

Abstract
Congenital diaphragmatic hernia (CDH) is characterized by incomplete formation of the diaphragm, occurring as either an isolated defect or in association with other anomalies. Genetic factors including aneuploidies and copy number variants are important in the pathogenesis of many cases of CDH, but few single genes have been definitively implicated in human CDH. In this study, we used whole exome sequencing (WES) to identify a paternally inherited novel missense GATA4 variant (c. 754C>T, p. R252W) in a familial case of CDH with incomplete penetrance. Phenotypic characterization of the family included magnetic resonance imaging (MRI) of the chest and abdomen demonstrating asymptomatic defects in the diaphragm in the two “unaffected” missense variant carriers. Screening 96 additional CDH patients identified a de novo heterozygous GATA4 variant (c.848G>A; p.R283H) in a non-isolated CDH patient. In summary, GATA4 is implicated in both familial and sporadic CDH, and our data suggests that WES may be a powerful tool to discover rare variants for CDH.
Keywords: Congenital diaphragmatic Hernia (CDH), whole exome sequencing (WES), incomplete penetrance, missense variant, GATA4
Introduction
Congenital diaphragmatic hernia (CDH) is characterized by incomplete formation of the posterolateral diaphragm, resulting in herniation of abdominal viscera into the chest cavity. It affects approximately 1 in 3,000 births (Pober et al. 1993) and is often associated with life-threatening lung hypoplasia and pulmonary hypertension. CDH can occur as either an isolated defect or in association with other anomalies. Rare monogenic forms of CDH in humans and mice and certain chromosomal anomalies associated with CDH suggest that genetic factors are important in the pathogenesis of many cases of CDH (Brady et al. 2011; Holder et al. 2007; Scott 2007; Yu et al. 2012)
Genes involved in the development of CDH have been identified largely through the analysis of recurrent chromosomal anomalies in humans with CDH and the characterization of mutant mouse models (Ackerman et al. 2005; Castiglia et al. 2005; Jay et al. 2007; Klaassens et al. 2005; Shimokawa et al. 2005; Wat et al. 2009; You et al. 2005). However, in light of the sporadic nature of most CDH cases and low rates of survival in previous generations allowing patients with CDH to reproduce, family based genetic methods of linkage analysis have been difficult due to the small number of familial cases and limited sizes of these families. Whole exome sequencing (WES) provides a powerful tool to detect pathogenic genetic variations in rare Mendelian disorders. In the present study, we report the identification of a novel GATA binding protein 4 (GATA4, NM_002052, NP_002043) variant in a familial case of CDH using WES and identification of a de novo GATA4 variant in sporadic CDH.
Methods
Subjects
Patients with CDH were recruited as part of the DHREAMS (Diaphragmatic Hernia Research & Exploration; Advancing Molecular Science) study (www.cdhgenetics.com/), approved by the Institutional Review Boards at Columbia University, Washington University Medical Center/St. Louis Children’s Hospital, University of Pittsburgh, Cincinnati Children’s Hospital and Medical Center / University of Cincinnati, Omaha Children’s Hospital / University of Nebraska, University of Michigan / CS Mott Children’s Hospital and Vanderbilt University (Yu et al. 2012). Clinical information was extracted from the medical record by study coordinators. Isolated CDH was defined as a CDH without an associated major birth defect. Pulmonary hypoplasia, cardiac displacement and intestinal herniation were considered to be part of the diaphragm anomaly sequence and not additional malformations. Informed consent was obtained from parents or guardians.
Genomic DNA was extracted from whole blood or saliva using a DNA extraction kit (Gentra DNA isolation kit; Qiagen, Valencia, CA) or Oragene.DNA (DNA genotek, Ontario, CA).
Chromosomal microarray analysis
Genome-wide SNP genotyping was performed in all probands using Affymetrix 6.0 and analyzed by PennCNV (Wang et al. 2007) to detect copy number variants (CNVs). Subjects with pathogenic CNVS or chromosomal anomalies were identified (Yu et al. 2012) and were excluded from this study.
Exome Analysis
Genomic DNA (3 μg) from proband IV.1 was fragmented and exons were captured using the Agilent SureSelect™ Human All Exon kit. The captured DNA was sequenced with 100 bp paired-end reads on an Illumina HiSeq 2000 according to the manufacturer’s protocol.
High quality sequencing reads were aligned to the human genome reference sequence human assembly hg19 using BWA (Version 0.5.9) (Li and Durbin 2009), allowing up to five mismatched, inserted or deleted bases (indels). GATK (Unified genotyper; version 1.0) was used to refine local alignment of reads, recalibrate base quality score, and call variants (single nucleotide variants (SNVs)/indels) within targeted regions. In addition to the default filters in GATK, variants were further filtered by genotype minimum quality of 30, minimum quality over depth of 5, minimum strand bias −0.10, and maximum fraction of reads with mapping quality of zero at 10%.
The identified variants were annotated based on dbSNP135 (http://www.genome.ucsc.edu/cgi-bin/hgGateway). The 1000 genome project (www.1000genomes.org/) and The NHLBI Exome Variant Server (EVS) (http://evs.gs.washington.edu/EVS/) were then used to filter polymorphisms. Homozygous variants were filtered based upon the assumption that the variant was autosomal dominantly inherited.
Functional impact predictions
The predicted functional impact of amino-acid substitutions in proteins and degree of cross-species conservation were used to filter and prioritize the novel/rare variants. For filtering purpose, we applied SIFT (Kumar et al. 2009) (http://sift.jcvi.org/, GRCh 37 Ensembl 63) and PolyPhen2 (Adzhubei et al. 2010) (http://genetics.bwh.harvard.edu/pph2/, GRCh37, HumVar classifier). Those variant sites predicted to be tolerated/benign by both of these tools were eliminated. After filtering, additional prediction tools were then applied to obtain more detailed pathogenicity scores for ranking purposes. These tools included, SeattleSeq Annotation 134 (http://snp.gs.washington.edu/SeattleSeqAnnotation134/, for PhastCons and GERP scores), ANNOVAR (Wang et al. 2010) (http://www.openbioinformatics.org/annovar/, for PhyloP, LRT and Mutation Taster Scores), Pmut (Ferrer-Costa et al. 2005) (http://mmb.pcb.ub.es/PMut/) and MutationAccessor (Reva et al. 2007) (http://mutationassessor.org/). Each tool gave a pathogenic score for each variant, and candidate variants were prioritized according to a weighted average of nine pathogenicity scores (15% for 1-SIFT score and 15% for PolyPhen2, and 10% each for PhyloP, LRT, Mutation Taster, GERP, PhastCons, Pmut and MutationAccessor).
Gene prioritization
Endeavour (Tranchevent et al. 2008) (http://homes.esat.kuleuven.be/~bioiuser/endeavour/tool/endeavourweb.php) and ToppGene (Chen et al. 2009) (http://toppgene.cchmc.org/prioritization.jsp) were used with the default training parameters to prioritize the genes. A list of 14 genes with variants identified in patients with diaphragm defect was created based on the PubMed and Online Mendelian inheritance in Man as the training set (Supplementary Table 1).
Mutation screening and variants validation
Sanger sequencing was performed to independently confirm the potentially pathogenic variants (Applied Biosystems, Foster City, CA) in the proband and genotype all the available family members. We sequenced 96 additional CDH patients for mutations in the coding exons and splice junctions of GATA4 using primers designed with Primer3 (http://frodo.wi.mit.edu/primer3/). The human gene mutation database (HGMD) was used to identify variants in GATA4 (Cooper et al. 1998).
Results
We studied one family with isolated CDH (Fig. 1a). The proband is a Caucasian male of Northern European descent who was diagnosed prenatally at 20 weeks gestation with an isolated, left sided CDH with the stomach, liver, spleen, small and large bowel herniated into the chest. Postnatally he was noted to have a small hemangioma on his foot, bilateral hydronephrosis with normal voiding cystourethrogram, and bilateral undescended testis that required orchiopexy. An echocardiogram on day 15 demonstrated a small patent foramen ovale and severe pulmonary hypertension but no structural cardiac defects. Echocardiogram at 12 months demonstrated resolution of the pulmonary hypertension. At 30 months he was developing appropriately by parental report. The family history is significant for a paternal uncle with an isolated left sided CDH with stomach, liver, spleen and intestine herniated into the chest. No echocardiogram was performed. He also had a left undescended testis. He died on day of life 2 from respiratory distress secondary to severe pulmonary hypertension. No autopsy was performed. A second paternal uncle died on the first day of life of unknown cause.
Fig. 1.

a Pedigree of the proband (arrow) with CDH. Squares indicate male subjects, and circles female subjects; slashes indicate deceased subjects; filled indicate affected subjects with CDH, filled with light gray indicate anatomical Bochdalek hernias who were clinically asymptomatic, unfilled clinically asymptomatic subjects. Genotypes of GATA4 c.C754T for all the available individuals are indicated. b Sequence chromatograms of GATA4 in proband, sibling, father and mother indicate the paternally inherited c. C754T (red arrow) in exon 3 resulting in an Arginine to Tryptophan amino acid substitution at amino acid 252. The sequence is the complementary strand.
A total of 13.1 Gb of sequence was generated and 58% of reads were mapped to the reference human coding exome (hg19) with a mean coverage 86.6-fold. The proportion of the targeted exome covered at 8× and 15× was 94.7% and 90.6%, respectively. A total of 30,502 SNVs and indels were detected.
Based upon the pedigree, we assumed an autosomal dominant mode of inheritance with incomplete penetrance (Fig. 1a). A total of 24,378 SNVs were detected. We kept only the 7,438 SNVs that were annotated by SIFT or SeattleSeq as non-synonymous or located in splice sites, or in 3′ or 5′ untranslated regions. We then removed 7,337 variants with an allele frequency of greater or equal to 0.1% in dbSNP135, 1000 Genomes, or ESP databases. Ninety-five variants were heterozygous. We further eliminated 24 variants that were predicted to be tolerated/benign by both SIFT and Polyphen2. The procedure resulted in 71 novel/rare and potentially deleterious heterozygous rare SNVs in 71 genes (Table 1, Supplementary Table 2). There were 6,124 indels that passed the quality filter. After the exclusion of variants that were present in dbSNP135, 1000 Genomes, or ESP databases and in introns or intergenic regions, 145 novel indels remained. 40 of the frameshift indels in 35 genes were predicted to cause nonsense-mediated decay (NMD) by both SIFT and Mutation Taster (Supplementary Table 3). A novel missense heterozygous SNV in GATA4 (c. 754C>T; p. R252W) was predicted to be pathogenic by all the algorithms (Supplementary Table 2). Using the gene prioritization tools Toppgene and Endeavor, GATA4 and LAMB1 were in top 1 and Top 2 of all the potential candidate genes (Supplementary Table 2, Table 2). LAMB1 (c. 613C>T; p. V205M) was predicted by the majority of the algorithms to be pathogenic. We sequenced the LAMB1 c. C613T and GATA4 c. 754C>T variants in all the available family members. Both LAMB1 c. C613T and GATA4 c. 754C>T variants were confirmed in the proband. However, three other family members (Paternal grandmother II.4, Father III.4, and Sibling IV.2) carry the LAMB1 c. C613T variant. The father and the paternal grandfather only carry the GATA4 c. 754C>T variant. All the seven other family members had the common CC genotype (Fig. 1a, b). The variant c. 754C>T p. R252W is highly conserved across species (Fig. 2).
Table 1.
Number of variants at each stage of the filtering process in Proband ( IV.1). SNV is single nucleotide variant. MAF is minor allele frequency. NMD is nonsense mediated decay.
| Filtering | Number of variants |
|---|---|
| Total SNVs | 24,378 |
| Functional SNVs (nonsense, missense, splice site or untranslated regions) | 7,438 |
| Novel/rare SNVs (<0.1% of MAF in dbSNP135, 1000 Genomes and EVS) | 101 |
| Heterozygous SNVs | 95 |
| Pathogenic SNVs by either SIFT or Polyphen2 | 71 |
|
| |
| Total Indels | 6,124 |
| Functional indels (frameshift) | 1,985 |
| Novel heterozygous indels | 145 |
| Frameshift and NMD | 40 |
Table 2.
The rank of the top 10 candidate genes by two gene prioritization algorithms
| Rank | Toppgene | Endeavour |
|---|---|---|
| 1 | GATA4 | LAMB1 |
| 2 | LAMB1 | GPX7 |
| 3 | SIRPA | GATA4 |
| 4 | SIRPB1 | ZNF263 |
| 5 | FOXA1 | DIAPH1 |
| 6 | PDE4DIP | ARHGEF17 |
| 7 | AEBP1 | CLASRP |
| 8 | BMP5 | ZFHX3 |
| 9 | ADCY8 | FOXA1 |
| 10 | ITIH5 | ADAMTS18 |
Fig. 2.
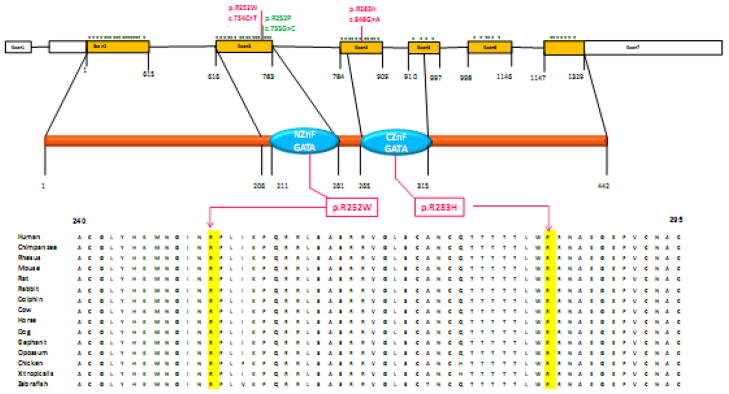
The 7 exons of GATA4 (Top). Filled squares with orange are coding exons of GATA4. Each * symbol represents a mutation in the coding exons from human genome mutation database. The c.C754T and c.G848A mutations are indicated in red, missense mutation c. G755C resulting in the Arginine to Proline at amino acid 252 is indicated in green; The protein of GATA4 (Middle). Two highly-conserved GATA-type zinc finger domains indicated as light blue. R252W indicated in red square. Amino acids alignment of a portion of GATA4 protein from different vertebrate species (Bottom). The Arginine at residue 252 is highlighted.
Because II.3 and III.4 were carriers for the GATA4 c. 754C>T variant yet were not clinically symptomatic (Fig. 1a), we screened both of them for subclinical diaphragmatic defects with chest x-ray and then thoracic and abdominal MRI. Chest x rays in both were normal, but both II.3 and III.4 had small left Bochdalek hernias (Fig. 3).
Fig. 3.

MRI of chest and abdomen in GATA4 R252W carriers III.4 and II.3. a–b III.4 (father) has a small left Bochdalek hernia indicated by white arrow. c–d II.3 (grandfather) has a small left Bochdalek hernia indicated by white arrow and arrowhead.
To independently confirm the role of GATA4 in other CDH families, we screened an additional 96 sporadic CDH patients (63 (65.6%) isolated CDH and 33 (34.4%) non-isolated CDH with congenital heart defect) without pathogenic CNVs or cytogenetic anomalies by sequencing all the coding exons of GATA4. Clinical characteristics were summarized in Table 3. There was one de novo variant (GATA4 c.848G>A p.R283H) identified in a patient with non-isolated CDH (Fig. 2). The patient is a Caucasian male with a CDH, atrial septal defect and ventricular septal defect. He had mild motor and cognitive delay at two years of age. The p.R283H variant is highly conservative and was predicted to be pathogenic by all the prediction tools (Table 4).
Table 3.
Clinical characteristics for the 96 sporadic CDH patients. Percentages of isolated and non-isolated cases are calculated as a percentage of all CDH patients.
| Isolated (%) | Non-isolated with CHD (%) | Total (%) | |
|---|---|---|---|
| Race (White / Black/ Asian/Other ) | 51 (53.1)/1 (1.0)/5 (5.2)/6 (6.3) | 28 (29.2)/0 (0)/2 (2.1)/3 (3.1) | 79 (82.3)/1 (1.0)/7 (7.3)/9 (9.4) |
| Gender (Male) | 38 (39.6) | 22 (22.9) | 60 (62.5) |
| Side of Lesion (Left/Right/Eventration) | 53 (55.2)/8 (8.3)/2 (2.1) | 28 (29.2)/4 (4.2)/1(1.0) | 81 (84.4)/12 (12.5)/3 (3.1) |
| Survival Status (Died) | 7 (7.3) | 6 (6.3) | 13 (13.5) |
Table 4.
Functional impact and conservation score of GATA4 p. R283H in different prediction tools
| GATA4 p.R283H | predictions (Score) |
|---|---|
| SIFT | Damaging (0) |
| Polyphen2 | Probably damaging (0.998) |
| pMUT | Pathological (0.88) |
| Mutation Taster | Disease causing (0.999996) |
| Mutation Accessor Functional impact | high (4.56) |
| LRT | Damaging (0.999999) |
| PhastCons | 1 |
| GERP | 5.38 |
| PhyloP | 0.999423 |
Discussion
We used WES and functional prediction and gene prioritization analysis to identify a novel variant (c.754C>T p.R252W) in GATA4 in a familial isolated CDH case. Although the CDH phenotype was initially thought to be incompletely penetrant based upon clinical symptoms, careful imaging of the diaphragm by MRI demonstrated that the CDH defect is fully penetrant although two members of the family were clinically asymptomatic. This is the first report to our knowledge demonstrating the utility of examination of the diaphragm in familial cases to define the affection status anatomically. We have confirmed that GATA4 is a relevant gene for CDH in humans by independently demonstrating a de novo c.848G>A p.R283H variant in a child with CDH and congenital heart disease.
GATA4 is a zinc finger transcription factor that controls gene expression and differentiation in a variety of cell types. It is located at 8p23.1-p22 where there is a recurrent microdeletion associated with both congenital heart disease and CDH (Wat et al. 2009). GATA4 is an important component of the retinoic signaling pathway. The retinoic pathway is implicated in numerous studies to be involved in diaphragm and lung development. A well-described animal model of CDH involves the administration of nitrofen, prenatal treatment of retinoic acid in this model results in elevated expression of GATA4 mRNA (Doi et al. 2009). Alternatively, disrupted Gata4 expression in mice results in both lung and diaphragm defects (Jay et al. 2007).
However, no mutations in GATA4 alone have been previously identified in either isolated CDH or CDH with cardiac defect patients although more than 60 missense mutations in GATA4 have been identified in patients with cardiac defects (data from HGMD® Professional 2011.4, Fig. 2). The p.R252W in exon 3 is within the N-terminal zinc finger (Fig. 2) known as the GATA motif and binds to the promoter region of many genes (Arceci et al. 1993). Residue R252 and the surrounding amino acids are highly conserved among vertebrates, suggesting that this amino acid may be functionally important. There are 17 missense mutations in exon 3 of GATA4 (Fig. 2), 15 of which were identified in patients with congenital heart defects (Bouchard et al. 2009; Nemer et al. 2006; Reamon-Buettner and Borlak 2005), c.G755C p.R252P was identified in a patient with atrioventricular septal defect (Reamon-Buettner and Borlak 2005). The N-terminal zinc finger of GATA4 interacts with FOG2 during cardiac morphogenesis (Crispino et al. 2001). FOG2 is an important gene in diaphragm development (Ackerman et al. 2005), and a de novo nonsense mutation of FOG2 was identified in a child with diaphragmatic eventration. Therefore, mutations in the N-terminal zinc finger may affect the interaction of GATA4 and FOG2, resulting the abnormal diaphragm development.
The p. R283H variant is located in the C-terminal zinc finger of GATA4 and was previously reported in a patient with atrioventricular septal defect (Reamon-Buettner and Borlak 2005). The C-terminal zinc finger is crucial for the interactions of GATA4 and NKX2-5 and TBX5, the two transcriptional regulators in human cardiac septal formation (Garg et al. 2003). Mutations in C-terminal zinc finger disrupt the protein interactions or DNA binding ability of GATA4, and are highly associated with septation defects (Reamon-Buettner and Borlak 2005). All the mutations identified in the C-terminal zinc finger (Fig.2) have been associated with cardiac defect (HGMD® Professional 2011.4). The patient with the R283H variant has both CDH and septal cardiac defects, suggesting that point mutations in C-terminal zinc finger of GATA4 are sufficient to cause cardiac defect but may not consistently cause clinically symptomatic CDH.
In summary, we provide data implicating GATA4 in a familial isolated CDH and sporadic CDH with congenital heart disease due to a de novo variant. Together with the murine evidence of the role of Gata4 in CDH development and the association of recurrent deletions of 8p23.1 including GATA4 in humans, we believe that mutations in GATA4 are an infrequent but definitive cause of CDH in human. This study also shows the power of WES to identify rare genetic variants associated with diseases in familial cases even using small numbers of affected individuals.
Supplementary Material
Acknowledgments
We greatly appreciate the families who participated in this study and all the clinical care teams who assisted with study coordination. We are grateful for the technical assistance provided by Patricia Lanzano, Jiancheng Guo, Liyong Deng and Josue Martinez from Columbia University. We thank Dr. Orpheus Kolokythas for assistance with obtaining and reading the abdominal and thoracic MRIs. We also thank Jeannie Kreutzman, and Robert Drongowski from University of Michigan; Trish Burns from Cincinnati Children’s Hospital Medical Center; Sheila Horak from University of Nebraska; Mary Dabrowiak from Monroe Carell Jr Children’s Hospital at Vanderbilt; Laurie Luther from University of Pittsburgh. Study data were collected and managed using Research Electronic Data Capture (REDCap) electronic data capture tools hosted at Columbia University. REDCap is a secure, web-based application designed to support data capture for research studies. This work was supported by NIH grant HD057036 and was supported in part by Columbia University’s CTSA grant UL1 RR024156 from NCATS-NCRR/NIH.
Footnotes
Ethical Standards
The authors declare that the experiments in this study comply with the current laws of the country in which they were performed.
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- Ackerman KG, Herron BJ, Vargas SO, Huang HL, Tevosian SG, Kochilas L, Rao C, Pober BR, Babiuk RP, Epstein JA, Greer JJ, Beier DR. Fog2 is required for normal diaphragm and lung development in mice and humans. Plos Genet. 2005;1:58–65. doi: 10.1371/journal.pgen.0010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arceci RJ, King AA, Simon MC, Orkin SH, Wilson DB. Mouse GATA-4: a retinoic acid-inducible GATA-binding transcription factor expressed in endodermally derived tissues and heart. Mol Cell Biol. 1993;13:2235–2246. doi: 10.1128/mcb.13.4.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard MF, Taniguchi H, Viger RS. The effect of human GATA4 gene mutations on the activity of target gonadal promoters. J Mol Endocrinol. 2009;42:149–160. doi: 10.1677/JME-08-0089. [DOI] [PubMed] [Google Scholar]
- Brady PD, Srisupundit K, Devriendt K, Fryns JP, Deprest JA, Vermeesch JR. Recent developments in the genetic factors underlying congenital diaphragmatic hernia. Fetal Diagn Ther. 2011;29:25–39. doi: 10.1159/000322422. [DOI] [PubMed] [Google Scholar]
- Castiglia L, Fichera M, Romano C, Galesi O, Grillo L, Sturnio M, Failla P. Narrowing the candidate region for congenital diaphragmatic hernia in chromosome 15q26: Contradictory results. Am J Hum Genet. 2005;77:892–894. doi: 10.1086/497082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37(Web Server issue):W305–311. doi: 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DN, Ball EV, Krawczak M. The human gene mutation database. Nucleic Acids Res. 1998;26:285–287. doi: 10.1093/nar/26.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crispino JD, Lodish MB, Thurberg BL, Litovsky SH, Collins T, Molkentin JD, Orkin SH. Proper coronary vascular development and heart morphogenesis depend on interaction of GATA-4 with FOG cofactors. Genes Dev. 2001;15:839–844. doi: 10.1101/gad.875201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi T, Sugimoto K, Puri P. Prenatal retinoic acid up-regulates pulmonary gene expression of COUP-TFII, FOG2, and GATA4 in pulmonary hypoplasia. J Pediatr Surg. 2009;44:1933–1937. doi: 10.1016/j.jpedsurg.2009.04.027. [DOI] [PubMed] [Google Scholar]
- Ferrer-Costa C, Gelpi JL, Zamakola L, Parraga I, de la Cruz X, Orozco M. PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics. 2005;21:3176–3178. doi: 10.1093/bioinformatics/bti486. [DOI] [PubMed] [Google Scholar]
- Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, Matsuoka R, Cohen JC, Srivastava D. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424:443–447. doi: 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- Holder AM, Klaassens M, Tibboel D, de Klein A, Lee B, Scott DA. Genetic factors in congenital diaphragmatic hernia. Am J Hum Genet. 2007;80:825–845. doi: 10.1086/513442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay PY, Bielinska M, Erlich JM, Mannisto S, Pu WT, Heikinheimo M, Wilson DB. Impaired mesenchymal cell function in Gata4 mutant mice leads to diaphragmatic hernias and primary lung defects. Dev Biol. 2007;301:602–614. doi: 10.1016/j.ydbio.2006.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaassens M, van Dooren M, Eussen HJ, Douben H, den Dekker AT, Lee C, Donahoe PK, Galjaard RJ, Goemaere N, de Krijger RR, Wouters C, Wauters J, Oostra BA, Tibboel D, de Klein A. Congenital Diaphragmatic hernia and chromosome 15q26: Determination of a candidate region by use of fluorescent in situ hybridization and array-based comparative genomic hybridization. American Journal of Human Genetics. 2005;76:877–882. doi: 10.1086/429842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protocols. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemer G, Fadlalah F, Usta J, Nemer M, Dbaibo G, Obeid M, Bitar F. A novel mutation in the GATA4 gene in patients with Tetralogy of Fallot. Hum Mutat. 2006;27:293–294. doi: 10.1002/humu.9410. [DOI] [PubMed] [Google Scholar]
- Pober BR, Russell MK, Ackerman KG. GeneReviews™ [Internet] Seattle (WA): University of Washington, Seattle; 1993. Congenital Diaphragmatic Hernia Overview. [PubMed] [Google Scholar]
- Reamon-Buettner SM, Borlak J. GATA4 zinc finger mutations as a molecular rationale for septation defects of the human heart. J Med Genet. 2005;42:e32. doi: 10.1136/jmg.2004.025395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reva B, Antipin Y, Sander C. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol. 2007;8:R232. doi: 10.1186/gb-2007-8-11-r232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DA. Genetics of congenital diaphragmatic hernia. Semin Pediatr Surg. 2007;16:88–93. doi: 10.1053/j.sempedsurg.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Shimokawa O, Miyake N, Yoshimura T, Sosonkina N, Harada N, Mizuguchi T, Kondoh S, Kishino T, Ohta T, Remco V, Takashima T, Kinoshita A, Yoshiura K, Niikawa N, Matsumoto N. Molecular characterization of del(8)(p23.1p23.1) in a case of congenital diaphragmatic hernia. Am J Med Genet A. 2005;136A:49–51. doi: 10.1002/ajmg.a.30778. [DOI] [PubMed] [Google Scholar]
- Tranchevent LC, Barriot R, Yu S, Van Vooren S, Van Loo P, Coessens B, De Moor B, Aerts S, Moreau Y. ENDEAVOUR update: a web resource for gene prioritization in multiple species. Nucleic Acids Res. 2008;36(Web Server issue):W377–384. doi: 10.1093/nar/gkn325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li MY, Hadley D, Liu R, Glessner J, Grant SFA, Hakonarson H, Bucan M. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wat MJ, Shchelochkov OA, Holder AM, Breman AM, Dagli A, Bacino C, Scaglia F, Zori RT, Cheung SW, Scott DA, Kang SHL. Chromosome 8p23.1 Deletions as a Cause of Complex Congenital Heart Defects and Diaphragmatic Hernia. Am J Med Genet A. 2009;149A:1661–1677. doi: 10.1002/ajmg.a.32896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You LR, Takamoto N, Yu CT, Tanaka T, Kodama T, DeMayo FJ, Tsai SY, Tsai MJ. Mouse lacking COUP-TFII as an animal model of Bochdalek-type congenital diaphragmatic hernia. P Natl Acad Sci USA. 2005;102:16351–16356. doi: 10.1073/pnas.0507832102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Wynn J, Ma L, Guha S, Mychaliska GB, Crombleholme TM, Azarow KS, Lim FY, Chung DH, Potoka D, Warner BW, Buncher B, LeDuc CA, Costa K, Stolar C, Aspelund G, Arkovitz M, Chung WK. De novo copy number variants are associated with congenital diaphragmatic hernia. J Med Genet. 2012;49:650–659. doi: 10.1136/jmedgenet-2012-101135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.