

Abstract
Background
Tauopathies, including Alzheimer’s disease (AD) and frontotemporal dementia, are diseases characterized by the formation of pathological tau protein aggregates in the brain and progressive neurodegeneration. Presently no effective disease modifying treatments exist for tauopathies.
Methods
To identify drugs targeting tau neurotoxicity, we have used a C. elegans model of tauopathy to screen a drug library containing 1120 compounds approved for human use for the ability to suppress tau-induced behavioral effects.
Results
One compound, the typical antipsychotic azaperone, improved the motility of tau transgenic worms, reduced levels of insoluble tau, and was protective against neurodegeneration. We found that azaperone reduces insoluble tau in a human cell culture model of tau aggregation, and that other antipsychotic drugs (flupenthixol, perphenazine, and zotepine) also ameliorate the effects of tau expression in both models.
Conclusions
Reduction of dopamine signaling through the dopamine D2 receptor with the use of gene knockouts in C. elegans or RNAi knockdown in human cell culture have similar protective effects against tau toxicity. These results suggest dopamine D2 receptor antagonism holds promise as a potential neuroprotective strategy for targeting tau aggregation and neurotoxicity.
Keywords: Alzheimer’s disease, tau, drug screen, dopamine, antipsychotics, protein aggregation
Introduction
Tauopathies are a group of diseases characterized by accumulation of pathological tau protein in the brain leading to progressive neurodegeneration. Current drug interventions for tauopathies are limited to the treatment of symptoms without directly affecting tau pathology or the resultant dysfunction, underscoring the need for development of tau-targeted therapeutics. Furthermore, results from amyloid-targeted clinical trials in humans suggest that tau-targeted therapies in conjunction with removal of amyloid may be required for therapeutic benefit in treating AD (1).
Drug repositioning—the discovery of new uses for drugs already approved for use in humans—promises to accelerate drug discovery and clinical testing for new therapeutic targets. These pharmacological agents have known functions and toxicities with the potential to translate quickly from disease models to human disease relief. Here we employed a simple animal model of tauopathy as a platform to screen a library of FDA-approved compounds for the suppression of tau-induced movement defects. We made use of a transgenic C. elegans model, which expresses human tau in neurons and exhibits several pathological hallmarks of tauopathies including detergent-insoluble tau aggregates and neuronal degeneration (2). Our target-independent, phenotype-based approach allows for detection of compounds with therapeutic potential without any a priori assumption of how tau function would be altered. This also allows for detection of drugs with activities not currently known to be important for tau function or neuron survival.
Materials and Methods
Reagents and Strains
Drugs used in primary screens were obtained from Prestwick Chemical (Illkirch, France) in 2 mg/ml stock solutions in DMSO. Solid drugs were obtained of azaperone, aztreonam, clofazimine, dehydrocholic acid, digoxigenin, dihydrostreptomycin sulfate, epivincamine, isoniazid, lorglumide sodium, mefloquine hydrochloride, perphenazine, risperidone, trazodone hydrochloride, trifluoperazine dihydrochloride and zotepine from Sigma-Aldrich (St. Louis, MO); of 6-benzylaminopurine from Acros Organics (Morris Plains, NJ); of nefopam, rescinnamin, syrosingopine and verteporfin from Prestwick Chemical (Illkirch, France); and amisulpride, flupenthixol dihydrochloride and haloperidol hydrochloride from Tocris Bioscience (Ellisville, MO). Stock solutions of all drugs were prepared in DMSO, then diluted as appropriate for various assays to a final concentration of 0.5% DMSO.
Caenorhabditis elegans strains used in this study were: JT5010 wild type Bristol N2; CB6055 bus-8(e2698); CK10 bkIs10[Paex-3::T337, Pmyo-2::gfp]; CK1301 bkIs10[Paex-3::T337, Pmyo-2::GFP] bus-8(e2698); CK1310 juIs76[Punc-25::gfp, lin-15(+)] bkIs10[Paex-3::T337, Pmyo-2::gfp] bus-8(e2698); CK1350 bkIs10[Paex-3::T337, Pmyo-2::gfp] dop-2(vs105); CK1351 bkIs10[Paex-3::T337, Pmyo-2::gfp] dop-3(vs106); CK1352 bkIs10[Paex-3::T337, Pmyo-2::gfp] dop-2(vs105) dop-3(vs106); CK1355 bkIs10[Paex-3::T337, Pmyo-2::gfp] juIs76[Punc-25::gfp + lin-15(+)] dop-2(vs105) dop-3(vs106).
Primary Drug Screen
Three concentrations (20, 10, 3.5 ug/ml) of each Prestwick drug were prepared in 1 ml NGM agar and poured into 12-well culture plates seeded with 20 to 40x concentrated OP50 E. coli as food. Plates were covered by foil to minimize light exposure. The following day, 20–40 eggs of CK1301 were applied in a 20 ul drop of M9 buffer. Once dried, plates were inverted and transferred to a metal box and grown at 23°C. Animals were assayed for phenotypes on multiple days after hatching until starved. This characterization included assessing spontaneous locomotion and stimulated locomotion in response to plate tap. While improved locomotion was the primary phenotype assayed, growth effects were also noted. Drug treatments improving locomotion were retested as described above except that candidate hits from the same 12-well plate in the initial screen were relocated to separate plates to avoid combinatorial drug effects. During this test, individual animals were counted for improved locomotion. Drugs that improved locomotion in ≥40% of animals at any concentration or that trended toward improving locomotion with increasing concentrations were further tested. In order to control for the individual variation in drug permeability of bus-8 animals, vehicle treated animals were examined as a control in all studies described.
Liquid Thrashing Assays
Responses of CK1301 to drugs identified in the primary screen were measured by comparing the number of thrashes per minute under various drug treatment concentrations. Several concentrations of each drug were prepared in 2 ml NGM agar and poured into 35mm petri dishes. All drug and control plates had a DMSO concentration of 0.5%. Freshly poured drug plates were seeded with concentrated OP50 E. coli and covered by foil to minimize light exposure. The following day 40–60 eggs of CK1301 were applied in a 20 ul drop of M9. Once dried, plates were inverted and transferred to a metal box and grown at 23°C. Three days later, individual L4 or day 1 adult animals were transferred to a Teflon-coated glass slide that contains shallow wells filled with M9 buffer. Each animal was allowed to adjust to the new environment for 30 seconds before the number of thrashes was counted for one minute. A single thrash was defined strictly as C-shaped, head-to-tail movement.
Neurodegeneration
Timed egg lays were arranged to produce synchronized populations. To score neuronal loss or nerve cords gaps, 30–40 animals (CK1310) were transferred onto 5% agarose pads containing 0.05% sodium azide. Using 60x magnification, the 19 GABAergic neurons along the ventral nerve cord were counted. Any gaps in GFP along the ventral nerve cord or the dorsal nerve cord were recorded separately. Twenty animals were counted for each condition tested. Imaging of live worms was done on a Nikon Eclipse TE300 epi-fluorescent microscope. Images were acquired using a Photometrics SenSys™ cooled CCD camera and IPLab image acquisition software (BD Biosciences Bioimaging). Images were deconvolved using MicroTome™ deconvolution software (BD Biosciences Bioimaging).
Cell Culture, Drug, and siRNA Treatments
A stable cell line (HEK/tau) overexpressing wild type human tau (1N4R) was constructed as described previously (3). HEK293 cells were used as the parental cell line due to their ease of use and the fact that they share many properties with immature neurons (4). HEK/tau cells were cultured under standard culture conditions (DMEM, 10% defined fetal bovine serum, Penicillin (50 IU/mL)-Streptomycin (50μg/mL) + Zeocin (100μg/mL)) to maintain selection. For azaperone, flupenthixol, or zotepine treatment of cultured cells, test concentrations of drug were added and a final concentration of 0.05% DMSO as a vehicle was maintained across samples. RNAi experiments were carried out as per protocol in the TriFECTa Dicer-Substrate RNAi manual (Integrated DNA Technologies) using human drd2 specific siRNAs to knockdown dopamine D2 receptor expression.
tau Biochemistry
tau fractions were obtained as described (2). The total fraction was analyzed by immunoblotting against tau and tubulin or actin (ab17025). The insoluble tau (FA) fraction was analyzed by immunoblotting against tau (ab17025).
Protein samples were diluted in buffer (10 mM Tris, pH 6.8, 1 mM EDTA, 40 mM DTT, 1% SDS, 10% sucrose) by addition of 5x sample buffer boiled 5 minutes and loaded onto 4–15% pre-cast criterion SDS-PAGE gradient gels (Bio-Rad). For immunoblotting, we detected human tau using antibody 17025 at a dilution of 1:6000 (a generous gift from Virginia Lee) as described previously (5). We used anti-tubulin antibody E7 at a dilution of 1:1000 (Developmental Studies Hybridoma Bank) and anti-Drd2 antibody ab88074 (Abcam) at 1:1000. Signals were measured by densitometry using Adobe Photoshop and normalized to endogenous actin for human cells and to tubulin for C. elegans.
Lifespan Assay
Lifespan plates were prepared the day before use by mixing standard NGM agar with 50 ug/ml (152.7 uM) azaperone and 0.5% DMSO, and seeded with 20x OP-50 bacteria after agar solidification. Plates were covered with foil to prevent potential light-induced degradation of the drug. Parent worms were allowed to lay eggs for 6 hours, and then removed to obtain a synchronized population of progeny. Experimental animals were grown at 20°C, and transferred to 35mm plates at a density of 25 animals per plate on day 2 of adulthood. Experimental animals were subsequently transferred away from progeny onto fresh drug plates at adult days 4 and 6, and were monitored every 1–2 days throughout their lifespan for movement. Animals were scored as dead for failure to respond to a gentle touch to the head or body. Total days of survival were calculated from hatching. Animals that ruptured or had progeny hatch internally were censored from analysis. Statistical analysis was performed using GraphPad Prism software.
Results
Cuticle-compromised bus-8(e2698) has increased drug sensitivities
To facilitate screening of anti-tau drugs in C. elegans, we utilized a drug sensitive strain. Prohibitively high doses of most drugs are required to generate visible phenotypes in C. elegans due to the relative impermeability of the worm cuticle to many drug-like small molecules. bus-8 mutations cause cuticle defects that allow multiple drugs to invade the worm cuticle. We chose the bus-8 allele e2698 because it has high cuticle permeability to diverse small molecules and lectins (6), yet the animals are grossly phenotypically normal (7). We confirmed that motor function in the bus-8(e2698) animals is similar to wild type using a standard liquid thrashing assay (Figure S1), and that bus-8(e2698) has increased sensitivity to several different toxic compounds (Figure S2). We crossed bus-8 (e2698) with previously characterized tau transgenic animals (T337) (2), to generate a tau transgenic strain sensitized to small molecules. The T337;bus-8 animals have a similar phenotype as compared to T337 transgenic animals in a wild type genetic background (Figure S1).
A small molecule screen for chemical suppressors of tau induced defects
The Prestwick chemical library contains 1120 compounds with diverse drug-like chemical characteristics. Most are approved off-patent drugs with a history of use as human therapeutics for a wide variety of disorders. We tested the ability of these compounds to suppress the severe, tau-induced, lethargic crawling defect of T337;bus-8 by exposing animals to each drug chronically throughout their development (Figure S3). To avoid potentially confounding maternal effects, we applied sterile batches of worm eggs onto drug-containing agar directly, and after hatching animals developed into adulthood in the presence of drug. Worms were exposed to three concentrations of each drug (20, 10 and 3.5 ug/ml) in multiwell plates, and the animals were assessed visually for any effects on the phenotype. This testing was conducted in duplicate and we found reproducible improvement of the tauopathy phenotype for 16 candidate compounds in greater than 40% of individual animals in each treated population. The variability of treatment effectiveness likely arises from variation in drug penetration in bus-8 animals resulting from variable morphological defects in the cuticle giving rise to drug permeability (7). These 16 initial hits from the Prestwick drug library yield an overall hit rate of ~1.4% (16/1120) in our phenotype based screen.
To ascertain the optimal concentration of these 16 compounds, we generated a dose response curve for each using a liquid thrashing assay. Only 6 of 16 compounds—azaperone, clofazimine, isoniazid, lorglumide, nefopam and trazodone—showed a significant improvement in swimming (thrashing in liquid) (Figure 1A). Drugs that failed to improve this measure of tau related behavioral phenotypes at multiple concentrations of drug were not evaluated further. These effects appear to be tau-specific, since none of these six compounds was observed to have any effects on the locomotion of wildtype C. elegans (data not shown).
Fig 1. Azaperone partially suppresses tau related phenotypes in C. elegans.

A. Dose response curves of T337;bus-8 to drug candidates from the Prestwick Library screen in liquid thrashing assays. Statistical analyses (Kruskal-Wallis test, followed by Dunns post test, *P<0.05, **P<0.01, ***P<0.001) found significant effects of azaperone, clofazimine, isoniazid, lorglumide NaCl, nefopam HCl and trazodone HCl at optimal doses. Chemical structure diagrams illustrate the diversity among suppressors of tau-induced movement defects. B. Azaperone or isoniazid treatment reduces accumulation of detergent insoluble tau in worms. Mixed populations of tau transgenic worms were grown in the presence of each drug at the optimal dosage for improving locomotion. Treated worms were subjected to a sequential extraction of tau using buffers of increasing solubilizing strength. Each treatment condition altered insoluble tau levels when normalized to soluble tau as follows: Azaperone (40%), Clofazamine (186%), Isoniazid (55%), Nefopam (111%), Lorglumide (121%), Trazadone (97%) of vehicle treated animals. C. Azaperone suppresses tau-induced neurodegeneration (p<0.01). CK1310 animals were scored for retention or loss of 19 GABAergic motor neurons. Clofazimine and lorglumide were also tested on a small number of animals and found not to have altered neurodegeneration (data not shown). Statistical analysis by Kruskal-Wallis test, followed by Dunns post test.
Azaperone treatment decreases deposition of aggregated tau
The appearance of insoluble aggregates of tau protein characterizes human tauopathy disorders and appears to be a relatively early event in the process of neurofibrillary degeneration (reviewed in (8)). To address whether any of the drugs identified above modulate tau related behavioral phenotypes by ameliorating tau aggregation, we examined the accumulation of detergent insoluble tau in tau transgenic animals with or without drug treatment (Fig 1B). Optimal drug concentrations (Table S1) for ameliorating behavioral defects for each of the drugs were applied chronically to the eggs of tau transgenic animals. The worms were then allowed to develop into adulthood in the presence of drug. Transgenic animals were harvested and subjected to sequential extraction of tau as previously described (2). Only those animals treated with azaperone or isoniazid exhibited visibly reduced levels of insoluble tau. This is consistent with drug modification of tau pathology in our C. elegans model.
Azaperone suppresses tau-induced GABAergic neuronal degeneration
T337 transgenic animals exhibit progressive neurodegeneration of GABAergic neurons (2,9). To test if this effect is ameliorated by drugs from our screen, we constructed a strain (CK1310) that allows us to visualize the GABAergic neurons in drug-sensitive T337;bus-8 animals. CK1310 carries an unc-25::GFP reporter construct which expresses GFP in 19 GABAergic neuron cell bodies positioned along the ventral nerve cord and brightly fluoresces in the dorsal and ventral nerve cords (10). CK1310 animals exhibited a loss of 1–4 out of 19 GABAergic motor neurons (Figure 1C). We scored cohorts of animals after 4 days of drug exposure at the optimal concentrations for ameliorating locomotion defects (Table S1). Only azaperone showed significant protection against neurodegeneration (Figure 1C). However, azaperone failed to extend lifespan of tau transgenic animals (Fig S4), suggesting that the neuroprotective effects of azaperone are not mediated by ameliorating aging related changes, but rather by reduction of tau aggregation and/or neurotoxicity.
Antipsychotic drugs suppress tau-induced defects in C. elegans
Azaperone is a butyrophenone antipsychotic drug. To test if antipsychotic drugs generally prevent tau-induced degenerative changes, we chose to characterize eight compounds from six structural classes as outlined in Table 1. To test suppression of tau-induced motor dysfunction, T337;bus-8 worms were exposed chronically to a range of drug concentrations and observed as young adults. Animals were assessed visually for their ability to crawl on the solid agar surface. With the exception of risperidone and haloperidol (which were not tested at high concentrations due to solubility constraints), 20 concentrations of each antipsychotic drug were tested. For each drug, we determined the optimal concentration for improving tau-induced behavioral defects, and this concentration was used for subsequent experiments. We found that in addition to azaperone (152.7 uM), the drugs flupenthixol (98.5 uM), perphenazine (123.8 uM) and zotepine (37.7 uM) greatly increased T337;bus-8 motility (Table 1).
Table 1.
Antipsychotics tested for the suppression of tau-induced lethargy in C. elegans
| Chemical | Class | Suppression of worm tauopathy | HEK/tau dose (nM) | Human D2 receptor Ki (nM) | Reference |
|---|---|---|---|---|---|
| AzaperoneT | Butyrophenone | +++ | 30 | 10 | Burt DR et al., 1976 |
| HaloperidolT | Butyrophenone | + | 30 | 0.7 | Seeman, Philip 2001 |
| RisperidoneA | Benzisoxazole | − | ND | 1 | Seeman, Philip 2001 |
| ZotepineA | Dibenzothiepine | ++ | 30 | 8 | Richelson E, Souder T. 2000 |
| PerphenazineT | Phenothiazine | ++ | - | 0.26 | Seeman, Philip 2001 |
| AmisulprideA | Benzamide | − | ND | 3 | Abbas AI et. al., 2009 |
| FlupenthixolT/A | Thioxanthene | ++ | 30 | 0.38 | Seeman, 2002 |
| TrifluoperazineT | Phenothiazine | + | ND | 1.3 | Kroeze WK, et. al., 2002 |
Abbreviations as follows: “T” superscript represents typical whereas “A” represents atypical antipsychotics; D2, dopamine receptor and Ki, dissociation constant. Antipsychotic drug inhibition of human D2 receptors as a measure of its dissociation (in nM) in competition with a radioligand (see references). Note that Ki values have not been determined for these drugs with C. elegans dopamine receptors, and thus we would not anticipate a correlation between their effectiveness at ameliorating worm tauopathy and their calculated affinity for human dopamine receptors. Suppression of worm tauopathy was assessed visually as the ability to crawl on a solid agar surface.
+++= greatly improved movement of the majority of animals on a plate; ++ = moderate improvement in movement of the majority of animals on a plate; + = minimal detectible improvement in movement; − = no detectible difference in movement versus untreated worms. HEK/tau dose shown is the optimal dosage in HEK/tau cells leading to reduced tau aggregation. ND= not determined, − = negligible effect on tau aggregation.
We further characterized these four antipsychotics to compare how they alter tauopathy phenotypes. We first tested their effects on neurotoxicity. As shown in Figure 2A, neurons are lost in untreated CK1310 animals whereas azaperone treatment suppresses this loss (Figure 2B). This neuroprotective ability was shared by flupenthixol, perphenazine and zotepine (Figure 2C). We next examined the accumulation of detergent insoluble tau in tau transgenic worms chronically exposed to azaperone, flupenthixol, perphenazine or zotepine at the concentrations listed above. We found that flupenthixol, perphenazine, and azaperone clearly reduce tau aggregation while zotepine’s effect on insoluble tau was more modest (Figure 2D).
Figure 2. Antipsychotic treatment limits neurodegenerative changes.

A. Picture of a segment along the ventral nerve cord of an untreated CK1310 worm indicating loss of neuron DD5 (arrow). B. Picture of an azaperone-treated CK1310 worm along the same segment of ventral nerve cord with no loss of neurons. C. Typical (azaperone, flupenthixol, perphenazine) and atypical (zotepine) antipsychotics significantly protect against neuron loss. Statistical analysis by Kruskal-Wallis test, followed by Dunns post test (***p<0.001). Graph shows number of GABAergic neurons lost in 20 animals on and off drug. D. Reduced tau accumulation in worms under treatment with antipsychotics. Each treatment condition reduced insoluble tau normalized to soluble tau as follows: Azaperone 96% reduction, Flupenthixol 92% reduction, Perphenazine 80%reduction, and Zotepine 38% reduction.
Antipsychotic treatment decreases pathological tau species in human cells
To determine if the protective effect of antipsychotic drugs against tau is conserved from worms to human cells, we used a simple cellular model for deposition of detergent insoluble tau based on HEK293 cells overexpressing wild type human 1N4R Tau (HEK/tau) (3). We tested azaperone for reduction of detergent insoluble tau over a wide range of drug concentrations (Fig 3A). We observed a U-shaped dose response of azaperone treatment on the accumulation of detergent insoluble tau in HEK/tau cells (Fig 3B). Similar U-shaped dose response profiles were obtained by treating HEK/tau cells with the antipsychotic drugs flupenthixol and zotepine (Fig S5). The maximal inhibitory concentration of azaperone for tau aggregation is approximately 30 nM. After 3 days of treatment with 30 nM azaperone, HEK/tau cells exhibit a significant reduction in the level of detergent insoluble tau (p = 0.04; Fig 3B).
Fig 3. Antipsychotic drugs decrease tau aggregation in HEK/Tau cells.
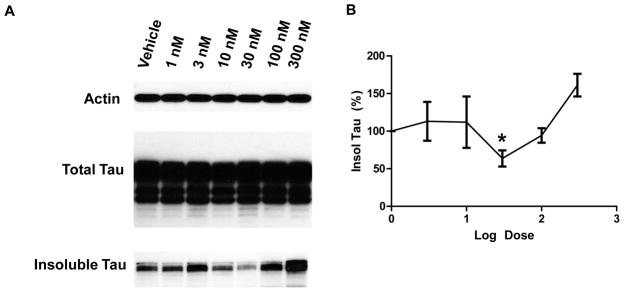
A. HEK/tau cells treated with azaperone exhibit decreased insoluble but not total tau. B. Azaperone treatment over a dosage range of 1 to 300 nM was conducted in triplicate. Shown is the resulting U-shaped dose response curve for azaperone treatment concentration (log) vs. level of insoluble tau normalized to vehicle treated cells. The azaperone concentration of maximal effect on insoluble tau is ~30nM (p=0.04 by Student’s t-test)
D2 receptor antagonism as a candidate neuroprotective strategy for tauopathy
Since typical antipsychotics share an ability to antagonize dopamine D2 receptors (D2R) (11), we investigated whether improvement of tau-induced defects is mediated via these receptors. The C. elegans genome encodes two D2-like receptors, DOP-2 and DOP-3 (12). We generated strains containing lesions within each of these genes combined with tau transgene: T337;dop-2 and T337;dop-3 as well as T337;dop-2;dop-3. We tested the locomotory responses of these strains compared to T337 in thrashing assays. Animals that lacked both worm D2 receptors exhibited significantly improved motility (Figure 4A) similar to that seen in azaperone-treated animals (Figure 1A).
Fig 4. Mechanism of anti-tau activity is D2 receptor antagonism.

A. Loss of DOP-2 and DOP-3 partially suppresses tauopathy phenotype. The motility of T337, T337;dop-2, T337;dop-3, and T337;dop-2;dop-3 young adult animals were compared in a liquid thrashing assay. T337;dop-2;dop-3 significantly improved (p<0.001) swimming compared to T337. Statistical analysis conducted was the Kruskal-Wallis test, followed by Dunns post-test. B. Loss of DOP-2 and DOP-3 suppresses neuron loss (p=0.0009) Statistical analysis was conducted using a Mann-Whitney, two-tailed test. C. Loss of both DOP-2 and DOP-3 reduces tau aggregation. Individual mixed populations of T337 (100% insol tau), T337;dop-2 (140% insol tau), T337;dop-3 (265% insol tau), and T337;dop-2;dop-3 (54% insol tau) worms were grown. Worms were subjected to a sequential extraction of tau using buffers of increasing solubilizing strength. Only T337;dop-2;dop-3 reduced the levels of detergent insoluble tau. D. Knockdown of D2 receptor reduces tau aggregation in HEK/tau cells. Equivalent pellets of HEK/tau cells with or without D2 receptor targeting RNAi were subjected to sequential extraction of tau using buffers of increasing solubilizing strength. Drd2 siRNA treatment reduced insoluble tau (normalized to soluble tau) relative to a scrambled siRNA control by 55%.
To look for effects on neurodegeneration, we added the unc-25::GFP transgene to T337;dop-2;dop-3, generating strain CK1355. Counting the presence or absence of the 19 GABAergic neurons along the ventral nerve cord in day 4 adult worms, we found that D2R-deficient animals had nearly complete protection against tau-induced GABAergic neuronal loss as compared to controls (Figure 4B). Azaperone treatment yielded comparable neuroprotection against tau toxicity (Fig 1C). As this suggests that tau-induced phenotypes and neuronal loss are mediated through D2 receptors, we then asked whether levels of insoluble tau itself are directly affected by the loss of these receptors. We harvested populations of T337, T337;dop-2, T337;dop-3, and T337;dop-2;dop-3 and compared levels of detergent insoluble tau in these samples. We found a surprising increase in tau aggregation in T337;dop-3 as compared to either T337 alone or T337;dop-2. Remarkably, with both D2 receptors missing T337;dop-2;dop-3 showed a significant decrease in insoluble tau (Figure 4C) consistent with its improved motility and neuroprotection. HEK293 cells are considered neuron-like, and are known to express D2 receptors (4). Furthermore, although HEK293 cells are not known to produce dopamine, there is dopamine present in the culture media (13), suggesting that some signaling through D2 receptors does occur in these cells. We knocked down D2 receptor levels by siRNA treatment in HEK/tau cells and observed a striking decrease in insoluble tau, consistent with the hypothesis that antipsychotic drugs reduce tau aggregation in human cultured cells by a D2 receptor mediated mechanism (Figure 4D).
Discussion
We have screened a collection of previously approved drugs to identify compounds with activity against tau neurotoxicity. Our goal was to enhance the possibility of finding suitable tau targeting drugs by using a chemical library both structurally complex and rich in bioactive drugs used for treatment of human disease. We have used both C. elegans and human cell culture to identify a previously unknown mechanism for suppression of tau pathology through D2 dopamine receptor antagonism.
A phenotype-based screen for compounds ameliorating tau induced behavioral dysfunction
Overexpression of human tau in C. elegans causes progressive motility defects, neurodegeneration and accumulation of insoluble tau (2). To screen for drugs ameliorating the effects of tau on the nervous system, we chose a chemically diverse library containing 1120 compounds composed of 90% FDA-approved, off-patent drugs and 10% alkaloid small molecules. We assayed each drug for suppression of tau-induced lethargy. The main benefit of a phenotype-based approach is that it does not presuppose mechanism. Our screen isolated six structurally distinct drugs with diverse pharmacological actions (Figure 1, Table S1). Two of these, azaperone and isoniazid, also reduced insoluble tau. Isoniazid is used as an anti-mycoplasma drug, but is also known to generate free radicals which lead to an array of side activities; furthermore, related hydrazides have been demonstrated to have anti-tau aggregation activity, but ambiguous in vivo behavior (14). When we examined other tau-induced defects—neurotoxicity and accumulation of insoluble tau—only the typical antipsychotic azaperone produced significant neuroprotection (Figure 1). Thus, we focused subsequent work on the mechanistic nature of tau suppression exhibited by azaperone.
In both authentic human tauopathies and in models of these disorders, tau aggregation and neurotoxicity are correlated (15–17). Thus we investigated the effect of azaperone treatment on tau aggregation in human cellular and C. elegans transgenic models of tauopathy. In both systems, azaperone treatment reduces tau aggregation (Figure 1B and 3). Furthermore, in tau transgenic C. elegans, azaperone treatment also reduces neuronal loss (Figure 1C) and improves behavioral outcomes (Figure 1A). These findings suggest treatment with azaperone in transgenic mouse tauopathy models may be protective against tau aggregation and neurotoxicity—a hypothesis we aim to test in future studies.
The molecular mechanism of azaperone action is conserved between C. elegans and human cells
Azaperone modulates tau aggregation in both transgenic worms (Figures 1 and 2) and human cells overexpressing human tau (Figure 3). This suggests the mechanism of azaperone action is conserved between C. elegans and humans. Furthermore, the antipsychotics flupenthixol and zotepine also reduce tau aggregation in both worms and human cells (Table 1 and Figure 2) indicating that this mechanism is not specific to azaperone. These antipsychotic drugs share a high affinity for D2 receptors and inhibit D2 receptor mediated dopaminergic signaling (11). In C. elegans, there are two D2-like receptors—DOP-2 and DOP-3 (12). The observation that knockout of dop-3 alone increases tau aggregation is difficult to interpret. We hypothesize that in the absence of dop-3, there could be some compensatory up regulation of D2 signaling via dop-2. Nonetheless, we have demonstrated that simultaneous knockout of both receptors in tau transgenic animals reduced tau accumulation, improved motility and protected against neuronal loss (Figure 4). The degree of change induced by D2 receptor knockout was similar to the level of effect observed at the optimal concentration of azaperone treatment (Figure 1). This indicates that the effects of antipsychotic drugs on tau toxicity are mainly due to their D2 receptor antagonism, and not due to any other drug activity, such as at serotonin receptors. We hypothesize that downstream signaling effects of D2 receptor inhibition mediate protection from tau aggregation and toxicity in drug-treated or D2 receptor-deficient animals. However, whether this effect occurs directly via a D2 receptor downstream effector (such as PKA) modulation of tau phosphorylation or indirectly via a D2 receptor-responsive, transcription factor-mediated change (such as CREB) (11) remains unclear and will require further investigation. Regardless, we have demonstrated that amelioration of tauopathy phenotypes by azaperone and related drugs occurs via inhibition of signaling through D2-like receptors (Table 1).
Potential pitfalls: High doses of antipsychotics may drive tau pathology
Previous studies of antipsychotic treatment of AD patients for agitation and aggression suggest that standard clinical doses of antipsychotics used to treat agitation in dementia patients are not beneficial for long term survival, and may even increase the risk of death (18–20). Furthermore, elderly schizophrenic patients treated chronically with neuroleptic drugs had an increased incidence of neurofibrillary pathology relative to schizophrenic patients not treated with neuroleptics (21). Consistent with these findings, we provide data from a human cell model of tau aggregation demonstrating high dose azaperone treatment promotes tau aggregation while intermediate doses protect against tau aggregation (Figure 3). Although this biphasic U-shaped dose response curve is difficult to interpret, one possible explanation is that these drugs are affecting a second receptor type at higher concentrations. This type of dose response curve has been described previously (22). The dopamine D1 receptor is not a likely candidate, since it is not known to be expressed by HEK293 cells (4). Another possible complicating factor in interpreting dose response curves in HEK/tau cells is the presence of dopamine in the serum in culture media, since fetal bovine serum is known to contain ~125 nM dopamine (13).
We propose that antipsychotic drugs may have potential benefit as tau targeted therapeutics when they are given at doses far lower than those currently used for clinical treatment of psychosis. In addition, there may be variation in the effectiveness of D2 antagonism for treatment of different types of tauopathy. For example, in FTDP-17 and PSP, dopaminergic and dopaminoreceptive neurons are susceptible to neurodegeneration and patients exhibit Parkinsonism (23). Additional preclinical work will be required in order to determine which subcategories of tauopathy will be the most promising candidates for treatment with low dose D2 antagonists.
Potential of C. elegans disease models for whole organism target independent drug discovery
C. elegans has great potential in future drug discovery efforts. Due to their small size and short, three-day life cycle, high-throughput drug screens are feasible in worms. In addition, most neuronal proteins are conserved between humans and C. elegans (24) making it a relevant model for the study of neurodegenerative diseases. Previous attempts at drug screening in C. elegans have been hindered by the relatively drug impermeable cuticle of C. elegans, but we have shown permeability can be increased by using a cuticle defective mutant. Using this model, we demonstrated pharmacological inhibition or genetic ablation of D2 dopamine receptors can protect against tau induced neuronal dysfunction, neurodegeneration, and protein aggregation. The complex dose response relationship between the dose of the antipsychotic drugs tested and tau aggregation suggests that the therapeutic window for this drug class may be narrow. Future studies using mouse models of tauopathy will determine whether D2 receptor antagonism may be a viable therapeutic approach for the prevention of tau pathology in the mammalian brain.
Supplementary Material
Acknowledgments
This work was supported by a grant from the Alzheimer’s Association (IIRG-08-90627, made possible in part by a generous gift from Sherrill Miller). Support was also provided by the Department of Veterans Affairs, and a National Institutes of Health grant [R01NS064131] to B.K. We thank the reviewers for helpful comments and suggestions. We thank Elaine Loomis, Aleen Saxton, Tobin Martin, and Susan Danner for outstanding technical assistance. We thank Virginia Lee, Peter Seubert, and Peter Davies for tau antibodies and the Developmental Studies Hybridoma Bank (NICHD) for the β-tubulin antibody E7.
Footnotes
Conflict of Interest
The authors report no biomedical financial interests or potential conflicts of interest.
Supplementary information is available at Molecular Psychiatry’s website.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sigurdsson EM. Immunotherapy targeting pathological tau protein in Alzheimer’s disease and related tauopathies. J Alzheimers Dis. 2008;15(2):157–168. doi: 10.3233/jad-2008-15202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kraemer BC, Zhang B, Leverenz JB, Thomas JH, Trojanowski JQ, Schellenberg GD. Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc Natl Acad Sci U S A. 2003;100(17):9980–9985. doi: 10.1073/pnas.1533448100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guthrie CR, Kraemer BC. Proteasome Inhibition Drives HDAC6-Dependent Recruitment of Tau to Aggresomes. J Mol Neurosci. 2011 doi: 10.1007/s12031-011-9502-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shaw G, Morse S, Ararat M, Graham FL. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 2002;16(8):869–871. doi: 10.1096/fj.01-0995fje. [DOI] [PubMed] [Google Scholar]
- 5.Guthrie CR, Schellenberg GD, Kraemer BC. SUT-2 potentiates tau-induced neurotoxicity in Caenorhabditis elegans. Hum Mol Genet. 2009;18(10):1825–1838. doi: 10.1093/hmg/ddp099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gravato-Nobre MJ, Hodgkin J. The acyltransferase gene bus-1 exhibits conserved and specific expression in nematode rectal cells and reveals pathogen-induced cell swelling. Dev Dyn. 2008;237(12):3762–3776. doi: 10.1002/dvdy.21792. [DOI] [PubMed] [Google Scholar]
- 7.Partridge FA, Tearle AW, Gravato-Nobre MJ, Schafer WR, Hodgkin J. The C. elegans glycosyltransferase BUS-8 has two distinct and essential roles in epidermal morphogenesis. Dev Biol. 2008;317(2):549–559. doi: 10.1016/j.ydbio.2008.02.060. [DOI] [PubMed] [Google Scholar]
- 8.Buee L, Troquier L, Burnouf S, Belarbi K, Van der Jeugd A, Ahmed T, et al. From tau phosphorylation to tau aggregation: what about neuronal death? Biochem Soc Trans. 2010;38(4):967–972. doi: 10.1042/BST0380967. [DOI] [PubMed] [Google Scholar]
- 9.Kraemer BC, Schellenberg GD. SUT-1 enables tau-induced neurotoxicity in C. elegans. Hum Mol Genet. 2007;16(16):1959–1971. doi: 10.1093/hmg/ddm143. [DOI] [PubMed] [Google Scholar]
- 10.Huang X, Cheng HJ, Tessier-Lavigne M, Jin Y. MAX-1, a novel PH/MyTH4/FERM domain cytoplasmic protein implicated in netrin-mediated axon repulsion. Neuron. 2002;34(4):563–576. doi: 10.1016/s0896-6273(02)00672-4. [DOI] [PubMed] [Google Scholar]
- 11.Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63(1):182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- 12.Chase DL, Pepper JS, Koelle MR. Mechanism of extrasynaptic dopamine signaling in Caenorhabditis elegans. Nat Neurosci. 2004;7(10):1096–1103. doi: 10.1038/nn1316. [DOI] [PubMed] [Google Scholar]
- 13.Little KY, Elmer LW, Zhong H, Scheys JO, Zhang L. Cocaine induction of dopamine transporter trafficking to the plasma membrane. Mol Pharmacol. 2002;61(2):436–445. doi: 10.1124/mol.61.2.436. [DOI] [PubMed] [Google Scholar]
- 14.Bulic B, Pickhardt M, Mandelkow EM, Mandelkow E. Tau Protein and Tau Aggregation Inhibitors. Neuropharmacology. 2010;59(4):276–289. doi: 10.1016/j.neuropharm.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 15.Nagy Z, Esiri MM, Jobst KA, Morris JH, King EM, McDonald B, et al. Relative roles of plaques and tangles in the dementia of Alzheimer’s disease: correlations using three sets of neuropathological criteria. Dementia. 1995;6(1):21–31. doi: 10.1159/000106918. [DOI] [PubMed] [Google Scholar]
- 16.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42(3 Pt 1):631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 17.Noble W, Hanger DP, Gallo JM. Transgenic mouse models of tauopathy in drug discovery. CNS Neurol Disord Drug Targets. 2010;9(4):403–428. doi: 10.2174/187152710791556131. [DOI] [PubMed] [Google Scholar]
- 18.Vigen CL, Mack WJ, Keefe RS, Sano M, Sultzer DL, Stroup TS, et al. Cognitive effects of atypical antipsychotic medications in patients with Alzheimer’s disease: outcomes from CATIE-AD. Am J Psychiatry. 2011;168(8):831–839. doi: 10.1176/appi.ajp.2011.08121844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schneider LS, Dagerman KS, Insel P. Risk of death with atypical antipsychotic drug treatment for dementia: meta-analysis of randomized placebo-controlled trials. JAMA. 2005;294(15):1934–1943. doi: 10.1001/jama.294.15.1934. [DOI] [PubMed] [Google Scholar]
- 20.Schneider LS, Dagerman K, Insel PS. Efficacy and adverse effects of atypical antipsychotics for dementia: meta-analysis of randomized, placebo-controlled trials. Am J Geriatr Psychiatry. 2006;14(3):191–210. doi: 10.1097/01.JGP.0000200589.01396.6d. [DOI] [PubMed] [Google Scholar]
- 21.Wisniewski HM, Constantinidis J, Wegiel J, Bobinski M, Tarnawski M. Neurofibrillary pathology in brains of elderly schizophrenics treated with neuroleptics. Alzheimer Dis Assoc Disord. 1994;8(4):211–227. doi: 10.1097/00002093-199408040-00001. [DOI] [PubMed] [Google Scholar]
- 22.Conolly RB. Nonmonotonic dose-response relationships: mechanistic basis, kinetic modeling, and implications for risk assessment. Toxicol Sci. 2004;77(1):151–157. doi: 10.1093/toxsci/kfh007. [DOI] [PubMed] [Google Scholar]
- 23.Chiba S, Takada E, Tadokoro M, Taniguchi T, Kadoyama K, Takenokuchi M, Kato S, Suzuki N. Loss of dopaminoreceptive neuron causes L-dopa resistant parkinsonism in tauopathy. Neurobiol Aging. 2012;33(10):2491–2505. doi: 10.1016/j.neurobiolaging.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 24.Bargmann CI. Neurobiology of the Caenorhabditis elegans genome. Science. 1998;282(5396):2028–2033. doi: 10.1126/science.282.5396.2028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.