detecting the incident effect of the TNF(−308) and IL10(−819) SNPs on the outcome of critical illnesses. COPD, chronic obstructive pulmonary diseases; VAP, ventilator associated pneumonia; RRT, renal replacement therapy; DIC, disseminated intravascular coagulation; DVT, deep venous thrombosis. GI; gastrointestinal, TNF(−308), SNP at position −308 nucleotides 5′ of the first exon of tumor necrosis factor; IL10(−819), SNP at position −819 nucleotides before the first exon of IL-10. Diamonds to the right of the vertical line indicate an increased risk of death from critical illnesses in patients carrying each covariate. The diamonds represent the odds ratios from the logistic regression analyses. Horizontal lines through the diamonds represent 95% confidence intervals (CIs).
As we expected, TNF(−308)* AA genotype carriers resulted in having worse outcome than the other genotype with recessive model after normalization by the selected heritable and acquired factors (OR(95% CI) = 2.67(1.29–5.55), P = 0.008). Hence, there is evidence that TNF(−308) is significantly associated with adverse outcome both on its own and also when adjusted for potential confounders including the number of organ failures, gastrointestinal bleeding as well as severe sepsis occurrence. IL10(−819)* TT genotype carriers were also susceptible to poor outcome than the other IL10 genotype group, although the genetic influence was weaker (OR(95% CI) = 1.89(1.00–3.57), P = 0.050, Fig. 2).
3.4. Genotypic distributions of the six inflammation gene polymorphisms and the ventilator free days of the critically ill in the genotype categories
Six hundred and twenty-five out of the 1057 Caucasian patients received ventilatory support (Appendix 3). The TNF(−308)* AA homozygote again showed the shortest ventilator free days of 5.56 (1.95) (mean(SE)) among the 18 genotypes evaluated (Fig. 3) and recessive model of TNF(−308) showed marginally significant association with the ventilator free days of the critically ill (P = 0.050 with the correlation/trend test, Fig. 3, Appendix 3). However, this results might be affected by the worse outcome of TNF(−308)* A presences of the critically ill patient population (Table 2). Therefore, we performed a further evaluation on their ventilator duration by Cox regression analysis because correction for potential confounding factors due to baseline imbalance of covariates indicated in Table 1 was thought to be important.
Fig. 3.
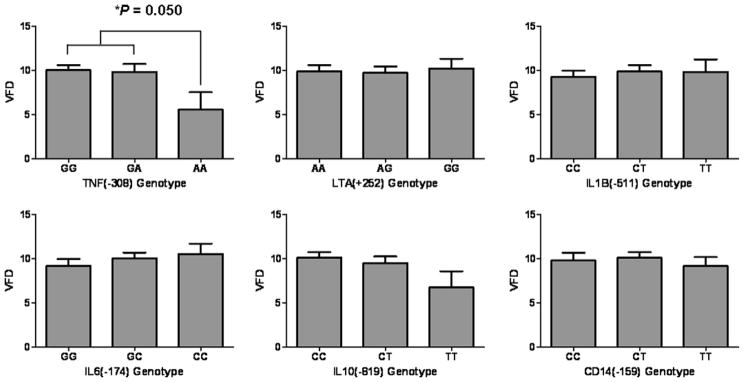
Ventilator free days of the critically ill in genotype categories of the six single nucleotide polymorphisms VFD, ventilator free day; SNP, single nucleotide polymorphism; TNF(−308), SNP at position −308 nucleotides 5′ of the first exon of tumor necrosis factor; LTA(+252), SNP at position 252 site of lymphotoxin-α; IL1B(−511), SNP at position −511 nucleotides 5′of the first exon of interleukin-1β; IL6(−174), SNP at position −174 nucleotides before the first exon of IL-6; IL10(−819), SNP at position −819 nucleotides before the first exon of IL-10, CD14(−159), SNP at position −159 nucleotides before the first exon of CD14. Y-axes for all graphs show the ventilator free days of the critically ill in each genotype category. The TNF(−308)* AA homozygote showed the shortest ventilator free days of 5.6 (2.0) (mean (SE)) among the 18 genotypes evaluated (P = 0.050 with recessive model of the correlation/trend test; AA v (GG + GA), see Appendix 3).
3.5. TNF(−308) SNP in relation to ventilator duration of the critically iII
Fig. 4 shows the cumulative frequency curve on ventilator duration of the ICU patients by TNF(−308) genotype (AA v non-AA). The TNF(−308)* AA genotype significantly diverged from the other genotypes (Adjusted HR(95%CI) = 2.53(1.11–5.79) with Cox regression, P = 0.028). This deviation meant that TNF(−308)* AA genotypes needed longer mechanical ventilation among this acute phase, in the 28 days after the start of mechanical ventilation, of their ICU stay. Therefore, genetic testing for this TNF(−308) SNP is thought to be useful for predicting the predisposition to longer ventilation, i.e. longer occupation of ICU resources.
Fig. 4.

Kaplan–Meier analysis of 28-day ventilator duration of the critically ill by TNF(−308) genotype (AA v non-AA). TNF(−308), SNP at position −308 nucleotides 5′ of the first exon of tumor necrosis factor. Statistical significance was assessed using the Cox regression analysis, censored cases; ventilator removal because of decease.
4. Discussion
Genomic approaches to study injury and critical illness still have high hurdles, unique to these methods, regarding resources, experimental design, and data analysis. Hence, it is difficult to determine which factors largely affect the outcome of the multifactorial disorders in clinical settings. Over the period of history, TNF(−308) has hold center stage of critical care genomics since McGuire et al. reported the association between the SNP and susceptibility to cerebral malaria [18]. In the present study, we derived associations between TNF(−308) or IL10(−819) and inhospital mortality with univariate genetic association analyses (Table 2). Thereafter, only the influence of TNF(−308) on both clinical outcome (Fig. 2) and ventilator duration (Fig. 4) was validated with multivariate analysis.
Recently meta-analysis on the TNF(−308) was performed and an association between development of sepsis and TNF(−308)* A presence was recognized, although TNF(−308)* A was not associated with sepsis mortality except for Asian population [3]. In this regard, immunosuppression due to severe sepsis [19] is considered to be predisposed to cytomegalovirus (CMV) reactivation [20]. Patients with severe sepsis who developed active CMV infection were found to have significantly prolonged mechanical ventilation [20], therefore the longer ventilator duration of TNF(−308)* AA carriers in the present study might have been caused partially by the CMV reactivation. On the other hand, a recent article evaluated with an Asian population insisted that APACHE II score combined with TNF(−308) genotyping was more accurate than APACHE II score alone in predisposing to clinical outcomes [21]. It is widely recognized that there is a substantial genotypic difference in TNF(−308) among various racial populations [22]. As part of this trend, the present study is one of the largest population genetic studies in relation to TNF(−308) with Caucasian patient population, and as we expected, the association between TNF(−308) and outcome of the critically ill in this large and heterogeneous population was also recognized.
Our previous report did not reveal any association between TNF(−308) and susceptibility to severe sepsis in the US patients of the GenPSS archive while LTA(+252) influenced predisposition to the disease [10]. Because the two markers on the same chromosome TNF(−308) and LTA(+252) were in a linkage disequilibrium, diplotype of these two SNPs may have a stronger association with clinical outcome of critically ill patients. Actually, a forward stepwise logistic haplotype trend regression test [23] for occurrence of severe sepsis on the two marker haplotype composed of TNF(−308) and LTA(+252) markers determined that the best predictor for severe sepsis was the (G, G) haplotype with a P-value of 6.47e-06 [10], even though the sepsis risk allele for the TNF(−308) marker is generally the minor A allele [3,24,25]. TNF(−308)* A shows high TNF producer phenotype [26], and an important role of TNF in controlling M.tuberculosis and many other intracellular infections in humans has been recognized [27]. Therefore, high TNF production does not always lead to vulnerability to sepsis but potentially deterioration of the inflammatory disorder due to extreme hypercytokinemia [28]. On the contrary, Foster et al. demonstrated that histone methylation inhibits TNF gene transcription [29], hence the epigenetic modification of chromatin and DNA is thought to be an important level of gene regulation in SIRS and sepsis pathophysiology. As the results, inflammatory gene silencing, i.e. endotoxin tolerance in sepsis could lead to deleterious effects on ICU patients [30]. In this regard, the rarity of the mutant allele homozygotes of TNF(−308) in both the US populations is noteworthy. Moreover, there was not even a single TNF(−308)* AA individual in the Japanese ICU patients [21,24,31]. The genetic effect on outcome of critical illnesses is thought to be substantial among most racial groups, and this may strongly select against mutant homozygote individuals and act to reduce the TNF(−308)* A allele frequency.
The present report begs the question of whether it is useful to introduce genetic testing in an acute phase of critical care illnesses. The answer might be ‘yes’, but only in selected and limited situations. Genetic testing has least utility in the severest patients who typically die soon after admission. Their care would not likely be affected by their genotypes. Hence, genetic testing for the patients with SAPS II PS of <0.2 in ICU settings, whom we eliminated in the present study, would incur expense with little promise of benefit. On the other hand, the genetic information may be useful in ‘milder’ critically ill patients to more rapidly institute appropriate treatments. TNF(−308)* AA homozygotes needed longer duration of mechanical ventilation for the other genotypes, TNF(−308)* (GG + GA), in their 28 ICU days (Fig. 4). This information might serve as a useful reference suggesting earlier tracheostomy and planning for long-term ventilator support for TNF(−308)* AA homozygotes.
The present study has several important limitations. First, 22 genetic markers of more than millions of genetic polymorphisms were evaluated preliminarily and only six were further analyzed in detail. Needless to say, sepsis pathophysiology is complex web of interactions and the disease advances through many physiologic pathway, i.e. inflammation, innate immunity, adoptive immunity, coagulation–fibrinolysis [25,32], which are closely involved in various types of cell death of vital organs [33]. However, even among these limited SNPs, we found statistically significant association between the genetic factors and mortality as well as ventilator duration. Hence, biological pathways related to TNF(−308) are suggested to affect both outcome of critical illness and use of ICU resources. Secondly, the LTA(+252) was deviated from HWE in the study population. There was evidence to suggest that the association between the outcome and these markers was not driven by departures from HWE. The TNF(−308) and LTA(+252) were shown in previous studies to be associated with both sepsis and respiratory failure [10,34]. The samples were not randomly drawn from a population but instead were taken from patients admitted to the ICU (Fig. 1, Table 1). The likelihood of cases and controls suffering from either sepsis, respiratory failure or both is greater than would normally be found in the general “healthy” population, plausibly accounting for the departures from HWE as discussed at our previous GenPSS study [10]. Longevity of ventilator duration is thought to be affected by susceptibility to severe sepsis [35]. Therefore, it is also striking that TNF(−308) was associated with longer mechanical ventilation even though this SNP did not affect sepsis occurrence [10]. A linkage disequilibrium between TNF(−308) and LTA(+252) might have caused the association between TNF(−308) and longer mechanical ventilation as well as susceptibility to poor outcome. Lastly, the assignment of race is known to be error-prone. We used the assignments recorded in the PI database. These self-assignments were made by patients and families at the time of admission.
The GenPSS project launched in 2001, and we selected more than 20 functional and candidate polymorphisms related to systemic inflammation [10] and the genes whose products had a known functional role in the inflammatory response had been focused. These SNPs were analyzed with TDI-FP, a strategy for high-throughput, cost-effective genotyping of samples [10]. With current technologies, information on up to more than a million unique sequence variants can be provided on a single chip with lower cost. Actually, oligonucleotide arrays can be used to identify presence of specific alleles in individuals, and a CYP chip and the microarray instrumentation system have been commercially available in the pharmacogenomics field (FDA K042259; http://www.fda.gov/, accessed 16 April 2012). This sort of DNA chip-based genotyping technique is more suitable for profiling of the genetic background of a particular individual than the present genotyping assay, i.e. TDI-FP, in the critical care field [36].
Genetic testings, to ultimately investigate the inherent order of biology, is thought to compete with the usefulness of biomarkers in prediction and prevention. One advantage of genetic testings is that their information remains unchanged in any clinical conditions in contrast to a variety of biomarkers from biological samples. Therefore, physicians will be able to perform prepared response to each biological reaction to stress on the basis of genomic markers in the critical care settings. Furthermore, the classic central dogma of molecular biology states that “DNA makes RNA makes protein”, and functional genomics explores to determine the significance of the observed changes in gene expression [37]. In terms of this concept, multiple-gene biomarkers, e.g. cDNA microarrays, are likely to be more sensitive and discriminating than single genes. Reaping the maximum fruit from these technologies, Ashley et al. lately provided an approach to comprehensive analysis of a human genome in a defined clinical context [38]. They assessed whole-genome genetic risk, focusing on variants in genes that are associated with Mendelian disease, novel and rare variants across the genome, and variants of pharmacogenomic importance [38]. This kind of whole-genome assessment for sepsis and the critical illnesses may come true in near future.
5. Conclusions
Heredity likely affects both predisposition to severe inflammation and to poor outcome of the disease. The specific effects remain opaque. Further analyses of haplotypes in candidate regions, e.g. MHC region including TNF and LTA in sepsis, and genome-wide analysis for the critical illnesses may prove to be a more productive approach. Although the genetic influences are likely overwhelmed by comorbid factors and acute illness in individual cases, population studies continue to suggest biological pathways that affect both critical illness and use of ICU resources.
Supplementary Material
Acknowledgments
This research was supported by NIH/GM (062809) and the Pfizer Travel Fellowship 2006 by Japanese Association for Acute Medicine. The funding sources had no role in study design, collection, analysis, and interpretation of data.
Abbreviations
- ICU
intensive care unit
- SNP
single nucleotide polymorphism
- GenPSS
Genetic Predisposition to Severe Sepsis
- SAPS
simplified acute physiology score
- TNF
tumor necrosis factor, LT, lymphotoxin
- IL
interleukin
- OR
odds ratio
- CI
confidence interval
- HR
hazard ratio
- SE
standard error
- SD
standard deviation
- PI
Project IMPACT™
- ICD
international classification of diseases
- VFD
ventilator free day
- COPD
chronic obstructive pulmonary diseases
- DIC
disseminated intravascular coagulation
- DVT
deep venous thrombosis
- PCR
polymerase chain reaction
- TDI-FP
template-directed, dye-terminator incorporation assay with fluorescence polarization detection
- HWE
Hardy–Weinberg equilibrium
- LD
linkage disequilibrium
- SIRS
systemic inflammatory response syndrome
Appendix A. Supplementary material
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cyto.2012.06.016.
References
- 1.Faist E, Baue AE, Dittmer H, Heberer G. Multiple organ failure in polytrauma patients. J Trauma. 1983;23:775–87. doi: 10.1097/00005373-198309000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–10. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Teuffel O, Ethier MC, Beyene J, Sung L. Association between tumor necrosis factor-alpha promoter −308 A/G polymorphism and susceptibility to sepsis and sepsis mortality: a systematic review and meta-analysis. Crit Care Med. 2010;38:276–82. doi: 10.1097/CCM.0b013e3181b42af0. [DOI] [PubMed] [Google Scholar]
- 4.Lin MT, Albertson TE. Genomic polymorphisms in sepsis. Crit Care Med. 2004;32:569–79. doi: 10.1097/01.CCM.0000110878.49476.42. [DOI] [PubMed] [Google Scholar]
- 5.Freeman BD, Kennedy CR, Frankel HL, Clarridge B, Bolcic-Jankovic D, Iverson E, et al. Ethical considerations in the collection of genetic data from critically ill patients: what do published studies reveal about potential directions for empirical ethics research? Pharmacogenomics J. 2010;10:77–85. doi: 10.1038/tpj.2009.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arcaroli J, Fessler MB, Abraham E. Genetic polymorphisms and sepsis. Shock. 2005;24:300–12. doi: 10.1097/01.shk.0000180621.52058.e1. [DOI] [PubMed] [Google Scholar]
- 7.Terwilliger JD, Haghighi F, Hiekkalinna TS, Goring HH. A bias-ed assessment of the use of SNPs in human complex traits. Curr Opin Genet Dev. 2002;12:726–34. doi: 10.1016/s0959-437x(02)00357-x. [DOI] [PubMed] [Google Scholar]
- 8.Clark MF, Baudouin SV. A systematic review of the quality of genetic association studies in human sepsis. Intensive Care Med. 2006;32:1706–12. doi: 10.1007/s00134-006-0327-y. [DOI] [PubMed] [Google Scholar]
- 9.Watanabe E, Zehnbauer B, Deutschman CS. Genetic variability and outcome in the critically ill: avoiding SNP judgments. Crit Care Med. 2009;37:357–60. doi: 10.1097/CCM.0b013e31819311c0. [DOI] [PubMed] [Google Scholar]
- 10.Watanabe E, Buchman TG, Hirasawa H, Zehnbauer BA. Association between lymphotoxin-alpha (tumor necrosis factor-beta) intron polymorphism and predisposition to severe sepsis is modified by gender and age. Crit Care Med. 2010;38:181–93. doi: 10.1097/CCM.0b013e3181bc805d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kilgannon JH, Jones AE, Shapiro NI, Angelos MG, Milcarek B, Hunter K, et al. Association between arterial hyperoxia following resuscitation from cardiac arrest and in-hospital mortality. JAMA. 2010;303:2165–71. doi: 10.1001/jama.2010.707. [DOI] [PubMed] [Google Scholar]
- 12.Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM consensus conference committee. American college of chest physicians/society of critical care medicine. Chest. 1992;101:1644–55. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- 13.American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med. 1992;20:864–74. [PubMed] [Google Scholar]
- 14.Le Gall JR, Lemeshow S, Saulnier F. A new Simplified Acute Physiology Score (SAPS II) based on a European/North American multicenter study. JAMA. 1993;270:2957–63. doi: 10.1001/jama.270.24.2957. [DOI] [PubMed] [Google Scholar]
- 15.Schoenfeld DA, Bernard GR. Statistical evaluation of ventilator-free days as an efficacy measure in clinical trials of treatments for acute respiratory distress syndrome. Crit Care Med. 2002;30:1772–7. doi: 10.1097/00003246-200208000-00016. [DOI] [PubMed] [Google Scholar]
- 16.Weycker D, Akhras KS, Edelsberg J, Angus DC, Oster G. Long-term mortality and medical care charges in patients with severe sepsis. Crit Care Med. 2003;31:2316–23. doi: 10.1097/01.CCM.0000085178.80226.0B. [DOI] [PubMed] [Google Scholar]
- 17.Guo SW, Thompson EA. Performing the exact test of Hardy–Weinberg proportion for multiple alleles. Biometrics. 1992;48:361–72. [PubMed] [Google Scholar]
- 18.McGuire W, Hill AV, Allsopp CE, Kwiatkowski D. Variation in the TNF-alpha promoter region associated with susceptibility to cerebral malaria. Nature. 1994;371:508–10. doi: 10.1038/371508a0. [DOI] [PubMed] [Google Scholar]
- 19.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–50. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 20.Heininger A, Haeberle H, Fischer I, Beck R, Riessen R, Rohde F, et al. Cytomegalovirus reactivation and associated outcome of critically ill patients with severe sepsis. Crit Care. 2011;15:R77. doi: 10.1186/cc10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimada T, Oda S, Sadahiro T, Nakamura M, Hirayama Y, Watanabe E, et al. Outcome prediction in sepsis combined use of genetic polymorphisms – a study in Japanese population. Cytokine. 2010;54:79–84. doi: 10.1016/j.cyto.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 22.Zehnbauer B. Population genetics in critical illness. Crit Care Med. 2005;33:242–3. doi: 10.1097/01.ccm.0000150763.21694.8a. [DOI] [PubMed] [Google Scholar]
- 23.Zaykin DV, Westfall PH, Young SS, Karnoub MA, Wagner MJ, Ehm MG. Testing association of statistically inferred haplotypes with discrete and continuous traits in samples of unrelated individuals. Hum Hered. 2002;53:79–91. doi: 10.1159/000057986. [DOI] [PubMed] [Google Scholar]
- 24.Watanabe E, Hirasawa H, Oda S, Shiga H, Matsuda K, Nakamura M, et al. Cytokine-related genotypic differences in peak interleukin-6 blood levels of patients with SIRS and septic complications. J Trauma. 2005;59:1181–9. doi: 10.1097/00005373-200511000-00025. [Discussion 1189–1190] [DOI] [PubMed] [Google Scholar]
- 25.Winning J, Claus RA, Huse K, Bauer M. Molecular biology on the ICU. From understanding to treating sepsis. Minerva Anestesiol. 2006;72:255–67. [PubMed] [Google Scholar]
- 26.Wilson AG, Symons JA, McDowell TL, McDevitt HO, Duff GW. Effects of a polymorphism in the human tumor necrosis factor alpha promoter on transcriptional activation. Proc Natl Acad Sci USA. 1997;94:3195–9. doi: 10.1073/pnas.94.7.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med. 2001;345:1098–104. doi: 10.1056/NEJMoa011110. [DOI] [PubMed] [Google Scholar]
- 28.Oda S, Hirasawa H, Shiga H, Nakanishi K, Matsuda K, Nakamua M. Sequential measurement of IL-6 blood levels in patients with systemic inflammatory response syndrome (SIRS)/sepsis. Cytokine. 2005;29:169–75. doi: 10.1016/j.cyto.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 29.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447:972–8. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 30.Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009;30:475–87. doi: 10.1016/j.it.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 31.Watanabe E, Hirasawa H, Oda S, Matsuda K, Hatano M, Tokuhisa T. Extremely high interleukin-6 blood levels and outcome in the critically ill are associated with tumor necrosis factor- and interleukin-1-related gene polymorphisms. Crit Care Med. 2005;33:89–97. doi: 10.1097/01.ccm.0000150025.79100.7d. [Discussion 242–243] [DOI] [PubMed] [Google Scholar]
- 32.Russell JA. Management of sepsis. N Engl J Med. 2006;355:1699–713. doi: 10.1056/NEJMra043632. [DOI] [PubMed] [Google Scholar]
- 33.Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361:1570–83. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Waterer GW, Quasney MW, Cantor RM, Wunderink RG. Septic shock and respiratory failure in community-acquired pneumonia have different TNF polymorphism associations. Am J Respir Crit Care Med. 2001;163:1599–604. doi: 10.1164/ajrccm.163.7.2011088. [DOI] [PubMed] [Google Scholar]
- 35.Shalhub S, Junker CE, Imahara SD, Mindrinos MN, Dissanaike S, O’Keefe GE. Variation in the TLR4 gene influences the risk of organ failure and shock post trauma: a cohort study. J Trauma. 2009;66:115–22. doi: 10.1097/TA.0b013e3181938d50. [Discussion 122–113] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otani S, Oda S, Sadahiro T, Nakamura M, Watanabe E, Nakada TA, et al. Clinical application of cytokine-related gene polymorphism analysis using a newly developed DNA chip in critically ill patients. Clin Biochem. 2009;42:1387–93. doi: 10.1016/j.clinbiochem.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 37.Cobb JP, Brownstein BH, Watson MA, Shannon WD, Laramie JM, Qiu Y, et al. Injury in the era of genomics. Shock. 2001;15:165–70. doi: 10.1097/00024382-200115030-00001. [DOI] [PubMed] [Google Scholar]
- 38.Ashley EA, Butte AJ, Wheeler MT, Chen R, Klein TE, Dewey FE, et al. Clinical assessment incorporating a personal genome. Lancet. 2010;375:1525–35. doi: 10.1016/S0140-6736(10)60452-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.