

Abstract
The cellular prion protein PrPC consists of two domains – a flexible N-terminal domain, which participates in copper and zinc regulation, and a largely helical C-terminal domain that converts to β-sheet in the course of prion disease. These two domains are thought to be fully independent and non-interacting. Compelling cellular and biophysical studies, however, suggest a higher order structure that is relevant to both PrPC function, as well as misfolding in disease. Here we identify a novel Zn2+ driven N-terminal – C-terminal tertiary interaction in PrPC. The C-terminal surface participating in this interaction carries the majority of the point mutations that confer familial prion disease. Investigation of mutant PrPs finds a systematic relationship between the type of mutation and the apparent strength of this newly identified domain structure. The novel structural features identified here suggest new mechanisms by which physiologic metal ions trigger PrPC trafficking and control prion disease.
Introduction
The transmissible spongiform encephalopathies (TSEs), or prion diseases, arise from misfolding of the cellular prion protein (PrPC) to its β-sheet rich scrapie form (PrPSc) (Prusiner, 1998, 2003). Among the TSEs are mad cow disease and, in humans, Creutzfeldt-Jakob disease (CJD) (Prusiner, 1997). Mature human PrPC is a 209 amino acid, membrane-anchored glycoprotein, possessing two domains: a flexible N-terminal segment (up to residue 124) (Donne et al., 1997), that selectively binds copper and zinc, and a predominantly helical C-terminal domain. Metal ion regulation is one of several functions ascribed to PrPC in its role in neuron development and maintenance (Aguzzi et al., 2008; Millhauser, 2007).
The prion protein ’s two domains are often thought to be non-interacting – a structured C-terminus with an N-terminal tail. This scheme cannot be correct in light of current cellular and biophysical observations. For example, studies with monoclonal antibodies (mAbs), aimed at identifying reactive surface sites that catalyze the PrPC to PrPSc conversion, find that mAbs with N-terminal epitopes block C-terminal mAbs access and result in partial C-terminal unfolding (Li et al., 2009). In addition, fibroblasts expressing mouse PrPC with the C-terminal E199K mutation (E200K in human), that causes familial CJD, when compared to wild type, exhibit reduced N-terminal copper uptake and are more susceptible to copper toxicity (Canello et al., 2012). Two separate studies show that elimination of a linker segment between the N- and C-terminal domains in transgenic mice results in an embryonic lethal phenotype (Baumann et al., 2007; Li et al., 2007). The developmental derangements in these experiments were interpreted as a consequence of altered PrPC binding to an unidentified partner protein; however, it is also possible that elimination of the linker disrupts higher order PrPC structure. In structural investigations, nuclear magnetic resonance (NMR) shows that the N-terminal domain increases helix order in the C-terminal domain (Zahn et al., 2000). EXAFS work finds that a distal mutation Q212P alter s copper coordination geometry at an N-terminal site localized to His111(D'Angelo et al., 2012). Finally, addition of copper drives association between the N-terminal domain and helix 2 of the C-terminus, possibly leading to a compact structure that facilitates PrPSc formation (Thakur et al., 2011).
The PrPC N-terminal domain binds both copper and zinc in vivo and participates in metal ion homeostasis (Brown et al., 1997). Cu2+ and Zn2+ induce PrP endocytosis (Pauly and Harris, 1998; Sumudhu et al., 2001), upregulate PrP expression (Bellingham et al., 2009; Varela-Nallar et al., 2006), inhibit in vitro fibril formation (Bocharova et al., 2005) and suppress PrPSc amplification (Orem et al., 2006). Mutations in the copper transport protein Atp7a, leading to reduced brain copper content, slows prion disease progression after PrPSc inoculation (Siggs et al., 2012). Moreover, copper, zinc and iron distribution in the mouse brain depends on PrP expression levels (Pushie et al., 2011). PrP is evolutionarily linked to the ZIP metal ion transport family, further supporting its role as a metal binding protein (Schmitt-Ulms et al., 2009). The ways in which Cu2+ and Zn2+ coordinate to N-terminal PrP are distinct. Cu2+ interacts with the octarepeat domain, residues 60 – 91 with the sequence (PHGGGWGQ)4 (Burns et al., 2002; Burns et al., 2003), and also segments at His96 and His111 (human sequence; Figure 1a) (Walter et al., 2009). The specific coordination features depend on Cu2+ to protein ratio (Chattopadhyay et al., 2005; Millhauser, 2007). In contrast, Zn2+ binding is restricted to the octarepeat domain, in which all four histidines coordinate a single Zn2+ ion (Walter et al., 2007), as shown in Figure 1b. The dissociation constant of this coordination mode is approximately 200 µM (Walter et al., 2007).
Figure 1. Sequence and Structure of Human PrPC.
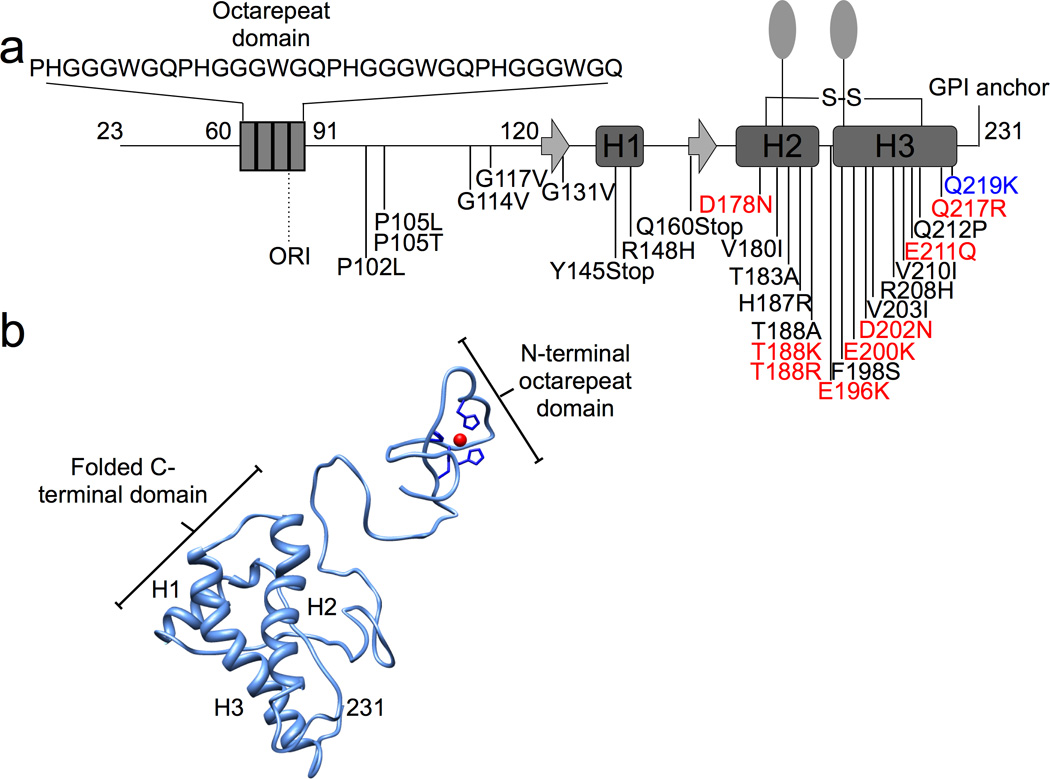
a) Schematic diagram of PrP (human sequence) with numbering indicating secondary structure, post-translational modifications, and pathogenic/ protective mutations. The N-terminus, which contains the octarepeat domain, is unstructured in the absence of Zn2+. The structured C-terminal domain possesses two N-linked carbohydrates (ovals), a disulfide bond, and a GPI anchor. The inherited pathogenic mutations that involve amino acid substitutions resulting in a change of charge are indicated in red. The protective inherited mutation is in blue. b) Zn2+ binds to the histidines within the octarepeat domain, as shown in the preliminary three-dimensional model.
A very recent study shows that PrPC facilitates zinc uptake into neuronal cells mediated by α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors (Watt et al., 2012). Zinc transport by this mechanism is independent of PrPC endocytosis, but does require the PrP octarepeat domain. Moreover, disease associated PrP mutants, or misfolding to PrPSc, inhibits this novel metal ion transport mechanism. These findings may explain why zinc is reduced in the hippocampus and other brain regions in prion disease.
Here we use magnetic resonance to evaluate how metal-ions affect the structure of full-length mouse PrP (MoPrP(23–230)). We use Zn2+ because it is a natural coordinating species that stimulates well-characterized, PrP-linked physiological processes (Brown et al., 1997; Pauly and Harris, 1998; Sumudhu et al., 2001; Watt et al., 2012). Moreover, Zn2+ is diamagnetic thus facilitating the interpretation of the NMR spectra. Using 1H- 15N HSQC NMR, we find that Zn2+ drives a tertiary contact between the octarepeat domain and a specific surface on the C-terminal domain. These findings are supported by direct distance measurements using double electron -electron resonance (DEER) EPR. Analysis of the cumulative data shows that the relevant C-terminal surface participating in this interaction carries the majority of PrP mutations that give rise to familial prion disease. We then examine several disease-associated PrP mutants, as well as a dominant negative mutation, and find a systematic relationship between the type of mutation and the NMR features of the Zn2+-driven inter-domain interaction. Consideration of this relationship suggests that electrostatics play an important role in stabilizing the global PrP-Zn2+ structure, and this finding is supported by direct surface potential calculations. This work identifies a previously unseen higher order structure in PrPC, and provides new insight in to the protein’s fu n ction as w ell as conversion to PrPSc.
Results
Zinc Drives an N-terminal – C-terminal Interaction in MoPrPC
To test whether the N- and C- termini of MoPrP physically interact upon Zn2+ binding, we titrated the metal ion into full-length murine prion protein, MoPrP(23–230), (~ 50 µM) at pH ~7.4 in 50 mM MOPS buffer and acquired 1H- 15N HSQC spectra (Figure 2, Supplementary Figure 1). Spectra of the C-terminal structured region were assigned using the data deposited for MoPrP(90–231) at pH 7.0 (BMRB ID: 16071). HSQC cross peaks derived from PrP-Zn2+ relative to protein alone were assigned to one of three categories: no change, change in chemical shift, or complete disappearance (Supplementary Table 1). The addition of three equivalents of zinc resulted in disappearance of 21 of the 101 assigned backbone amides in the C-terminal domain, and significant shifts of 5 cross peaks. Loss of these cross peaks is consistent with a chemical environment in intermediate exchange. Residues affected by the addition of Zn2+ are mapped onto the secondary structure in Figure 3a. We observed three primary regions of interaction: the short loop just before helix 1, the solvent exposed surface of helix 2 and the N-terminal, exposed surface of helix 3. Amino acids marking the boundaries of these regions are indicated. Figure 3b, a surface plot, shows that these residues form two proximal and nearly contiguous patches located primarily on helices 2 and 3.
Figure 2. Selected region of the 1H-15N HSQC NMR spectra of wild type MoPrP.
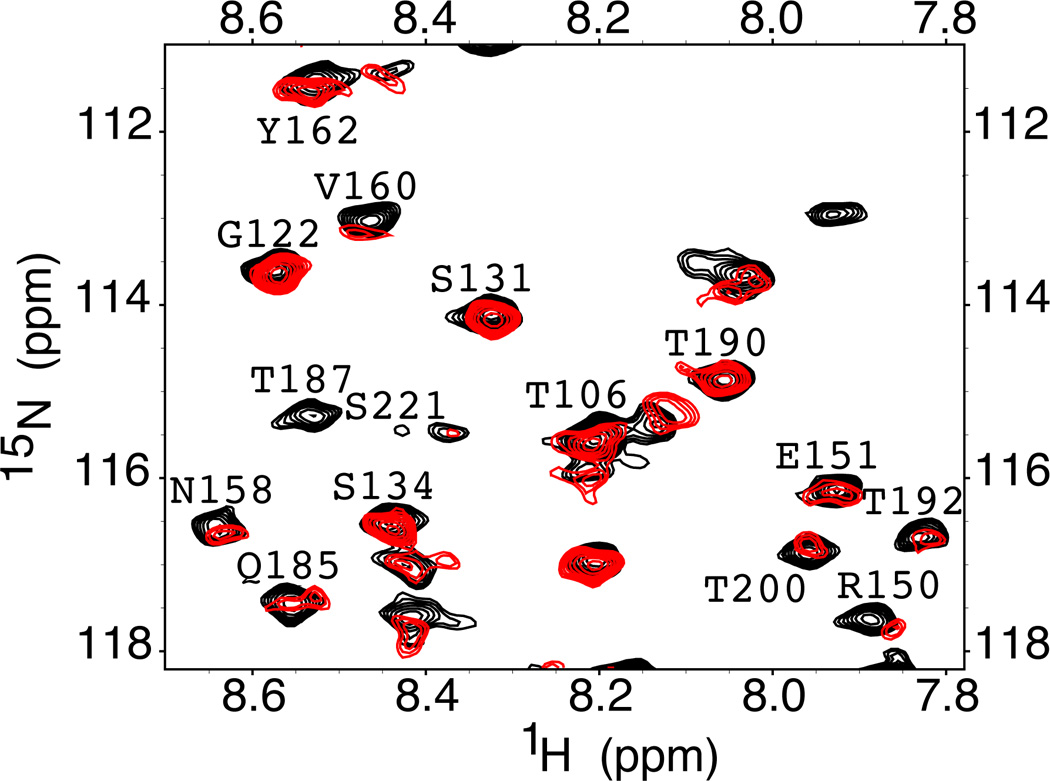
The black spectrum is from the protein in the absence of Zn2+. The red spectrum is from addition of 3 equivalents of ZnCl2 and shows chemical shift changes, and loss or broadening of select cross peaks.
Figure 3. Zn2+ affects specific C-terminal residues.
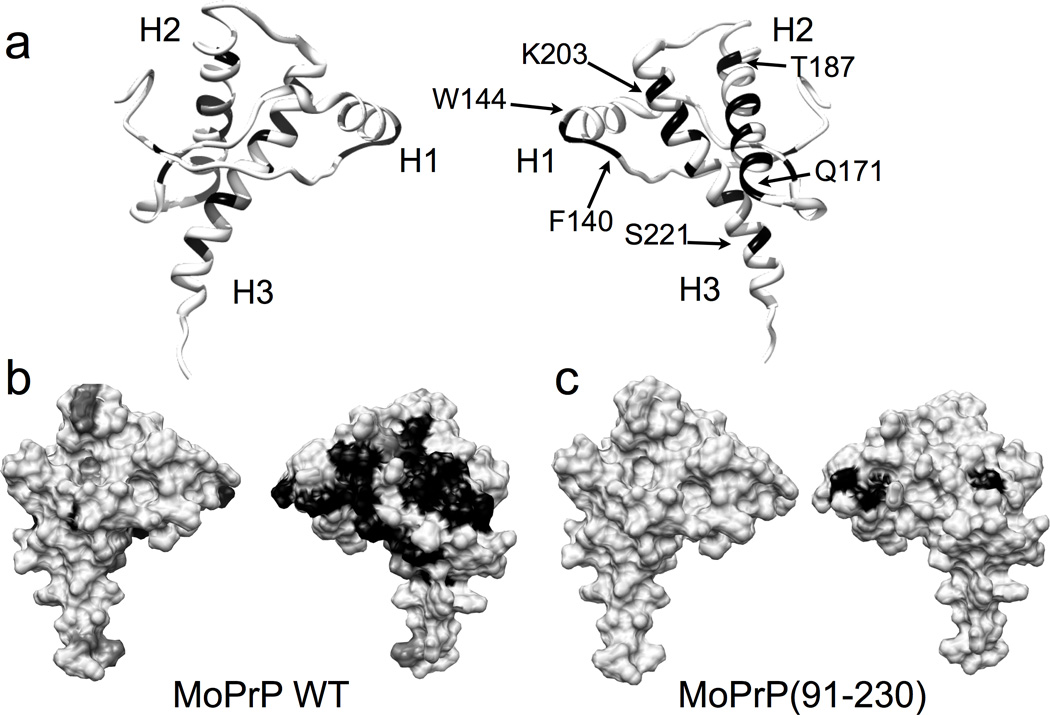
a) Ribbon diagrams showing residues that correspond to the disappeared (black) and significantly shifted (dark gray) peaks in the wild type MoPrP. b) The corresponding surface diagram for the full-length, wildtype MoPrP and c) the C-terminal construct MoPrP(91–230), lacking the Zn2+ binding N-terminal octarepeat domain.
The observed influence of added Zn2+ can arise from either its direct interaction with the C-terminal domain, or by binding first to the N-terminal octarepeat domain, which then associates with the C-terminal domain. To distinguish between these two possibilities, we repeated HSQC titration experiments with MoPrP(91–230), lacking most of the N-terminal region including the octarepeat domain, and the result is shown in Figure 3c. In contrast to full-length PrPC, only three resonances disappeared, while all others remained unaffected. Consequently, the octarepeat domain is required for the observed influence of Zn2+ on the HSQC cross peaks in C-terminal domain. Specifically, Zn2+ itself does not significantly perturb the C-terminal resonances but, instead, coordinates to the octarepeat histidines, partially ordering this domain, which then folds onto the C-terminal domain forming a tertiary contact with the exposed surfaces of helices 2 and 3. Moreover, because MoPrP(91–230) contains histidines 95 and 110, we conclude that the imidazoles from these residues are not sufficient for driving the observed N-terminal—C-terminal interaction found in full-length PrPC. In full-length PrPC, the lack of enhanced chemical shift d ispersion from addition of Zn2+ for residues in the octarepeat domain suggests that the peptid e segments between the coordinating His residues do not take on a specific structure even when docked to the PrP C-terminal domain.
To explore the strength of the N-terminal – C-terminal interaction, the two domains were added in trans, along with Zn2+. Specifically, samples were prepared with 15N MoPrP(91–230), and increasing concentrations of PrP(23–28, 56–90) (50 to 500 µM) and Zn2+ (150 µM to 2.0 mM). HSQC experiments failed to identify significantly shifted or broadened peaks in the C-terminal domain. We conclude, therefore, that the linker region between the N- and C-termini of MoPrP is essential for limiting PrP’s conformational freedom, thereby facilitating the metal-driven inter-domain interaction.
DEER EPR Demonstrates a Zn2+-Dependent Conformational Change
Although the HSQC experiments clearly demonstrate an interaction between PrP’s two domains, they do not provide detail on whether this ordered state is in equilibrium with extended conformations. To directly assess the conformational distribution between the N-terminal and C-terminal domains, we measured select inter-domain distances using four -pulse double electron-electron resonance (DEER) EPR spectroscopy. The DEER technique, in combination with nitroxide site-directed spin labeling (SDSL), is capable of accurately measuring macromolecular distances in the range of 20Å to 80Å and has been successfully applied in the determination of ligand - induced conformational changes in proteins (Jeschke and Polyhach, 2007; Smirnova et al., 2007).
Doubly spin labeled MoPrP constructs were generated using two different site - directed spin labeling techniques. In the N-terminal domain, we used the traditional method of incorporating a Cys at the desired label position, followed by reaction with the MTSSL reagent to give the R1 spin label side chain. Labeling in the C-terminal domain is more challenging given the essential disulfide bond between helices 2 and 3. To avoid non-native disulfides that might arise from a third Cys residue, we used genetic methods to incorporate the unnatural amino acid p -acetyl phenylalanine, followed by reaction with a hydroxylamine nitroxide reagent to produce the ketoxime - linked K1 side chain (Fleissner et al., 2009). Detailed reactions are given in Supplementary Figure 2. To test whether DEER with the K1 label would report meaningful distances, we produced three doubly K1 labeled PrP constructs with both labels in the structured C-terminal domain. As shown in the Supplementary Figure 2, DEER measurements agreed with the expected distances to within 5Å.
We developed three R1/ K1 doubly labeled PrPC constructs, and performed DEER measurements with and without Zn2+ (Figure 4). The first contained the R1 side chain at position 66, within the octarepeat domain, and K1 at position 200, which is a surface site in the N-terminal region of helix 3. The HSQC experiments suggest a Zn2+ driven interaction between these two regions (Figure 3). In the absence of Zn2+, the dipolar evolution for PrP(Q66R1/ T200K1) is shallow and the resulting distance distribution between the two labels is broad, consistent with an ensemble of distances between 20Å and 60Å, as shown in Figure 4. However, upon addition of excess Zn2+, the dipolar evolution deepens and the distance distribution sharpens with a pronounced peak at approximately 22 Å. Along with the NMR experiments, these data demonstrate a close contact between the Zn2+ occupied octarepeat domain and the N-terminal segment of helix 3.
Figure 4. Zn2+-mediated inter-domain interaction measured by DEER.
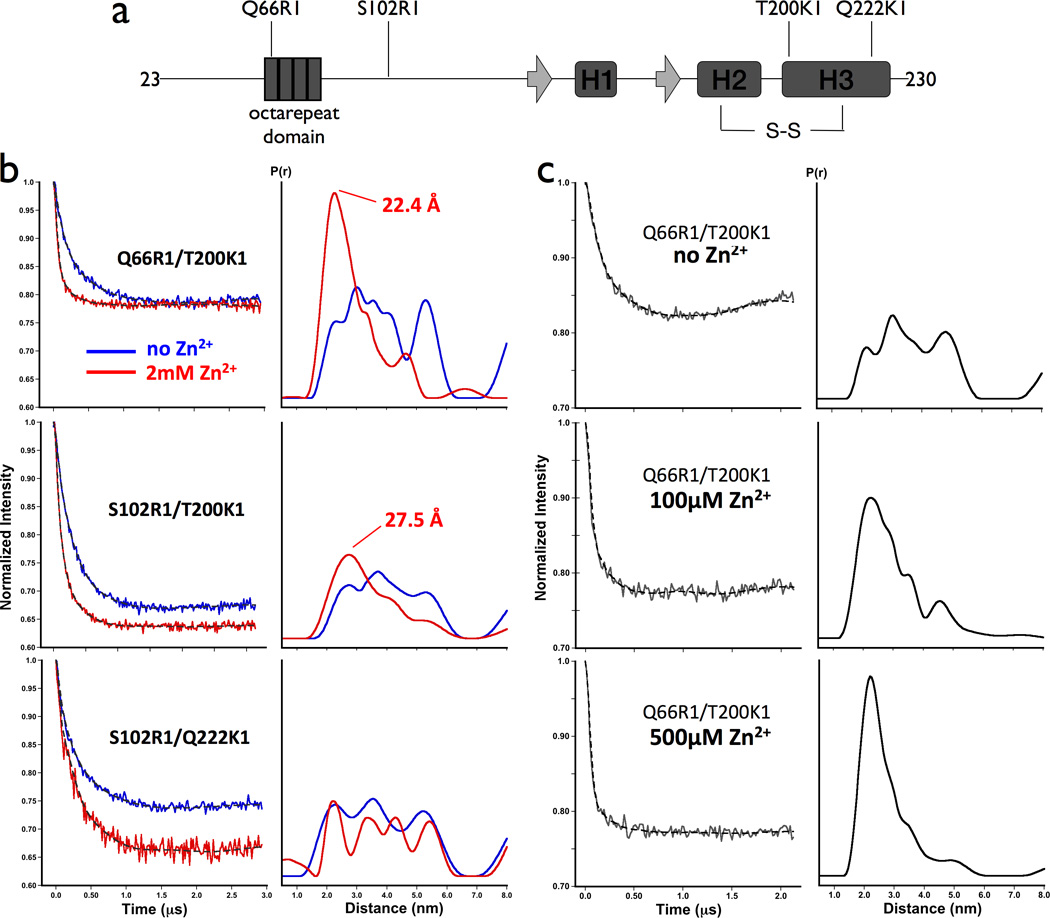
a) Schematic representation of MoPrP showing the location of spin labeled side chains in relation to known secondary structures. b) Background -corrected dipolar evolution spectra and distance distributions obtained by Tikonov regularization for the three doubly-labeled constructs in the absence of Zn2+ (blue) and in the presence of 2mM ZnCl2 (red). c) Dipolar evolution spectra and distance distributions for MoPrP Q66R1/ T200K1 with 0, 100, and 500µM ZnCl2. Protein concentrations were between 80 and 100 µM.
The next construct, PrP(S102R1/ T200K1), has the R1 label in the ~30 amino acid linker region between the octarepeat domain and the folded C-terminus. Addition of Zn2+ also sharpens the distance distribution, with a peak at 28Å, but less so than observed for PrP(Q66R1/ T200K1). This result suggests that the linker retains partial conformational heterogeneity. Finally, we examined PrP(S102R1/ Q222K1), with labels in the linker region and at the C-terminus of helix 3, which is just outside of the interaction domain identified by the NMR experiments. Here we observe no sharpening of the distribution upon the addition of Zn2+. Taken together, these data support the formation of an organized structure in the presence of Zn2+ where the octarepeat domain docks against the surface of PrPC’s C-terminal domain identified by the NMR experiments.
The strongly interacting PrP(Q66R1/ T200K1) was further examined with a Zn2+ titration experiment, as shown in Figure 4c. The distribution derived from the DEER experiment sharpens between 100 µM and 500 µM Zn2+, consistent with the Zn2+ dissociation constant of 200 µM.
Pathogenic Mutants Alter the Inter-domain Tertiary Contact
We noticed that the region that forms the C-terminal docking surface for the Zn2+ occupied octarepeats (Figure 3) also carries the majority of the PrP point mutations that confer inherited disease (Mead, 2006) (Figure 1). To test whether familial mutations alter the Zn2+-driven tertiary structure, we repeated our HSQC titration experiments with MoPrP(E199K) and MoPrP(D177N), which correspond to the human mutations E200K and D178N, respectively. The results, shown in Figure 5a and b, reveal a significant decrease, relative to wild type, in the number of affected C-terminal residues. Specifically, MoPrP(E199K) shows a decrease in the size of the affected patches on both helices 2 and 3, with 13 peaks of 99 assigned peaks broadened to the point of being unobservable. With MoPrP(D177N), much of the interaction with helix 2, as observed in the wild type protein is lost, with 17 of 94 peaks total C-terminal peaks unobservable due to broadening.
Figure 5. C-Terminal mutations that affect the course of prion disease show different interaction patterns from wild type.
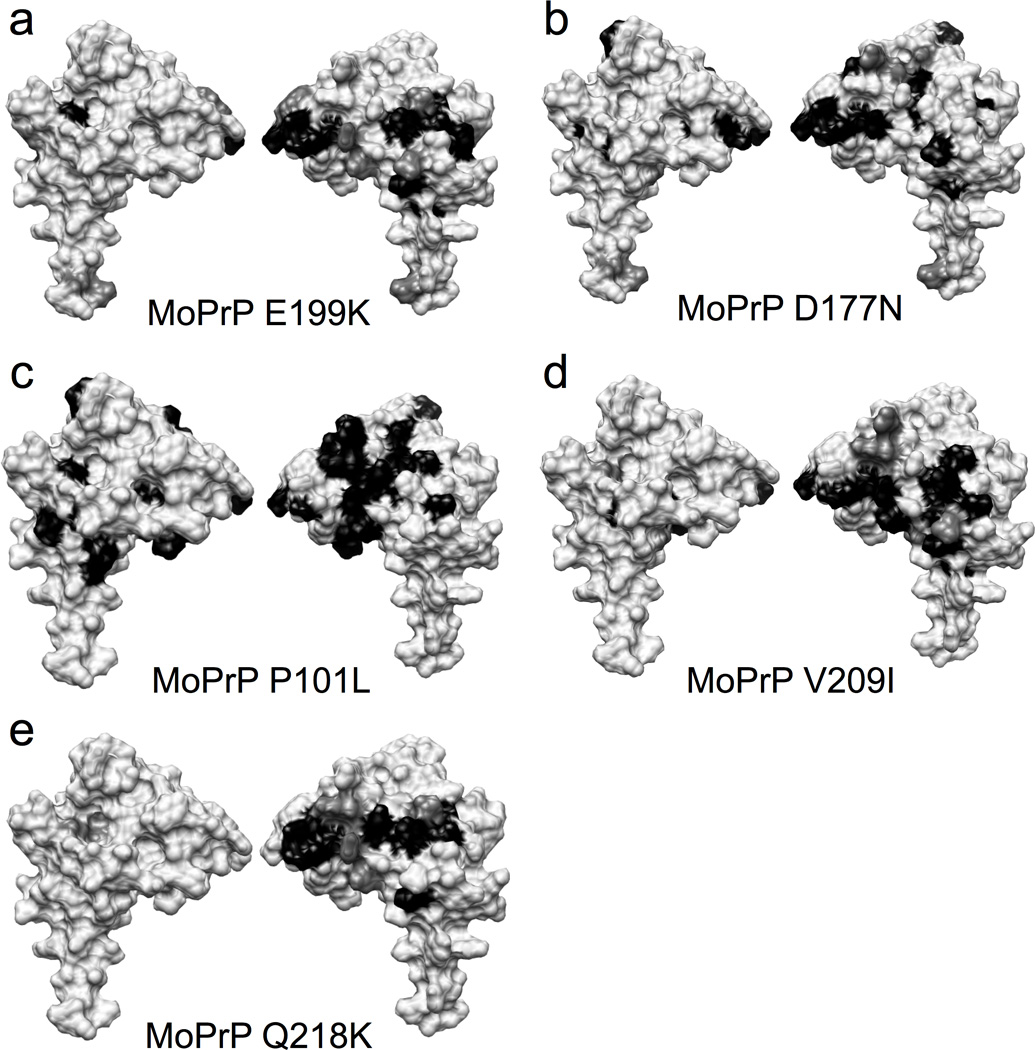
Surface diagrams showing the disappeared (black) and significantly shifted (dark gray) peaks in the path ogenic mutants (a–d), and the dominant negative mutant (e).
We also examined MoPrP(P101L), corresponding to the P102L mutation in humans that causes Gerstmann-Straussler-Scheinker disease (GSS) (Mead, 2006). This mutation, which is in the linker between the octarepeat and C-terminal domains, results in loss of 26 of 97 peaks due to broadening (Figure 5c). However, whereas most of the affected residues are in helices 2 and 3, consistent with wild type, we also observed broadening or disappearance of peaks from residues on the opposite side of the C-terminal domain, suggesting interaction with the antiparallel β-sheet.
We also investigated MoPrP(V209I), which corresponds to the human V210I mutation that results in CJD (Prusiner, 2003). The introduction of bulkier hydrophobic isoleucine residue to helix 3 caused 15 of 89 assigned peaks to broaden to the point of disappearing. The patch of affected residues on the surface of helices 2 and 3 in this mutant was more compact as compared to the wild type protein.
Finally, we performed experiments on MoPrP(Q218K), which is the dominant negative Q219K polymorphism in humans. As demonstrated in statistical analyses of Asian populations, individuals with this allele resist the development of sporadic CJD (Shibuya et al., 1998). Our results show that the patches of interaction on helices 2 and 3 are largely preserved, although several peaks that were completely broadened in wild type are now visible and only partially broadened.
The results above suggest that pathogenic mutations lead to a systematic weakening of the Zn2+ driven association between PrPC’s N - and C-terminal domains. To test this hypothesis in a more thorough and quantitative fashion, we determined the peak intensities of all observable HSQC cross peaks for wild type and mutant proteins. Peak heights decrease with an increase in line width and therefore serve as a convenient metric for residues affected by association with the Zn2+-occupied N-terminal domain (Panchal et al., 2003). We determined I/ Io for all assigned residues in the C-terminal helices, where I and Io are the peak intensities in the presence and absence of Zn2+, respectively. The results are shown in Figure 6. MoPrP(91–230), lacking the N-terminal domain, shows only a minimal effect, as expected. Approximately half of the r esidues in helices 1 and 3 are near I/ Io ≈ 1. Helix 2 shows some systematic broadening near residues 178 to 180. Full-length MoPrP, however, shows a dramatic decrease in I/ Io along all helical segments, with the greatest effect at helix 2. In addition to residues broadened to zero and thus unobservable (I/ Io = 0, Figure 3), a significant number of residues are reduced to < 0.25 of their original intensity. The greatest influence is observed at helix 2.
Figure 6. Normalized peak intensities show different patterns for disease mutants, relative to wild type and the dominant negative.
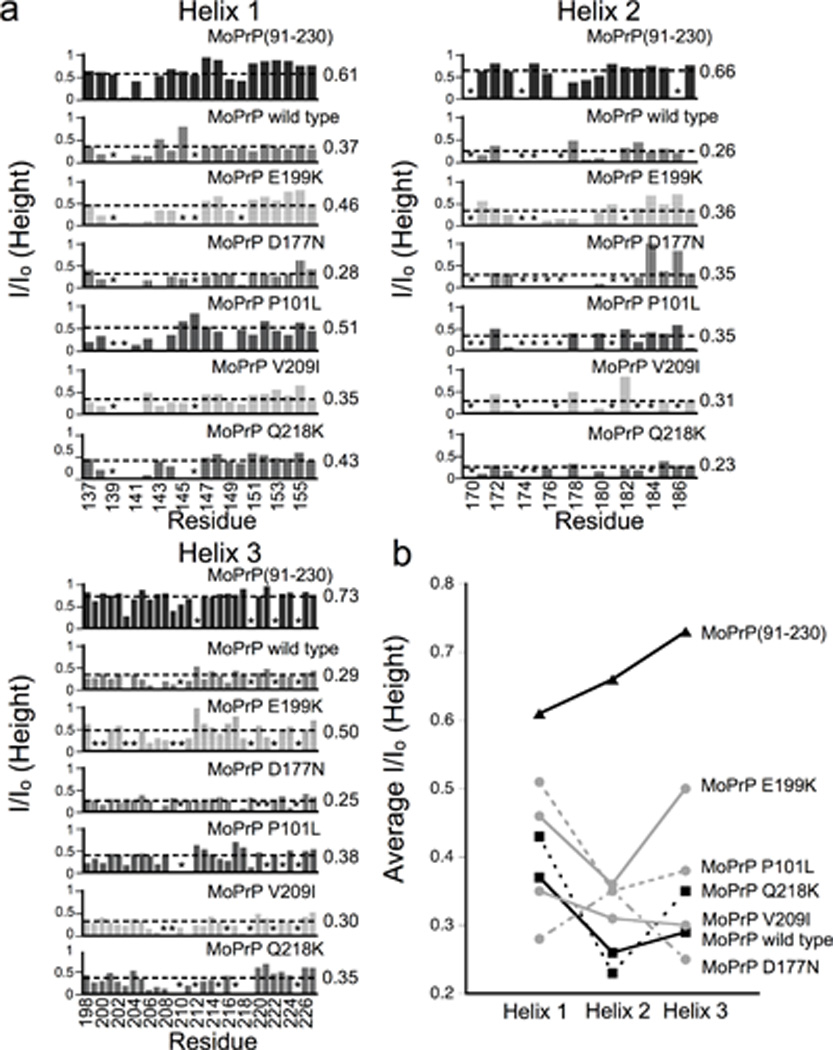
a) Cross peak intensity profiles for helices 1, 2 and 3 are compared for all proteins in this study. Asterisks mark unassigned peaks and the dashed lines represent the average I/ Io values. b) A plot of the average I/ Io for each helix comparing wild type and protective mutant (black squares) with pathogenic mutant s (gray circles). Residues in helix 2 exhibit lower I/ Io (greater broadening) for wild type and dominant negative compared to familial mutants.
Each disease-associated mutant MoPrPC gives a unique I/ Io profile. However, we observe a distinct pattern for residues 182 – 187 at the end helix 2, which show a systematic increase in I/ Io relative to wild type. To extract composite values, thus smoothing out site-by-site variations, we determined the average I/ Io value for each helix from the set of proteins. These values are reported to the right of each helix profile in Figure 6 and plotted in Figure 6d. Average I/ Io values for helices 1 and 3 appear to be randomly distributed and show no clear pattern. However, the average I/ Io values for helix 2 cluster around two values. Wild type and the dominant negative Q218K show the greatest line broadening, with average I/ Io values of 0.26 and 0.23, respectively. Disease associated mutants cluster at approximately 0.35, consistent with a systematic change of the interaction with the Zn2+-occupied octarepeat domain.
Electrostatics Show a C-terminal Negative Patch at the Interaction Surface
Of the 17 pathogenic mutations in helices 2 and 3, eight involve a shift in charge, and without exception, all of these specific mutations increase positive charge by either loss of an acidic side chain, gain of a basic side chain, or both (Shen and Ji, 2011). Among these charge increasing mutations are those responsible for the most prevalent inherited diseases: E200K (change of +2) that results in the development of CJD (Mead, 2006) and D178N (change of +1), which confers either fatal familial insomnia or CJD, depending on the Met/ Val polymorphism at position 129 (Goldfarb et al., 1992).
Based on this observation, we hypothesized that the local increase in C-terminal positive charge presented on helices 2 and 3 would electrostatically weaken the interaction with the Zn2+-occupied octarepeat domain. We therefore examined the surface charge properties of these constructs using electrostatic calculations. The NMR structure of MoPrP(121–230) (PDB ID:1XYX) was used for a surface potential calculation and the Adaptive Poisson-Boltzmann Solver (APBS) (Baker et al., 2001; Dolinsky et al., 2007) produced the electrostatic surface. The results show an intensely negative region centered between helices 2 and 3 that is composed of E199, E206, E210, and D177 (Figure 7A). This negative patch, previously described by Zhang et al. (Zhang et al., 2000) is the same area that experiences the greatest change in the 1H- 15N HSQC spectra upon the addition of zinc (Figure 3). Interestingly, other negative regions exist on the surface of the protein, for example at the C-terminus of helix 3, but HSQC peaks corresponding to this region are not greatly affected in the NMR experiments.
Figure 7. Electrostatic surface diagrams of wild type MoPrP and mutants.
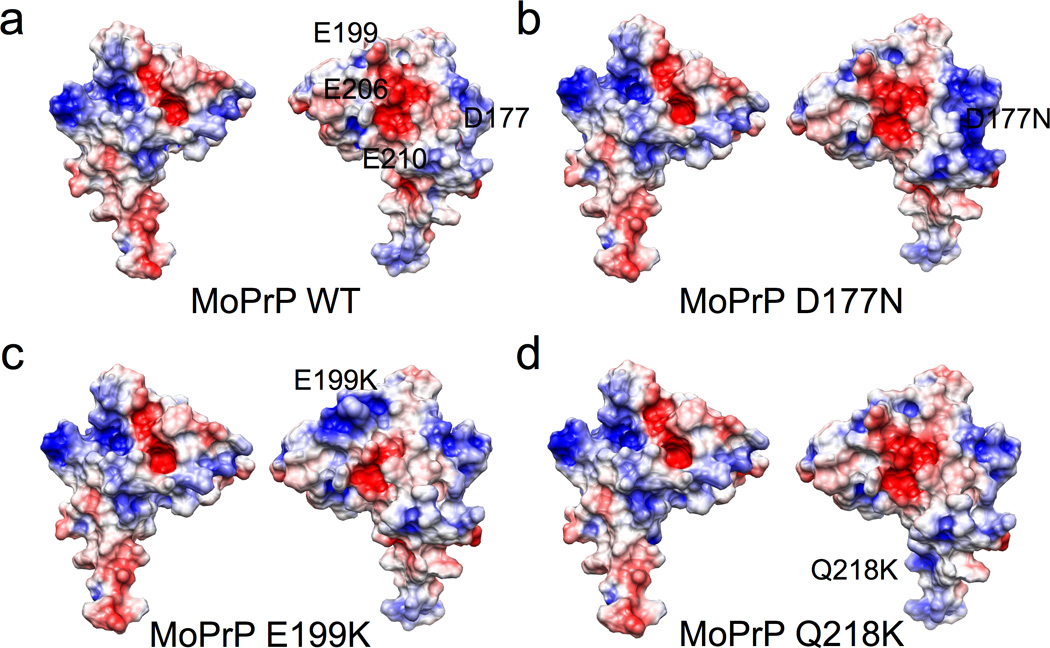
a) wild type as compared to the pathogenic mutants in b) and c). d) is the dominant negative. Blue denotes positive charge and red is negative charge.
We then used computational techniques to produce structural models of the pathogenic and dominant negative mutants, and then examined their surface electrostatics with APBS. D177N shows a slightly attenuated negative patch due to the loss of charge (Figure 7b). E199K has a greater change in the negative region owing to replacement of a negative charged glutamate with a positively charged lysine at the N-terminus of helix 3 (Figure 7C). Q218K showed a more diffuse negative electric potential between helices 2 and 3 (Figure 7D). These changes in the electrostatics of the surface of the protein are consistent with the peak broadening/ shifting observed in the NMR spectra. We hypothesize that the N - and C-terminal interaction may be weakened in the two pathogenic mutants due to the loss of negative charge in either helix 2 or the N-terminus of helix 3.
The surface electrostatics of helices 2 and 3 are not changed in the pathogenic mutants MoPrP(P101L) and MoPrP(V209I), and therefore do not explain these con stru ct’s weaken ed N-terminal – C-terminal interaction. For MoPrP(P101L), the loss of the conformationally rigid proline residue may alter the ability of the Zn2+ occupied N-terminus to wrap around and interact with the C-terminus upon zinc binding.
Given the electrostatic nature of the Zn2+ driven association between the octarepeat and C-terminal domains, we wanted to determine whether the interaction is enhanced by the relatively low ionic strength solution in the NMR samples. All experiments above were performed in 50 mM MOPS buffer (added as a 1:1 sodium salt), which has an approximate ionic strength of 25 mM. PrPC precipitates with the addition of simple monovalent salts so, to increase ionic strength, we increased the MOPS concentration. HSQC experiments performed at a four -fold higher MOPS concentration, ionic strength ≈ 100 mM, gave results with line broadening and profiles similar to those presented in Figures 5 and 6. Consequently, ionic strengths up to physiological levels do not appear to fundamentally alter the PrPC inter-domain interaction.
The concept that emerges from our findings is that the Zn2+ coordinated octarepeat domain docks to the surface of PrPC’s C-terminal domain formed by helices 2 and 3. This tertiary interaction is likely assisted and localized by an electrostatic attraction between the Zn2+ ion and the negatively charged C-terminal patch arising from a cluster of acidic residues. To explore the feasibility and geometric features of this higher order organization in PrPC, we used molecular dynamics to simulate the docking of Zn2+-bound N-terminus to the negative patch on the C-terminus of PrP. The Zn2+ ion was coordinated by the four histidine epsilon nitrogens within the octarepeat domain at a set bond distance of 1.99 Angstroms, and defined inter-atom angles to maintain tetrahedral geometry. Beyond this, the N-terminal domain up to residue 125 was left otherwise unrestrained. The C-terminal domain backbone was confined to its NMR structure (PDB 1XYX). The solvated starting structure was energy minimized and then subjected to 600 picoseconds of simulated annealing with the two DEER-derived distance restraints, S102 to T200 and Q66 to T200, both set at 10 to 20 Angstroms. In addition, we included two distance restraints suggested by the HSQC spectra and electrostatic calculations: D177 to Zn2+ and E199 to Zn2+, both at 5 to 15 Angstroms. Next, the distance restraints were removed, and the protein was subjected to 400 picoseconds of constant pressure and volume simulation, which yielded no significant changes to the protein structure. The resulting model, Figure 8, shows that the Zn2+- bound octarepeat region (purple) docks at the C-terminal end of helix 2 and the N-terminal end of helix 3. The C-terminal docking surface is formed primarily by the contiguous sequence from residue 177 to 210 (mouse numbering), which contains the sites of the majority of mutations in familial prion disease. The flexible Zn2+ occupied octarepeat domain fully encompasses this surface.
Figure 8. Model of the Zn2+ mediated N-terminal–C-terminal interaction.
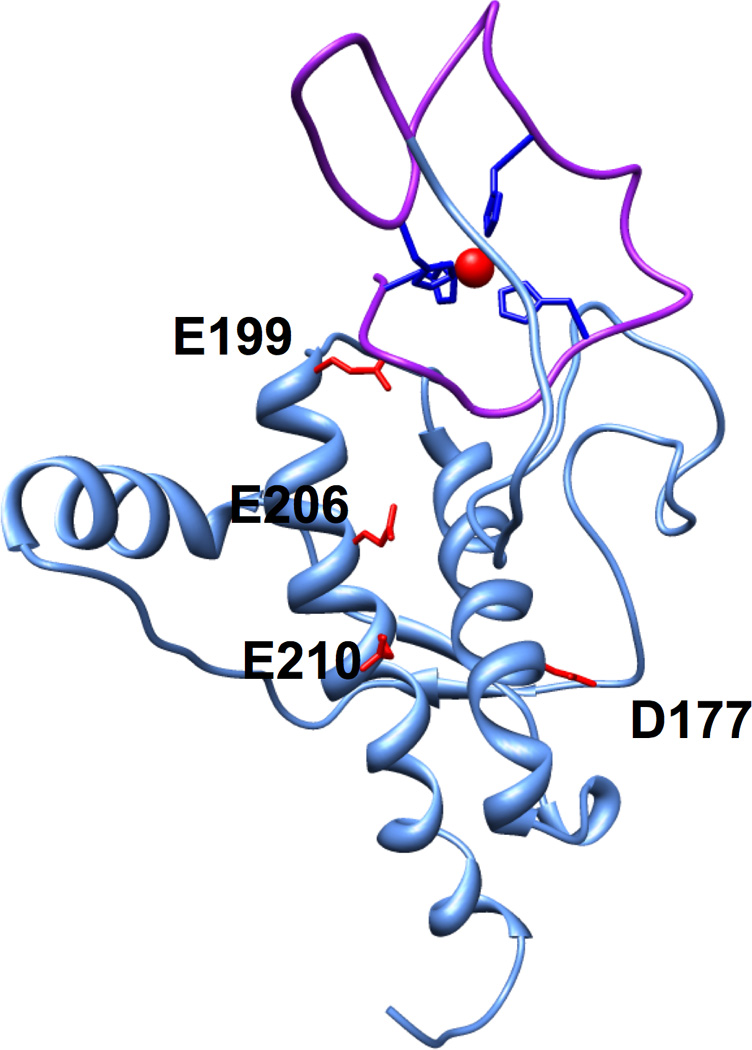
The octarepeat domain (purple) containing the bound Zn2+ (red) was docked against the relevant C-terminal surface using restraints from DEER and NMR. The acidic residues that define the negative patch on helices 2 and 3 are highlighted.
Discussion
Prior NMR studies on full-length PrPC found little order in the N-terminal region proceeding residue 120; treatment of PrPSc with proteinase K cleaves away the N-terminus around residue 90, but without loss of infectivity (Flechsig et al., 2000; Prusiner, 1989). As such, PrPC’s N-terminal region has often been considered an appendage – an “N -terminal tail” – with little role in structure or misfolding to PrPSc. Our results motivate a reconsideration of this scheme. The higher order structure and new tertiary contact identified here requires a divalent metal ion cofactor to coordinate to the four conserved, octarepeat His residues. Both NMR and EPR show consistently that the Zn2+ occupied octarepeat domain makes a tertiary contact encompassing the surface on the C-terminal domain formed by helices 2 and 3.
HSQC experiments with full-length wild type protein in the presence of Zn2+ show the greatest influence on the C-terminal residues, as indicated by peak broadening. In contrast, parallel NMR experiments with PrP(91–230) lacking the octarepeat domain show almost no change in C-terminal line shapes or chemical shift. PrPC mutants are intermediate between these two extremes. It is logical, therefore, to propose that the N-terminal – C-terminal interaction is weakened relative to wild type in the familial mutant proteins. However, because the spectra for all full -length proteins are in intermediate exchange, we treat this interpretation with caution. The mutations may influence the dynamics of the loops between the N-terminal histidines that coordinate Zn2+, chemical exchange between N-terminal – C-terminal open and closed structures, or the chemical shifts at the interface. Nevertheless, the clustering of the average linewidth of the helix 2 resides among wildtype and the Q218K versus that observed for the familial mutants (Figure 6d) strongly suggests that the latter proteins possess systematically altered structures or dynamics.
Our results reveal a previously unseen correlation between inherited prion disease mutations and an in vitro structural change in PrP. Early investigations hypothesized that mutations would thermodynamically destabilize the C-terminal structure of PrPC rendering the protein susceptible to misfolding (Cohen et al., 1994; Huang et al., 1994). However, structure and denaturation studies reveal only minimal changes in three-dimensional fold, and no systematic variation in stability (Bae et al., 2009; Biljan et al., 2011; Ilc et al., 2010; Lee et al., 2010; Liemann and Glockshuber, 1999; Swietnicki et al., 1998; Zhang et al., 2000). Our investigation of the pathogenic mutants shows that the D177N and E199K mutations systematically alter the N-terminal – C-terminal tertiary interaction, likely by reducing the negative surface potential on helices 2 and 3 that attracts the Zn2+-bound N-terminus. Thus, this class of charge altering mutations may reduce stability by affecting the new tertiary fold in PrPC revealed here. It has been noted that familial disease arises from mutation directional selection in which all mutations either increase positive charge or hydrophobicity (Shen and Ji, 2011). Our findings provide a preliminary explanation for those mutations involving a change in charge. In contrast, the cases of P101L and V209I, surface electrostatics cannot explain the reduced interactions we observed at helix 2. The loss of proline may remove conformational restrictions in the linker region, resulting in inhibition of the inter-domain interaction. Data in Figure 5 do suggest that the Zn2+ occupied octarepeats in MoPrPC(P101L) make contact with the C-terminal domain on the surface opposite to that formed by helices 2 and 3, possibly due to alteration in linker conformation. In contrast to findings with the familial disease mutations, the dominant negative mutant Q218K, located near the end of helix 3, results in a modest strengthening of the Zn2+ driven tertiary interaction when compared to the wild type and pathogenic mutants. From this perspective, a weakening of the N- and C-terminal interaction may contribute to disease while a strengthening of the interaction may be protective.
This zinc mediated N-terminal – C-terminal interaction may contribute to the understanding of PrP’s function and trafficking in the cell. Zn2+ initiates PrPC internalization through endocytosis (Pauly and Harris, 1998; Sumudhu et al., 2001). Coordination to PrPC is hypothesized to cause a conformational transition that moves the protein laterally out of lipid rafts to be endocytosed via clathrin-coated pits (Hooper et al., 2008). The structural change induced by zinc binding identified here offers an explanation. The Zn2+ driven tertiary association could either displace a lipid raft targeting protein, or create a bin d in g site for proteins that facilitate PrP’s endocytosis, such as LRP1 (LDL receptor-related protein 1). Similarly, PrP is hypothesized to act as a zinc sensor, in which it detects extracellular levels up to a threshold (Watt and Hooper, 2003). Exceeding this threshold initiates a signal transduction cascade that enables the cell to protect itself against toxic amounts of metal. The conformational change caused by the inter-domain interaction may act as the trigger for this signal cascade. To this juncture, we’ve considered the N-terminal – C-terminal interaction as an intramolecular property. However, given the length of the linker region between the PrPC domains, it is also possible that proximal PrPC copies domain swap, with the N-terminal domain of one PrPC docking to the C-terminal domain of its neighbor. In this scenario, Zn2+ facilitates non-covalent crosslinking that could stimulate transmembrane processes.
It is well established that PrPC takes up both Cu2+ and Zn2+ (Chattopadhyay et al., 2005; Jackson et al., 2001; Millhauser, 2007; Walter et al., 2007). Our initial experiments here focused on zinc because it binds with only one coordination mode and is diamagnetic, thus avoiding paramagnetic line broadening. However, a recently published NMR study with PrPC in the presence of excess copper showed an interaction at helix 2, residues 174–185 (Thakur et al., 2011), which is supported by our results here. At full Cu2+ saturation, however, the N-terminal domain can take up to six equivalents, so developing a three-dimensional structure based on paramagnetic line broadening is challenging. Nevertheless, it is clearly important to evaluate how metal driven tertiary structure varies with concentration, and for example, whether low versus high Cu2+ levels d rive additional conformational transitions.
In summary, our findings here reveal a fundamentally new tertiary structure in PrPC, assisted by Zn2+ coordination in the octarepeat domain. The correlation we find with familial disease mutants suggest that stabilization of this tertiary fold creates a barrier to misfolding and the formation of PrPSc. In support of this, both Cu2+ and Zn2+ inhibit in vitro amplification of prions (Orem et al., 2006). Small molecules or accessory proteins that support this newly identified fold could serve in the treatment of the TSEs.
Materials and Methods
15N-labeled Protein Expression
MoPrP variants (created with the GeneArt Site Directed Mutagenesis kit (Invitrogen)) were constructed using the template plasmid pET101 MoPrP (Invitrogen) containing full-length Mus musculus PrP(23–230). The C-terminal fragment, MoPrP(91–230), was cloned into the pET101 vector using EcoRI and XhoI restriction sites. All constructs and mutations were confirmed by DNA sequencing. Protein expression was executed in E.coli BL21Star (DE3) (Invitrogen). All 15N labeled proteins were grown in M9 minimal media supplemented with 1 g L−1 15N ammonium chloride. Cells were grown at 37°C until they reached an OD600 of 0.6, at which they were then transferred to the M9 minimal media and protein expression was induced with 1mM isopropyl-1-thio- D-galactopyranoside (IPTG) for three hours. The full-length constructs were solubilized from inclusion bodies in 8M urea and run over a Ni2+ charged immobilized metal affinity column (IMAC) on an AKTA purifier system (GE Healthcare). MoPrP(91–230) was purified from inclusion bodies prior to running over a Q-Sepharose anion exchange column. All of the proteins were then further purified using reverse phase high performance liquid chromatography (HPLC), and subsequently identified by mass spectrometry. The proteins were then lyophilized and quantified in guanidinium chloride (GdHCl) by UV-vis spectrophotometry prior to use in the NMR experiments.
Spin Labeled PrP Sample Preparation
Expression of mutant PrP for dual spin labeling was achieved by a method similar to that described by Fleissner et al (Fleissner et al., 2009). Briefly, PrP containing site specific cysteine and amber (TAG) codon mutations were co-transformed into BL21(DE3) cells along with the pEVOL vector specific for pAcPhe incorporation (Young et al., 2010). The growth media was supplemented with 400mg/ L pAcPhe (Synchem) and the cells were grown to an OD600 of 0.6 – 0.8. Expression was induced with 1mM IPTG and 300mg/ L L-Arabinose and the cells were incubated for an additional 5-7 hrs at 37°C.
Ni2+ affinity purification and HPLC were performed as described above. HPLC purified protein was then buffer exchanged into 10mM sodium acetate pH 4.5 and concentrated through a 10kDa MWCO membrane (Amicon). A 10-fold molar excess of MTSSL spin label (Toronto Research Chemicals) was added and allowed to react for 6 – 12 hrs at room temperature. The reaction mixture was again purified by HPLC and lyophilized. The dried protein was dissolved in 10 mM sodium acetate buffer at pH 4 to a concentration of approximately 100 µM. A 10-fold molar excess of the hydroxylamine spin label HO-4120, synthesized in-house using published procedures (Fleissner et al., 2009; Hankovszky, 1981; Powell, 2000), was added and the reaction was incubated at 37°C for 24 – 28 hrs before a final round of HPLC purification.
Samples for EPR were made in 25 mM MOPS pH 7.4 in D2O containing 30% v/ v d8-glycerol. Samples containing Zn2+ were made using a stock solution of ZnCl2 in D2O. Double spin labeled samples were loaded into 4 mm (o.d.) quartz EPR tubes (Wilmad 707-SQ) and flash frozen in liquid nitrogen p rior to spectral analysis.
Nuclear Magnetic Resonance (NMR) Spectroscopy
All samples were prepared in a buffer containing 50mM MOPS hemisodium salt (Sigma), 10% D2O, 0.1% NaN3, and 0.2mM TMSP at pH 7.39, except in the case of varying salt concentrations where 25mM and 200mM MOPS was used . Protein was added to final concentration of 50µM and either 50µM or 150µM ZnCl2 was added to the samples containing zinc. Zn2+ concentrations beyond this tended to result in protein precipitation. 1H- 15N HSQC spectra were recorded at 25°C on an Avance II 900-MHz spectrometer (Bruker-Biospin, Boston, MA) at the Central California 900-MHz NMR facility (Berkeley, CA). NMR spectra were analyzed with NMR Pipe and Sparky. Structural analysis was done with UCSF Chimera.
Double Electron-Electron Resonance (DEER) Spectroscopy
4-pulse DEER data was collected on a Bruker ELEXSYS E580 X-band spectrometer equipped with an MD-5 dielectric resonator and a second frequency DEER module (Bruker). The pump pulse was fixed to the center peak in the field swept nitroxide spectrum and the probe frequency was chosen 65–75 MHz above this frequency. π/2 and π pulses were 16 and 32 ns respectively. The delay between the first and second probe pulses was 400 ns and dipolar evolution data was collected out to 2.5 – 3.5 µs. Experiments were run at 50 or 80 K, depending on the sample, and were signal averaged for 6 – 8 hrs. The raw data was background corrected and analyzed by Tikhonov regularization u sing the DeerAnalysis 2011 software package (Jeschke, 2006).
Electrostatic Calculations
The electrostatic calculations were performed on MoPrP(121–230) (PDB ID: 1XYX) with the Adaptive Poisson-Boltzmann Solver (APBS) plug-in in PyMol (Baker et al., 2001). The mutations in the pathogenic mutants were introduced with the swapaa command in Chimera (starting PDB ID: 1XYX), prior to calculating the surface electrostatics with APBS.
Molecular Dynamics Simulations
The molecular dynamics simulation package GROMACS (Berendsen, 1995; Lindahl, 2001) was used to create a molecular model of the inter-domain interaction using the OPLS-AA/ L force field (Jorgensen, 1996; Kaminski, 1994) and SPC water model (Berendsen, 1981; Berendsen, 1984). Dr M. Jake Pushie generously donated the starting structure with Zn2+ bound. The initial structure was subjected to 1000 steps of steepest-descent energy minimization in the presence of explicit solvent. The protein was then subjected to 600 picoseconds of simulated annealing with four additional distance restraints derived from the magnetic resonance experiments. After the protein structure stabilized, 400 picoseconds of production molecular dynamics simulation was run to produce the final model.
Supplementary Material
Highlights.
-
-
Zinc binds to the prion protein octarepeat domain
-
-
The zinc-bound octarepeat makes a tertiary contact to C-terminal helices 2 and 3
-
-
The encompassed surface carries a majority of the mutations in familial prion disease
-
-
Familial mutations weaken this zinc-driven tertiary contact
Acknowledgements
This work was funded by a grant from the National Institutes of Health (GM065790 awarded to GM). The 900 MHz NMR instrument was obtained from funds provided by the NIH (GM68933). The authors thank Ms. Audrian Howard for assistance in the preparation of several PrP mutants, Dr. Jake Pushie (University of Saskatchewan) for kindly providing the starting coordinates and advice on the molecular dynamics calculations, and Drs. Seth Rubin (UC Santa Cruz) and David Wemmer (UC Berkeley) for helpful comments on the manuscript. Drs. Wayne Hubbell and Mark Fleissner (UCLA) are gratefully acknowledged for their assistance with technologies for incorporation of the K1 label.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguzzi A, Baumann F, Bremer J. The prion's elusive reason for being. Annu Rev Neurosci. 2008;31:439–477. doi: 10.1146/annurev.neuro.31.060407.125620. [DOI] [PubMed] [Google Scholar]
- Bae SH, Legname G, Serban A, Prusiner SB, Wright PE, Dyson HJ. Prion proteins with pathogenic and protective mutations show similar structure and dynamics. Biochemistry. 2009;48:8120–8128. doi: 10.1021/bi900923b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann F, Tolnay M, Brabeck C, Pahnke J, Kloz U, Niemann HH, Heikenwalder M, Rulicke T, Burkle A, Aguzzi A. Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J. 2007;26:538–547. doi: 10.1038/sj.emboj.7601510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellingham SA, Coleman LA, Masters CL, Camakaris J, Hill AF. Regulation of prion gene expression by transcription factors SP1 and metal transcription factor-1. J Biol Chem. 2009;284:1291–1301. doi: 10.1074/jbc.M804755200. [DOI] [PubMed] [Google Scholar]
- Berendsen HC, Postma JPM, Fvan Gunsteren W, Hermans J. Interaction models for water in relation to protein hydration. In: Pullman B, editor. Intermolecular Forces. Dordrecht, Reidel: 1981. pp. 331–342. [Google Scholar]
- Berendsen HJC, van der Spoel D, van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput Phys Commun. 1995;91:43–56. [Google Scholar]
- Berendsen HJC, Postma JPM, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- Biljan I, Ilc G, Giachin G, Raspadori A, Zhukov I, Plavec J, Legname G. Toward the molecular basis of inherited prion diseases: NMR structure of the human prion protein with V210I mutation. J Mol Biol. 2011;412:660–673. doi: 10.1016/j.jmb.2011.07.067. [DOI] [PubMed] [Google Scholar]
- Bocharova OV, Breydo L, Salnikov VV, Baskakov IV. Copper(II) inhibits in vitro conversion of prion protein into amyloid fibrils. Biochemistry. 2005;44:6776–6787. doi: 10.1021/bi050251q. [DOI] [PubMed] [Google Scholar]
- Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R, Fraser PE, Kruck T, von Bohlen A, Schulz-Schaeffer W, et al. The cellular prion protein binds copper in vivo. Nature. 1997;390:684–687. doi: 10.1038/37783. [DOI] [PubMed] [Google Scholar]
- Burns CS, Aronoff-Spencer E, Dunham CM, Lario P, Avdievich NI, Antholine WE, Olmstead MM, Vrielink A, Gerfen GJ, Peisach J, et al. Molecular features of the copper binding sites in the octarepeat domain of the prion protein. Biochemistry. 2002;41:3991–4001. doi: 10.1021/bi011922x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns CS, Aronoff-Spencer E, Legname G, Prusiner SB, Antholine WE, Gerfen GJ, Peisach J, Millhauser GL. Copper coordination in the full-length, recombinant prion protein. Biochemistry. 2003;42:6794–6803. doi: 10.1021/bi027138+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canello T, Friedman-Levi Y, Mizrahi M, Binyamin O, Cohen E, Frid K, Gabizon R. Copper is toxic to PrP-ablated mice and exacerbates disease in a mouse model of E200K genetic prion disease. Neurobiol Dis. 2012;45:1010–1017. doi: 10.1016/j.nbd.2011.12.020. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay M, Walter ED, Newell DJ, Jackson PJ, Aronoff-Spencer E, Peisach J, Gerfen GJ, Bennett B, Antholine WE, Millhauser GL. The octarepeat domain of the prion protein binds Cu(II) with three distinct coordination modes at pH 7.4. J Am Chem Soc. 2005;127:12647–12656. doi: 10.1021/ja053254z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen FE, Pan KM, Huang Z, Baldwin M, Fletterick RJ, Prusiner SB. Structural clues to prion replication. Science. 1994;264:530–531. doi: 10.1126/science.7909169. [DOI] [PubMed] [Google Scholar]
- D'Angelo P, Della Longa S, Arcovito A, Mancini G, Zitolo A, Chillemi G, Giachin G, Legname G, Benetti F. Effects of the Pathological Q212P Mutation on Human Prion Protein Non-Octarepeat Copper-Binding Site. Biochemistry. 2012 doi: 10.1021/bi300233n. [DOI] [PubMed] [Google Scholar]
- Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, Baker NA. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007;35:W522–W525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donne DG, Viles JH, Groth D, Mehlhorn I, James TL, Cohen FE, Prusiner SB, Wright PE, Dyson HJ. Structure of the recombinant full-length hamster prion protein PrP(29–231): the N terminus is highly flexible. Proc Natl Acad Sci U S A. 1997;94:13452–13457. doi: 10.1073/pnas.94.25.13452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flechsig E, Shmerling D, Hegyi I, Raeber AJ, Fischer M, Cozzio A, von Mering C, Aguzzi A, Weissmann C. Prion protein devoid of the octapeptide repeat region restores susceptibility to scrapie in PrP knockout mice. Neuron. 2000;27:399–408. doi: 10.1016/s0896-6273(00)00046-5. [DOI] [PubMed] [Google Scholar]
- Fleissner MR, Brustad EM, Kalai T, Altenbach C, Cascio D, Peters FB, Hideg K, Peuker S, Schultz PG, Hubbell WL. Site-directed spin labeling of a genetically encoded unnatural amino acid. Proc Natl Acad Sci U S A. 2009;106:21637–21642. doi: 10.1073/pnas.0912009106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb LG, Petersen RB, Tabaton M, Brown P, LeBlanc AC, Montagna P, Cortelli P, Julien J, Vital C, Pendelbury WW, et al. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: disease phenotype determined by a DNA polymorphism. Science. 1992;258:806–808. doi: 10.1126/science.1439789. [DOI] [PubMed] [Google Scholar]
- Hankovszky HO, Hideg K, Lex L. Synthesis of nitroxylphosphinimines; a convenient route to amine, isothiocyanate, aminocarbonylaziridine, and carbodiimide nitroxyls. Synthesis. 1981:147–249. [Google Scholar]
- Hooper NM, Taylor DR, Watt NT. Mechanism of the metal-mediated endocytosis of the prion protein. Biochem Soc Trans. 2008;36:1272–1276. doi: 10.1042/BST0361272. [DOI] [PubMed] [Google Scholar]
- Huang Z, Gabriel JM, Baldwin MA, Fletterick RJ, Prusiner SB, Cohen FE. Proposed three-dimensional structure for the cellular prion protein. Proc Natl Acad Sci U S A. 1994;91:7139–7143. doi: 10.1073/pnas.91.15.7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilc G, Giachin G, Jaremko M, Jaremko L, Benetti F, Plavec J, Zhukov I, Legname G. NMR structure of the human prion protein with the pathological Q212P mutation reveals unique structural features. PLoS One. 2010;5:e11715. doi: 10.1371/journal.pone.0011715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson GS, Murray I, Hosszu LL, Gibbs N, Waltho JP, Clarke AR, Collinge J. Location and properties of metal-binding sites on the human prion protein. Proc Natl Acad Sci U S A. 2001;98:8531–8535. doi: 10.1073/pnas.151038498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeschke G, Chechik V, Ionita P, Godt A, Zimmermann H, Banham J, Timmel CR, Hilger D, Jung H. DeerAnalysis2006 - a comprehensive software package for analyzing pulsed ELDOR data. Applied Magnetic Resonance. 2006;30:473–498. [Google Scholar]
- Jeschke G, Polyhach Y. Distance measurements on spin-labelled biomacromolecules by pulsed electron paramagnetic resonance. Phys Chem Chem Phys. 2007;9:1895–1910. doi: 10.1039/b614920k. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- Kaminski G, Duffy EM, Matsui T, Jorgensen WL. Free energies of hydration and pure liquid properties of hydrocarbons from the OPLS all-atom model. J Phys Chem. 1994;98:13077–13082. [Google Scholar]
- Lee S, Antony L, Hartmann R, Knaus KJ, Surewicz K, Surewicz WK, Yee VC. Conformational diversity in prion protein variants influences intermolecular beta-sheet formation. EMBO J. 2010;29:251–262. doi: 10.1038/emboj.2009.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Christensen HM, Stewart LR, Roth KA, Chiesa R, Harris DA. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105–125. EMBO J. 2007;26:548–558. doi: 10.1038/sj.emboj.7601507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Guest W, Huang A, Plotkin SS, Cashman NR. Immunological mimicry of PrPC-PrPSc interactions: antibody-induced PrP misfolding. Protein Eng Des Sel. 2009;22:523–529. doi: 10.1093/protein/gzp038. [DOI] [PubMed] [Google Scholar]
- Liemann S, Glockshuber R. Influence of amino acid substitutions related to inherited human pr ion diseases on the thermodynamic stability of the cellular prion protein. Biochemistry. 1999;38:3258–3267. doi: 10.1021/bi982714g. [DOI] [PubMed] [Google Scholar]
- Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Model. 2001;7:306–317. [Google Scholar]
- Mead S. Prion disease genetics. Eur J Hum Genet. 2006;14:273–281. doi: 10.1038/sj.ejhg.5201544. [DOI] [PubMed] [Google Scholar]
- Millhauser GL. Copper and the prion protein: methods, structures, function, and disease. Annu Rev Phys Chem. 2007;58:299–320. doi: 10.1146/annurev.physchem.58.032806.104657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orem NR, Geoghegan JC, Deleault NR, Kascsak R, Supattapone S. Copper (II) ions potently inhibit purified PrPres amplification. J Neurochem. 2006;96:1409–1415. doi: 10.1111/j.1471-4159.2006.03650.x. [DOI] [PubMed] [Google Scholar]
- Panchal SC, Kaiser DA, Torres E, Pollard TD, Rosen MK. A conserved amphipathic helix in WASP/ Scar proteins is essential for activation of Arp2/3 complex. Nat Struct Biol. 2003;10:591–598. doi: 10.1038/nsb952. [DOI] [PubMed] [Google Scholar]
- Pauly PC, Harris DA. Copper stimulates endocytosis of the prion protein. J Biol Chem. 1998;273:33107–33110. doi: 10.1074/jbc.273.50.33107. [DOI] [PubMed] [Google Scholar]
- Powell JH, Johnson II EM, Gannett PM. Improvement of a critical intermediate step in the synthesis of a nitroxide-based spin-labeled deoxythymidine analog. Molecules. 2000;5:1244–1250. [Google Scholar]
- Prusiner SB. Scrapie prions. Annu Rev Microbiol. 1989;43:345–374. doi: 10.1146/annurev.mi.43.100189.002021. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Prion diseases and the BSE crisis. Science. 1997;278:245–251. doi: 10.1126/science.278.5336.245. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB. Prion Biology and Diseases. 2nd edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2003. [Google Scholar]
- Pushie MJ, Pickering IJ, Martin GR, Tsutsui S, Jirik FR, George GN. Prion protein expression level alters regional copper, iron and zinc content in the mouse brain. Metallomics. 2011;3:206–214. doi: 10.1039/c0mt00037j. [DOI] [PubMed] [Google Scholar]
- Schmitt-Ulms G, Ehsani S, Watts JC, Westaway D, Wille H. Evolutionary descent of prion genes from the ZIP family of metal ion transporters. PLoS One. 2009;4:e7208. doi: 10.1371/journal.pone.0007208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Ji HF. Mutation directional selection sheds light on prion pathogenesis. Biochem Biophys Res Commun. 2011;410:159–163. doi: 10.1016/j.bbrc.2011.06.007. [DOI] [PubMed] [Google Scholar]
- Shibuya S, Higuchi J, Shin RW, Tateishi J, Kitamoto T. Codon 219 Lys allele of PRNP is not found in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1998;43:826–828. doi: 10.1002/ana.410430618. [DOI] [PubMed] [Google Scholar]
- Siggs OM, Cruite JT, Du X, Rutschmann S, Masliah E, Beutler B, Oldstone MB. Disruption of copper homeostasis due to a mutation of Atp7a delays the onset of prion disease. Proc Natl Acad Sci U S A. 2012;109:13733–13738. doi: 10.1073/pnas.1211499109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova I, Kasho V, Choe JY, Altenbach C, Hubbell WL, Kaback HR. Sugar binding induces an outward facing conformation of LacY. Proc Natl Acad Sci U S A. 2007;104:16504–16509. doi: 10.1073/pnas.0708258104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumudhu W, Perera WS, Hooper NM. Ablation of the metal ion-induced endocytosis of the prion protein by disease-associated mutation of the octarepeat region. Curr Biol. 2001;11:519–523. doi: 10.1016/s0960-9822(01)00147-6. [DOI] [PubMed] [Google Scholar]
- Swietnicki W, Petersen RB, Gambetti P, Surewicz WK. Familial mutations and the thermodynamic stability of the recombinant human prion protein. J Biol Chem. 1998;273:31048–31052. doi: 10.1074/jbc.273.47.31048. [DOI] [PubMed] [Google Scholar]
- Thakur AK, Srivastava AK, Srinivas V, Chary KV, Rao CM. Copper alters aggregation behavior of prion protein and induces novel interactions between its N- and C-terminal regions. J Biol Chem. 2011;286:38533–38545. doi: 10.1074/jbc.M111.265645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela-Nallar L, Toledo EM, Larrondo LF, Cabral AL, Martins VR, Inestrosa NC. Induction of cellular prion protein gene expression by copper in neurons. Am J Physiol Cell Physiol. 2006;290:C271–C281. doi: 10.1152/ajpcell.00160.2005. [DOI] [PubMed] [Google Scholar]
- Walter ED, Stevens DJ, Spevacek AR, Visconte MP, Dei Rossi A, Millhauser GL. Copper binding extrinsic to the octarepeat region in the prion protein. Curr Protein Pept Sci. 2009;10:529–535. doi: 10.2174/138920309789352056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter ED, Stevens DJ, Visconte MP, Millhauser GL. The prion protein is a combined zinc and copper binding protein: Zn2+ alters the distribution of Cu2+ coordination modes. J Am Chem Soc. 2007;129:15440–15441. doi: 10.1021/ja077146j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt NT, Hooper NM. The prion protein and neuronal zinc homeostasis. Trends Biochem Sci. 2003;28:406–410. doi: 10.1016/S0968-0004(03)00166-X. [DOI] [PubMed] [Google Scholar]
- Watt NT, Taylor DR, Kerrigan TL, Griffiths HH, Rushworth JV, Whitehouse IJ, Hooper NM. Prion protein facilitates uptake of zinc into neuronal cells. Nat Commun. 2012;3:1134. doi: 10.1038/ncomms2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young TS, Ahmad I, Yin JA, Schultz PG. An enhanced system for unnatural amino acid mutagenesis in E. coli. J Mol Biol. 2010;395:361–374. doi: 10.1016/j.jmb.2009.10.030. [DOI] [PubMed] [Google Scholar]
- Zahn R, Liu A, Luhrs T, Riek R, von Schroetter C, Lopez Garcia F, Billeter M, Calzolai L, Wider G, Wuthrich K. NMR solution structure of the human prion protein. Proc Natl Acad Sci U S A. 2000;97:145–150. doi: 10.1073/pnas.97.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Swietnicki W, Zagorski MG, Surewicz WK, Sonnichsen FD. Solution structure of the E200K variant of human prion protein. Implications for the mechanism of pathogenesis in familial pr ion diseases. J Biol Chem. 2000;275:33650–33654. doi: 10.1074/jbc.C000483200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.