

Abstract
The pancreatic islet secretes the hormones insulin and glucagon to regulate glucose metabolism. To generate an adequate secretory response, islet endocrine cells must receive multiple regulatory signals relaying information about changes in the internal and external environments. Islet cells also need to be made aware about the functional status of neighboring cells through paracrine interactions. All this information is used to orchestrate a hormonal response that contributes to glucose homeostasis. Several neurotransmitters have been proposed to work as paracrine signals in the islet. Most of these, however, have yet to meet the criteria to be considered bona fide paracrine signals, in particular in human islets. Here, we review recent findings describing autocrine and paracrine signaling mechanisms in human islets. These recent results are showing an increasingly complex picture of paracrine interactions in the human islet and emphasize that results from other species cannot be readily extrapolated to the human context. Investigators are unveiling new signaling mechanisms or finding new roles for known paracrine signals in human islets. While it is too early to provide a synthesis, the field of islet research is defining the paracrine and autocrine components that will be used to generate models about how islet function is regulated. Meanwhile, the identified signaling pathways can be proposed as therapeutic targets for treating diabetes, a devastating disease affecting millions worldwide.
INTRODUCTION
Diabetes mellitus is a common, disabling, and life-threatening disease. According to the Centers for Disease Control and Prevention, if current trends continue, 1 of 3 adults in the US will have diabetes by 2050. Today there is no cure for diabetes, but treatments include replacing the pancreatic hormone insulin, stimulating pancreatic beta cells to produce more insulin, or transplanting pancreatic islets of Langerhans. Thus, preserving glucose homeostasis and preventing diabetes critically depend on a well-functioning endocrine pancreas, the islets of Langerhans. Not surprisingly, the pancreatic islet has been intensively investigated. We know a great deal about how islet endocrine cells respond to glucose and how they couple these responses to hormone secretion. The basic mechanisms of islet function, however, have mainly been elucidated using animal models. As a result, our understanding of islet biology reflects anatomical and physiological features of islets from species other than the human, and in particular mice. Recent studies have revealed that islets from different species are so different that it is difficult to generalize findings from any particular species. An emphasis of this review is that to be relevant to human health, models of islet biology need to be reassessed by taking into account new findings about human islet structure and function.
As a consequence of a major effort aimed at moving therapeutic islet transplantation into the clinic, the incidence and quality of human islet isolations increased in the last decade [1]. Laboratories began using human islets to develop quality assays for transplantable human islet preparations [2]. This made human islets increasingly available for research purposes and ignited a new wave of studies of their functional properties. In the past, results on human islets were anecdotal. Now, however, with islet distribution programs, many groups world-wide are able to perform detailed mechanistic studies of human islet physiology in vitro. This has generated results that are often at odds with previous information gained from studying non-human species. Indeed, in a recent keynote lecture a renowned islet physiologist questioned whether he had wasted 25 years of his career conducting research on mouse islets! But thus far, detailed studies of human islets are only possible in vitro, and manipulation is limited to pharmacological tools. Cell-specific genetic interventions in islets in living mice will still be the gold standard for years to come for studying the role of particular signaling pathways in glucose homeostasis. Nevertheless, there is an impressive amount of new data that is providing an image of human islet biology not foreseen from rodent studies.
From the expression of voltage-gated channels to islet cell composition, from the basic mechanisms of beta cell division to autonomic neural control, investigators are revisiting the human islet [3-11]. The newly acquired information is slowly permeating the field, but it may take years for these findings to become accepted common knowledge. Needless to say, many results need to be confirmed, and there is skepticism, too. The rodent islet model is so ingrained that there is reluctance to view discrepant findings as real differences. There is a tendency to dismiss structural differences between islets from different species by considering them variations of a prototype that is based on the mouse islet. Thus, the porcine islet has been described as consisting of several mouse-like islets, and the human islet is viewed as a mouse islet with invaginations [12, 13]. The mouse has been such a spectacular animal model in islet research that it has “murinized” our view of islet biology.
Yet human islets are different, and because diabetes is reaching epidemic levels they require special attention. One striking difference between human and mouse islets is that endocrine cells of different types intermingle more in the human islet. In the mouse islet, most insulin-producing beta cells only abut other beta cells, whereas most beta cells in the human islet are in contact with alpha cells, delta cells, or both [8, 9, 11]. These anatomical arrangements likely have consequences for cell-to-cell communication within the islet to the point where paracrine interactions may play a dominating role in orchestrating hormone secretion in the human islet.
The major theme of this review is autocrine and paracrine signaling mechanisms in human islets. We examine results showing how molecules known to work as paracrine or neural signals in rodent islets have unexpected roles in the human islet. While it is too early to provide a synthesis on how paracrine and autocrine signaling shapes human islet biology, the new results point at signaling pathways that could be interventional targets for the treatment of diabetes.
ANATOMY OF PANCREATIC ISLETS
Paracrine interactions in human islets
Pancreatic islets are islands of endocrine tissue dispersed in the exocrine pancreas. They are composed not only of hormone-secreting endocrine cells but also of vascular cells, resident immune cells, and, in many species, neurons and glial cells of the neuroinsular complex (Figure 1). Human islets are surrounded by a complex double basement membrane [14]. Each islet is a functional unit; it has all the elements to produce adequate responses to changes in glucose concentration. Indeed, in vitro hormone responses to glucose from isolated islets faithfully reflect the secretory activity of the endocrine pancreas in the organism [15]. When transplanted into diabetic individuals, islets take over glucose homeostasis and restore normoglycemia [1]. Examining the structure of islets provides important clues about how they perform this function.
Figure 1. Cellular composition of pancreatic islets.

A, Confocal image of a human pancreatic section containing an islet immunostained for insulin (red, beta cells), glucagon (green, alpha cells), and somatostatin (blue, delta cells). These endocrine cells are distributed throughout the islet. Scale bar = 20 μm. B, Schematic diagram depicting endocrine cells (colors as in A), vascular cells (pink), and sympathetic axons (yellow) in the human islet. The vasculature in the human islet possesses numerous vascular smooth muscle cells embedded deep within the islet. These vascular cells are the main targets for sympathetic axons. The endocrine cells are aligned along the vessel without apparent order.
Endocrine cells of the pancreas include insulin-secreting beta cells, glucagon-secreting alpha cells, somatostatin-secreting delta cells, and cells that secrete pancreatic polypeptide. The relative population of these cells varies from islet to islet, from individual to individual, from species to species, and from study to study [16, 17]. In most species studied to date, beta cells are predominant. Human islets have a larger proportion of alpha cells than mouse islets (38% versus 18%) [11]. These numbers are disputed in the field of islet research, but perhaps more relevant than the relative proportion of these cells is how they are distributed within the islet. In human islets, alpha and delta cells are not segregated to the periphery as they are in the mouse islet. This has profound implications for islet function, as discussed next.
First, in human islets most if not all beta cells directly appose alpha cells, delta cells, or both (Figure 1). The association of beta cells with alpha cells in human islets is so close that, after dispersion of islets into single cells, most beta cells remain attached to an alpha cell [9]. These intimate contacts have multiple effects for endocrine cell function. Interactions between membrane-bound molecules expressed in these cells promote function and survival [18]. Indeed, beta cells that are associated with alpha cells secrete more insulin when stimulated with glucose [19]. The proximity of beta cells with alpha cells further enables paracrine interactions. Paracrine signaling requires close contact between source and target cells to be effective because many signaling molecules are rapidly degraded in the interstitial space and in the bloodstream. Close proximity also ensures that paracrine regulation is locally restricted. As explained below, the cytoarchitecture of the human islet suggests that molecules secreted by alpha or beta cells act as paracrine signals.
Second, in human islets, endocrine cells are distributed along blood vessels (Figure 2) [11]. As a result of the interspersion of beta, alpha, and delta cells in human pancreatic islets, no region is composed exclusively of a single cell type. In contrast with in islets in mice where blood flows sequentially through distinct regions containing either beta cells or non-beta cells, this does not occur in human islets. Any given region of the human islet contains a heterogeneous cell population through which blood flows. It is therefore unlikely that there is a hierarchy in the sequence in which the different endocrine cells are perfused in the human islet. This implies that the direction of blood flow does not dictate whether a particular paracrine interaction is possible or not in human islets, as has been suggested [20]. For instance, somatostatin has been disregarded as a paracrine signal because delta cells in mouse islets were assumed to be located downstream of beta cells and thus would not be able influence beta cells through the vascular route [20, 21]. Given the cytoarchitecture of the human islet, paracrine interactions can occur via interstitial spaces and do not require vascular perfusion.
Figure 2. The cellular organization of human and rodent islets is strikingly different.

A, B, Confocal images of pancreatic islets from a human (A) and a mouse (B), immunostained for insulin (red), glucagon (green), and somatostatin (blue). Notice the species differences in cell composition and cytoarchitecture. In particular, mouse beta cells (red) mostly appose other beta cells, whereas human beta cells almost always are in contact with alpha cells (green), or delta cells (blue), or both. Scale bar = 50 μm. C, Confocal images of human islets stained as in A showing alignment of endocrine cells along blood vessels (seen as dark spaces). There is no apparent order or segregation of different endocrine cells to distinct regions within the islet. Scale bar = 10 μm.
Cells of the human islet vasculature are potential sites for paracrine interactions
Endocrine cells in islets detect glucose and other nutrients in the bloodstream and release their hormones into a rich network of capillaries. Despite its importance for islet function, there are no detailed studies of the human islet vasculature. Blood vessels in mouse islets are mainly capillary endothelial tubes connected to one or two feeding arterioles that penetrate the islet. Recent findings indicate that blood vessels in human islets may contain more smooth muscle than those in mouse islets [10]. These contractile cells seem to be present throughout the islet vasculature and are the preferred target of sympathetic axons in the human islet (Figure 1B). As in other organs, blood flow can be locally regulated by vascular smooth muscle cells acting as sphincters. The presence of such sphincters deep within the islet suggests that these cells could also be targets for paracrine signals released from endocrine cells. The possibility that endocrine cells regulate their own blood supply by influencing adjacent vascular smooth muscle cells needs to be tested experimentally, but so far it has not been possible to study vascularized human islets.
In short, from an anatomical point of view human islets seem to be predisposed for paracrine signaling. The cellular arrangement makes it possible for endocrine cells to interact with other endocrine cells as well as with contractile vascular cells. The potential for paracrine regulation of islet function is immense.
THE PHYSIOLOGY OF PANCREATIC ISLETS
Stimulus-secretion coupling in islet cells provides targets for paracrine modulation
Although its cells are able to detect other nutrients such as amino acids, the islet is considered a glucose sensor. Unlike other chemoreceptors in the body that use G protein-coupled receptors for detecting sugars, islet endocrine cells transduce glucose stimulation by a series of metabolic processes that lead to changes in the cells membrane potential [22]. These processes include glucose transport into the cells via the low-affinity glucose transporter type 1 (GluT1; mouse beta cells depend on GluT2); glucose phosphorylation by the enzyme glucokinase; and the subsequent metabolism of glucose that increases the intracellular ATP-to-ADP ratio. Elevated ATP closes KATP channels, depolarizes the membrane, and opens voltage-gated Ca2+ channels, triggering hormone secretion. This complex chain of events not only ensures that glucose metabolism is coupled to membrane electrical activity but also provides multiple points where external signals can modulate hormone secretion.
The various endocrine cells in the human islet share many of the molecular components for detecting glucose [3, 4, 23]. This likely reflects their common developmental origin, but the similarities in stimulus-secretion coupling are puzzling given that the secretion of each hormone is elicited under different circumstances. For instance, increases in glucose concentration stimulate insulin secretion but inhibit glucagon release. Instead of relying on different mechanisms to transduce increases in glucose concentration into decreased cellular activity, alpha cells have additional mechanisms beyond those they share with beta cells. Thus, like beta and delta cells, alpha cells are depolarized by glucose. However, in alpha cells voltage-gated Na+ channels become inactivated and/or they become inhibited by paracrine signals released from beta cells [23, 24]. Both proposed mechanisms invert the effects of glucose by inhibiting electrical activity in alpha cells.
Many signals impinging on islet endocrine cells affect membrane electrical activity. Islet endocrine cells express voltage-gated ion channels that make them excitable; they display complex electrical activity with bursts of action potentials. In a recently proposed model for electrical activity in the human beta cell [25], glucose-induced closure of KATP channels depolarizes the cell to a membrane potential (~55 mV) where T-type and L-type Ca2+ channels are activated to initiate the action potential. Voltage-gated Na+ channels also contribute to the action potential, leading to sufficient depolarization to activate P/Q type Ca2+ channels. Ca2+ influx through P/Q type Ca2+ channels triggers exocytosis of insulin granules. The beta cell is repolarized by the activation of Ca2+-activated potassium channels (BK channels). In mouse islets, as with other molecular mechanisms (e.g., glucose transporter), beta cells use a different complement of ion channels for Ca2+-mediated exocytosis. Human beta, alpha, and delta cells express similar ion channels, and their electrical activities depend on a delicate balance between hyperpolarizing and depolarizing influences.
Because electrical activity is closely associated with exocytosis, islet hormone secretion can be modulated by activating ligand-gated membrane channels (i.e. ionotropic neurotransmitter receptors) or via signaling pathways affecting ion channel conductances.
Function of islet endocrine cells can also be regulated at the level of the secretory pathway. Islet endocrine cells are packed with dense core granules containing hormones and neurotransmitters. In response to a rise in cytoplasmic Ca2+, these granules fuse with the plasma membrane in a process requiring soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) [26]. This mechanism is similar to that of vesicular transmitter release in neurons. Stimuli that increase intracellular Ca2+, either via Ca2+ influx or via Ca2+ release from intracellular stores, trigger granule fusion. Granule fusion is crucial for insulin secretion and glucose homeostasis. Single nucleotide polymorphisms (SNPs) and alterations in the abundance of SNARE proteins have been linked to type 2 diabetes [27]. In addition to fusion, secretion also involves the mobilization of granules to the plasma membrane. Trafficking of granules to the plasma membrane requires cytoskeletal reorganization, which is accomplished with the help of the small Rho-family GTPases Cdc42 and Rac1. Additional GTPases play a role in docking and priming hormone granules. From the point of view of paracrine signaling, the Ras-like GTPase Rap1 is particularly interesting because its guanine-nucleotide exchange factor Epac2 is directly activated by cAMP, thus making it a target for signaling molecules modulating intracellular cAMP levels [26]. Neuronal, paracrine, or autocrine signals can therefore use different intracellular signaling cascades to regulate islet hormone exocytosis.
Multiple signals converge on pancreatic islets to coordinate the hormonal output
In spite of their name, islets are not “isolated”. To produce an adequate secretory response, islets need to integrate signals from multiple sources that relay information from within and from outside the body. For instance, plasma glucose provides islet cells not only with information about alterations in blood glucose (e.g. during food absorption) but also with feedback about how efficiently they are working. Incretins (hormones secreted by endocrine cells of the small intestine epithelium) provide signals from the GI tract of an ensuing rise in plasma glucose and prepare islet cells accordingly. Islets receive input from the hypothalamus via autonomic innervation and these signals integrate sensory inputs from the internal and external environments. Paracrine molecules signal the functional status of neighboring islet cells and modify a cell’s activity to coordinate its hormone secretion. The islet processes all these signals as a multicellular unit to produce a concerted hormonal output that efficiently maintains homeostatic control over plasma glucose.
The relative importance of the different inputs appears to vary from species to species. Because most beta cells in mouse islets are not associated with alpha cells [11], paracrine interactions between these cells may not be as strong in the mouse islet as they are in the human islet. On the other hand, the endocrine parenchyma of the mouse islet is densely innervated by autonomic axons, whereas in the human islet autonomic axons predominantly contact vascular smooth muscle cells [10]. Thus, coordination of endocrine cell function in human islets may rely more on local paracrine regulation and less on autonomic control. Yet human endocrine cells express a variety of neurotransmitter receptors and their secretory activity can be modulated by application of neurotransmitter agonists. The sympathetic transmitter adrenaline released by the adrenal gland likely reaches islets via the bloodstream. However, it is also possible that, in view of the sparse innervation, islet endocrine cells themselves are an alternative source for these neurotransmitters in the human islet. As discussed below, recent results indicate that molecules that were proposed as neuronal signals may be paracrine signals derived from local endocrine cells.
PARACRINE AND AUTOCRINE SIGNALING IN HUMAN ISLETS
Defining paracrine and autocrine signaling in the pancreatic islet
Paracrine communication can be defined as cellular signaling in which a factor secreted by a cell affects other cells in the local environment. In the context of the pancreatic islet, it is convenient to consider secreted molecules as paracrine signals if they affect other cells within the same islet, either via diffusion through interstitial spaces or via local microcirculation. In autocrine signaling, a cell secretes a messenger molecule that binds to receptors on the same cell. This definition of autocrine signaling can be extended to reflect functional features of islet. Thus, released molecules that affect other cells of the same cell type (e.g. beta cell to beta cell communication) are also considered autocrine signals because these cells work together to produce a common, synchronized hormonal output.
No signaling molecule is inherently a paracrine signal. The same hormone or neurotransmitter can be used in multiple contexts such as endocrine, paracrine, autocrine, or synaptic signaling. It is therefore crucial to establish criteria for a molecule to be considered a paracrine signal. By analogy to the formal criteria used to establish that a molecule is a neurotransmitter [28], we propose that a molecule is a paracrine signal if (1) the molecule is present in the islet cell; (2) the molecule is released in response to stimulation of the cell; and (3) specific receptors for the molecule are present on target cells in the islet. Although these criteria seem obvious, establishing them experimentally is challenging.
Paracrine signaling molecules themselves can be detected in islet cells. However, it is usually easier to demonstrate that precursors and biosynthetic enzymes for the molecules are present. Moreover, a major confounding factor is that many of the signaling molecules are also part of the general metabolism in all cells (e.g., glutamate). Therefore, the presence of a candidate molecule or its biosynthetic pathway is not sufficient to identify it as a paracrine signal. A more rigorous approach is to show that the molecule is present at higher densities in secretory granules or vesicles. This requires methodologies such as post-embedding immunoelectron microscopy. Expression of vesicular transporters for transmitters has recently been used to define a cell’s secretory phenotype. For instance, neurons expressing the vesicular acetylcholine transporter (vAChT) are considered cholinergic [29]. Because it is likely that not all transporters have been identified and that non-vesicular release routes are used, this approach may fail to identify cells using alternative mechanisms to secrete paracrine transmitters.
The second criterion defining a paracrine signal is that it is released from the islet cell. Demonstrating release is challenging for two reasons. First, the molecule may be secreted at low concentrations, or degraded by enzymes, or taken up by membrane transporters, making it barely detectable with current analytical methods. Second, it is difficult to stimulate specific islet cells selectively. When isolated intact islets are used in in vitro experiments, stimulation may induce intercellular signaling within the islet, thus confounding the cellular source for the paracrine signal. To distinguish which cells secrete which paracrine signals, dispersed islet cells can be studied. Yet, most techniques lack the sensitivity to detect molecules secreted from single cells. Alternatively, islet cells of a given type can be enriched using cell sorting and examined for secretion. Such protocols have yet to be established for human islets. Because of these technical limitations, most molecules proposed to function as paracrine signals in the pancreatic islet have not met this criterion.
A third criterion is that the target cell must express receptors for the signaling molecule. Receptor expression in a particular cell can be examined by using single cell RT-PCR or immunohistochemistry. Single cell RT-PCR, however, may not detect transcripts expressed at low levels and few antibodies for immunohistochemical detection of neurotransmitter or hormone receptors are reliable. An alternative method that has been commonly used is to stimulate islets with pharmacological agonists of the signaling candidate and examine changes in hormone secretion. Calcium imaging and electrophysiological techniques have also been used to demonstrate functional receptors expressed in islet cells. Indeed, most of the molecules proposed as paracrine signals have been discovered with pharmacological stimulation of receptors on islet cells. Satisfying this criterion, however, is not sufficient to establish a molecule as a paracrine signal because the receptors may be receptors for humoral or neural communication.
Paracrine and autocrine signals are involved in regulatory circuits
Mechanisms regulating hormone secretion from the islet often depend on feedback regulation. Many of the paracrine and autocrine signals in the islet are involved in regulatory circuits that use feedback. Interestingly, all the autocrine signals described below provide positive feedback. That is, the autocrine signals reinforce the effects produced by the initial perturbation (i.e., a change in glucose concentration). As a result, a small perturbation at the input causes a much larger effect at the output. Small deviations in plasma glucose concentration (~10%) are thus counteracted by sharp increases in insulin and glucagon secretion (3-fold) [30]. Positive feedback is often used in the rising phase of a physiological response to a perturbation. Not surprisingly, autocrine feedback loops in the islet are activated near the threshold for hormone responses to changes in glucose concentration. This helps making hormonal responses fast and robust.
Autocrine loops with positive feedback, however, could make intra-islet signaling highly unstable and produce inappropriate, runaway responses. This is prevented by negative feedback mechanisms. The physiological response to secreted hormones (i.e., restoring normoglycemia) is in itself a negative feedback signal. In addition to this reduction of the stimulus, or perturbation, there may also be active inhibition of hormone secretion within the islet. Indeed, somatostatin secretion from delta cells has been proposed to play this role. Somatostatin inhibits both glucagon and insulin secretion [31]. Autocrine loops with positive feedback thus appear to be embedded within, and controlled by more extensive negative feedback loops. At present, we know little about the exact mechanisms, but these principles provide a conceptual framework to investigate the signaling pathways contributing to orchestrate hormone secretion from the human islet.
Glutamate can function as a positive autocrine signal for glucagon secretion
Glutamate is the major excitatory neurotransmitter in the central nervous system. Although it has been proposed as an islet paracrine signal for almost 20 years [32], the functional role of glutamate in the islet is still controversial. In islets, glutamate may potentially originate from different sources such as the plasma, nerve terminals, or endocrine cells. Glutamate can reach plasma levels that stimulate receptors expressed in islet cells. However, to be relevant for paracrine or autocrine signaling, glutamate must be released from islet cells. Glutamate is present in glucagon secretory granules [33], and vesicular glutamate transporters (vGlut1, 2) are expressed in alpha cells of rodent, monkey, and human islet cells [34, 35]. Initial studies of rodent islets indicated that glutamate is secreted together with glucagon [33]. Findings in human islets later confirmed that glutamate is released under conditions that also stimulate glucagon secretion [35]. Results from a recent study, however, suggest that glutamate derived from metabolic pools, and not from granules, is released via membrane glutamate transporters by uptake reversal [36]. These results need to be confirmed in human islets, but they suggest that multiple mechanisms for glutamate secretion may exist.
Once released, glutamate can act on three different types of membrane receptors: AMPA/kainate receptors, metabotropic receptors, and NMDA receptors. Results of the expression of glutamate receptors in islet cells are conflicting [37]. Glutamate has been reported to activate alpha cells via AMPA/kainate receptors [32, 35, 38]; inhibit alpha cells via metabotropic receptors [39]; stimulate delta cells via AMPA/kainate receptors; or activate AMPA/kainate and metabotropic receptors in beta cells to increase insulin secretion [40, 41]. In an effort to obtain a coherent picture, a recent study compared glutamate signaling in human, monkey, and mouse islets [35]. In these species, glutamate activated AMPA/kainate receptors on alpha cells and stimulated glucagon secretion. By contrast, beta cells and insulin secretion could not be stimulated with glutamate agonists, either at basal or stimulatory glucose concentrations. These results were recently confirmed for mouse islets [38]. Physiological results indicate that activation of AMPA/kainate receptors leads to membrane depolarization, opening of voltage-gated Ca2+ channels, and an increase in glucagon secretion due to a rise in intracellular Ca2+ [35, 38]. Blocking AMPA/kainate receptors in vivo reduces glucagon release and exacerbates insulin-induced hypoglycemia in mice [35]. Although beta cells may also release glutamate from its metabolic pool [36], these results suggest that glutamate, derived from alpha cells, potentiates the alpha cell’s secretory activity. Thus, glutamate represents an autocrine positive feedback loop. This autocrine positive feedback ensures that even small decreases in plasma glucose concentration elicit sufficient glucagon secretion to avoid further hypoglycemia.
It is important, however, to consider that the concentrations of glutamate in the islet may be strongly affected by reverse transport from a cell’s metabolic pool [36]. Glutamate derived from such a mechanism could act as an extracellular signal as well as an intracellular metabolite that affects hormone secretion, thereby producing multiple effects. Furthermore, involvement of metabotropic receptors cannot be dismissed [39], and other endocrine cells within the islet (e.g. delta cells) may also respond to glutamate [42]. Clearly, more research is needed to further define the function of glutamate in the islet. Given that extracellular glutamate signaling through AMPA/kainate receptors is rapid, techniques that allow measuring glutamate secretion in real time while detecting receptor activation in target cells will be useful. It will also be important to determine if the proposed autocrine signaling loop via AMPA/kainate receptors is activated in alpha cells in the human organism in vivo.
Glutamate and the control of hypoglycemia
Hypoglycemia is potentially life threatening but is prevented by a series of physiological mechanisms termed glucose counterregulation. Among the glucose counterregulatory factors that prevent hypoglycemia, increased glucagon release is pivotal. Most cases of hypoglycemia seen in clinical practice are diabetic patients over-treated with insulin or sulfonylureas. Insulin excess causes glucose concentrations to fall to low levels in type 1 diabetes. Were it not for the devastating effects of hypoglycemia on the brain, diabetes would be easier to treat. As glucose levels decrease, glucagon secretion is not stimulated in type 1 diabetes. Glucagon secretory responses to stimuli other than hypoglycemia, however, are intact, indicating that the alpha cells retain their potential to secrete glucagon. AMPA/kainate receptors expressed in the human alpha cell thus represent a putative target for pharmacological intervention to prevent hypoglycemia. Safe drugs that positively modulate AMPA receptors (e.g. Ampakines) are currently being developed for potential therapeutic applications to improve memory and cognition as well as to treat schizophrenia. Using these new drugs to activate AMPA/kainate receptors in alpha cells could be an adjuvant therapy in the management of drug-treated diabetes.
ATP is a positive feedback signal promoting human beta cell responses to glucose
ATP is an important extracellular messenger molecule in the brain as well as in the vasculature and in endocrine organs [43]. Although its role in islet function has been extensively investigated, the results in the literature are contradictory. This is likely due to substantial species differences in ATP signaling in the islet. Insulin granules contain ATP that is released during high glucose stimulation [44-46]. ATP is secreted by “kiss- and-run” exocytosis while insulin is retained in the granule [47, 48], suggesting that ATP release precedes that of insulin. Thus, although present in the same granule, ATP and insulin may be released under different circumstances. In isolated human islets, blocking ATP degradation leads to increases in insulin secretion, providing indirect evidence that ATP is endogenously secreted [49, 50]. To date, consensus is that ATP is released by conventional secretory mechanisms (i.e., Ca2+-dependent exocytosis). However, as with other signaling molecules (e.g., glutamate and GABA), metabolic pools of ATP are large and transport across the membrane could be a major source for extracellular ATP. These alternative mechanisms have not been explored.
There are major species differences related to the effects of ATP in pancreatic islets. In mouse beta cells, ATP mainly activates P2Y receptors and decreases insulin secretion [51-53]. In rat islets, ATP is excitatory or inhibitory for beta cells [54-56]. By contrast, in monkey and human beta cells, ATP predominantly activates P2X receptors [49, 57, 58]. Activation of these ionotropic purinoreceptors likely produces large inward currents and depolarizes the beta cell membrane. Thus, ATP signaling increases human beta cell electrical activity and promotes insulin secretion.
Because ATP is released from beta cells, it is likely that ATP establishes a positive feedback loop that is activated during glucose-induced insulin release [49]. ATP can be released at relatively low basal glucose concentrations through kiss-and-run exocytosis [47, 48], suggesting that ATP may modulate the beta cell’s responsiveness to glucose at levels near threshold. This would make the beta cell more sensitive to small changes in glucose concentration and would accelerate the secretory response. This autocrine mechanism allows the beta cell to respond to small changes in glucose concentration with robust insulin secretion, preventing further increases in glycemia. The effects of ATP on other endocrine cells in the human islet have barely been investigated.
Given the well-established role ATP plays in vascular function, it is plausible that ATP released from endocrine cells increases microvascular permeability and modulates vascular resistance by acting on vascular cells. In many blood vessels, endothelial cells, vascular smooth muscle cells, and pericytes express a variety of ATP (purinergic) receptors [59]. Because their release generally increases with cellular activity, ATP and other purinergic compounds are ideal signals to couple blood flow with tissue metabolism. Therefore, ATP secreted from beta cells in response to increases in glucose concentration could activate endothelial cells in the islet microvasculature to change their permeability and promote hormone release into the bloodstream. ATP also acts on purinergic receptors expressed in smooth muscle cells and induces vasoconstriction [59]. Given that blood vessels in human islets have a larger proportion of contractile smooth muscle cells [10], ATP could act on these cells to change blood flow locally. These intriguing possibilities remain to be explored. For instance, it is not known what type of purinergic receptors are expressed in vascular cells in human islets. Investigating vascular function in human islets, however, is challenging because islet vascular cells do not survive in vitro. Furthermore, thus far it is not possible to study blood flow in vascularized human islets. New experimental approaches are needed to address paracrine interactions between endocrine cells and vascular cells as these may turn out to be crucial for islet function.
Acetylcholine: a shift from autonomic signal to paracrine signal?
Numerous studies have shown that acetylcholine acting via muscarinic receptors is crucial for pancreatic beta cell function [60]. Outside the central nervous system, acetylcholine is mainly a neurotransmitter of the autonomic nervous system, both in preganglionic axonal endings and in postganglionic parasympathetic axons. In the rodent pancreatic islet, acetylcholine is likely released from parasympathetic axons innervating alpha and beta cells [60]. Although release has not been demonstrated directly, vesicular acetylcholine transporter (vAChT) is present in axons in the mouse islet, indicating that acetylcholine is secreted [10]. The mouse islet parenchyma is densely innervated but few if any vAChT-immunoreactive axons penetrate the human islet [10]. Instead, recent results indicate that the human alpha cell is cholinergic [61]. Human alpha cells not only express vAChT but also choline acetyltransferase (ChAT), the enzyme required to produce acetylcholine. Within the human alpha cell, immunostaining for vAChT does not overlap with glucagon staining, suggesting that acetylcholine may not be secreted from hormone-containing granules. Acetylcholine release is stimulated by kainate or by lowering glucose concentration. Both these stimuli are specific for alpha cells. In line with the notion that acetylcholine is endogenously released, blocking degradation of acetylcholine increases insulin secretion from isolated human islets. These results suggest that in human islets, acetylcholine is a paracrine signal secreted from alpha cells.
Once released, it is likely that acetylcholine acts only locally on adjacent endocrine cells. The quick degradation by cholinesterases in the extracellular space and in the blood stream prevents acetylcholine from diffusing within the islet or from being transported in the blood stream. However, given the arrangement of endocrine cells in human islets, acetylcholine released from alpha cells can readily activate neighboring beta cells. Because alpha cells release acetycholine in response to a lowering in glucose concentration, this paracrine transmitter will act on beta cells when these latter cells are not themselves stimulated. How can a signal derived from alpha cells affect beta cells under these circumstances? It is known that muscarinic receptor activation affects beta cell secretion for tens of minutes [62]. Acetylcholine does not necessarily produce immediate effects on insulin secretion but can activate signaling cascades that potentiate beta cell responses to forthcoming rises in glucose concentration. This makes sense in vivo. Independent of the glucose concentration, secretory pulses of insulin and glucagon alternate within less than 10 minutes [63]. Insulin secretion is therefore always preceded by a period of alpha cell activation. When the pulsatile pattern of hormone secretion is mimicked in vitro, endogenously released acetylcholine sensitizes beta cells to subsequent increases in glucose concentration [61]. Thus, acetylcholine in human islets may work as a feed forward signal to keep beta cells responsive to subsequent challenges.
Muscarinic signaling in beta cells has been extensively investigated in mouse islets, where it is essential for maintaining adequate insulin secretion and glucose homeostasis [64]. By contrast, cholinergic signaling has barely been investigated in human islets. In view of recent findings suggesting that acetylcholine is a paracrine signal and not derived from neural innervation [10, 61], it will be important to define the different components that contribute to cholinergic signaling in the human islet. For example, because they play a role in shaping the duration and magnitude of acetylcholine signals, the expression of cholinesterases and choline transporters must be explored. Moreover, initial studies suggest that human beta cells, like mouse beta cells, express muscarinic M3 receptors [61], but the full complement of muscarinic receptors in human beta cells, alpha cells, and delta cells is not known. Knowing the intracellular signaling pathways activated by acetylcholine will help understand the role this paracrine signal plays not only in islet cell function but also in long-term survival of human beta cells. All these cholinergic components are putative targets for pharmacological interventions and have implications for the treatment of diabetes.
Whereas the muscarinic signaling mechanisms may turn out to be similar in mouse and human beta cells, the finding that acetylcholine is a paracrine signal derived from human alpha cells can be considered a paradigm shift (Figure 3). If these recent results are confirmed in vivo, it is likely that cholinergic signaling is activated in human islets under circumstances that are very different from those in mouse islets. The cholinergic signal in the human islet would not convey information from central autonomic brain centers but transmit local information aimed at optimizing hormone secretion in the context of pulsatile hormone secretion.
Figure 3. Schematic diagram depicting cholinergic signaling in pancreatic islets.
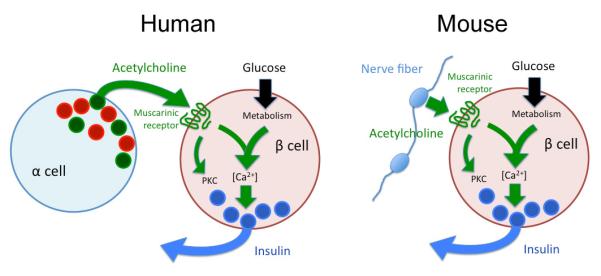
Whereas muscarinic signaling may be similar in human (left) and mouse (right) beta cells, acetylcholine originates from alpha cells in human islets and from parasympathetic axons in mouse islets.
Mysterious and magical GABA
GABA is a major inhibitory neurotransmitter in the central nervous system. This inhibitory neurotransmitter is also making headlines in the field of diabetes research. Since the initial findings showing that GABA released from beta cells inhibit alpha cells in rodent islets [65], new roles for GABA signaling have emerged. In a recent study, for instance, GABA was shown to enhance proliferation of mouse beta cells and reverse diabetes in mice [66]. Relevant for human islet function are recent findings suggesting that GABA signaling via GABAA receptors constitutes an autocrine positive feedback loop in human beta cells [5].
GABA levels in human islets are as high as in the brain [67]. Similar to glutamate, metabolic pools of GABA in islet endocrine cells are large. Immunogold electron microscopy studies show that GABA levels in the cytoplasm of human beta, alpha, and delta cells are high [5]. The high content of GABA in human islet endocrine cells is in agreement with strong expression in these cells of glutamic acid decarboxylase type 65 (GAD65) [68], the enzyme catalyzing decarboxylation of glutamate to GABA. the enzyme catalyzing conversion of glutamate to GABA. By contrast, in rodent islets only beta cells express GAD65, and GABA levels are lower in non-beta cells than in beta cells [69, 70]. GABA is also present in insulin granules, from which it is released upon stimulation with glucose [5]. Given that GABA is present in granules, it is puzzling that human beta cells do not appear to express the vesicular GABA transporter vGAT [71]. GABA is also secreted using non-vesicular, glucose-independent mechanisms that have not been examined in the human islet [72]. As a result, human islets have resting (background) levels of GABA that do not vary with glucose concentration. Background GABA secretion may in itself represent a paracrine signal that may interfere with GABA signaling from the vesicular pool. Once released, GABA can activate ionotropic GABAA and metabotropic GABAB receptors. In rodents, activation of GABAB receptors reduces insulin secretion [73], but the functional role of these receptors in human islets is not known. Human beta, alpha, and delta cells express functional GABAA receptors [5]. Activating these receptors increases a Cl− conductance that drives the membrane potential towards the equilibrium potential for Cl−. The Cl− equilibrium potential varies from cell to cell and depends on the expression and activity of Cl− transporters. Interestingly, in the human islet, GABA inhibits alpha cell electrical activity but it depolarizes delta cells and beta cells [5]. However, given that GABA moves the membrane potential of the beta cell to the cell’s Cl− equilibrium potential (estimated to be > −40 mV in human beta cells), GABA will depolarize beta cells at low glucose concentrations but may inhibit beta cells when they are stimulated by higher glucose concentrations. Accordingly, GABA was reported to contribute to insulin secretion at low glucose concentrations, suggesting that under these conditions GABA signaling forms an autocrine positive feedback loop in human beta cells [5]. Yet, higher GABA concentrations inhibited action potentials [5]. Thus, GABA could have an intriguing dual role in regulating beta cell function.
In addition to insulin secretion, GABA may also regulate the secretion of glucagon and somatostatin in human islets. Because somatostatin is a strong inhibitor of insulin and glucagon secretion, GABA released from beta cells may activate a negative feedback loop involving delta cells. If so, GABA would be working as an autocrine signal providing fast positive feedback as well as a paracrine signal mediating delayed negative feedback for beta cells. An arrangement in which the autocrine positive feedback loop is embedded within a loop with negative feedback would prevent exacerbated activation of the beta cell. To be involved in these mechanisms, however, the concentration of extracellular GABA must be tightly regulated and coupled to changes in plasma glucose concentration. This is unclear given the large background secretion of GABA. Thus, it will be important to determine the temporal and spatial profiles of GABA release and accumulation within the intact islet. It is also possible that, as in the brain [74], non-vesicular and vesicular GABA release participate in different physiological processes. To add to the complexity, GABA may also activate GABAB receptors expressed in different endocrine cells. Hence, it is likely that GABA will turn out to be a signal with multiple functions within the human islet.
Recent results in mice suggest that GABA may also have remarkable trophic effects for beta cells [66]. The actions of GABA are so beneficial that they reportedly restore beta cell mass in The actions of GABA are so beneficial that they reportedly restore beta cell mass in severely diabetic mice. Because they suggest an additional, important role for GABA in the islet, these effects need to be confirmed in human islets.
GABA and diabetes
In a twist of fate, GABA could be involved both in the pathogenesis and in the treatment of type 1 diabetes. Indeed, the enzymes producing GABA, GAD67 and GAD65, are targets of autoantibodies in people who later develop type 1 diabetes [75]. If applied clinically, the product of the enzymatic reaction these enzymes catalyze (i.e. GABA) would be used to treat a condition they help create.
The panoply of candidates for paracrine and autocrine interactions
The list of putative paracrine and autocrine signals in pancreatic islets keeps growing. Although not discussed here, the hormones glucagon, insulin, and somatostatin also serve as local signaling molecules. Somatostatin secreted from delta cells is likely a major regulator of human islet function that deserves attention. Little is known about how the function of delta cells and the secretion of somatostatin are controlled in the human islet. Because delta cells in human islets intermingle with beta and alpha cells, it is likely that delta cells are affected by paracrine signals. These signals are not yet known and how they would combine with glucose stimulation to modulate somatostatin release needs to be investigated. Furthermore, the effects of somatostatin on other endocrine cells need to be explored in more detail. We still do not understand how somatostatin shapes hormonal output from the human islet.
Other neurotransmitters and neuropeptides suggested to play a signaling role in the islet include serotonin [76], dopamine [77-79], and NPY [80-82]. Of these, serotonin is fascinating because it reportedly plays a role in promoting beta cell proliferation during pregnancy [83]. The expression of the different components involved in serotonin signaling is lower under normal physiological conditions, suggesting that serotonin signaling is restricted to islets in mammalian females during pregnancy. Thus, serotonin may be one of the few signaling molecules in the islet for which a very specific function (stimulate beta cell proliferation) within a very well defined physiological context (increased demand for insulin during pregnancy) has been identified. The situation may be more complex, however, because serotonin affects vascular permeability in other organs and may have similar effects in the islet [84]. Whether or not serotonin plays additional roles in human islet function remains to be explored.
Many molecules have been suggested to function as paracrine or autocrine signals. Most of these, however, have not yet met the standard to be considered bona fide signaling molecules in the islet. For instance, many receptors found in islet endocrine cells are still in search of an endogenous ligand. Nevertheless, receptor expression results can give a hint of what additional paracrine interactions may exist in the islet and thus could serve to guide future studies. While their involvement in intra-islet signaling is being determined, receptors expressed in islet endocrine cells can be used as targets for imaging probes aimed at detecting islet mass in vivo.
CONCLUSIONS
A potpourri of paracrine and autocrine signals in search for a function
The findings reviewed here stress two points: paracrine and autocrine signaling in human islets is complex, and results from other species cannot be readily extrapolated to the human context. As recent studies show, investigators are unveiling new signaling mechanisms or finding new roles for known paracrine signals in human islets (Figure 4). Because human islet biology has been investigated less extensively, we can expect more discoveries in the next years. This may add to the complexity or confusion. However, firmly establishing which candidate molecules are bona fide paracrine or autocrine signals can bring some light. Further, although challenging, it is critical to identify the biological contexts in which the paracrine and autocrine signals work. Because these signals may be involved in a variety of functions (e.g. shaping hormone secretion, local regulation of blood flow, controlling islet cell proliferation), conceptual frameworks specific for each function are needed to guide the research. To understand its functional role, we need to know precisely when and where, and not only whether and how, a molecule is released and acts.
Figure 4. Schematic diagram depicting putative paracrine and autocrine pathways in human pancreatic islets.

Arrows indicate the origin and target of a particular paracrine signal. Black arrows denote excitatory signaling, white arrows indicate inhibition.
These challenges require technological innovation. An important step towards elucidating the role of a paracrine signal is to determine its spatial and temporal profile of release and action within the islet. The use of biosensor cells placed in close vicinity to secreting cells is a promising approach to detect a paracrine signal locally and in real time [35, 47, 48, 61]. Biosensor cell technology has microscopic spatial resolution and can resolve signals in the order of seconds, sufficient to record paracrine events. A biosensor cell apposed to an islet, or an islet cell converted into a biosensor cell could be used to detect local changes in the concentration of the paracrine signal. Ultimately, however, to understand their biological function, paracrine interactions will need to be investigated in vivo. Examining in vivo islet biology with cellular resolution and in real time is currently not possible in human islets. As a compromise, transplanting human islets into mice can be used as a model to study vascularized human islets in a living organism. The recent approach in which islets are transplanted into the anterior chamber of the mouse eye for intravital imaging could be adapted to study human islets in vivo [85, 86]. With this model, selective stimulation of a particular cell type within the islet can be achieved noninvasively using optogenetics, and secretion can be measured using islet cells that are converted into biosensor cells [87]. At the same time, glucose homeostasis can be recorded by measuring glycemia and plasma hormone concentrations in the mouse. These in vivo studies will likely generate results not predicted by in vitro studies.
A new picture of human islet biology is emerging based on recent data, but it is too early to provide a synthesis of paracrine and autocrine signaling in the human islet. The recent findings emphasize that a model of islet biology has to include structural and functional features of the human islet. This is particularly important when it comes to translate this knowledge into the clinical setting. To propose a particular signaling pathway for pharmacological intervention in human beings will require that the specific pathway is present in human islets. It is likely that detailed knowledge of human islet biology will lead to effective therapies to treat diabetes.
HIGHLIGHTS.
The cellular arrangement predisposes human islets for paracrine interactions
Paracrine and autocrine signaling in human islets is complex
Results from other species cannot be readily extrapolated to the human context
Human islets have paracrine signaling mechanisms not described before
Known paracrine signals have new roles in human islets
ACKNOWLEDGEMENTS
The author thanks Drs. Per-Olof Berggren, Camillo Ricordi, and Stephen D. Roper for their mentorship and support. Work of the author was funded by the Diabetes Research Institute Foundation (DRIF), NIH grants R03DK075487, R56DK084321, and R01DK084321, and the Juvenile Diabetes Research Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Harlan D, Kenyon N, Korsgren O, Roep B, Society IoD. Current advances and travails in islet transplantation. Diabetes. 2009;58:2175–84. doi: 10.2337/db09-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Papas KK, Suszynski TM, Colton CK. Islet assessment for transplantation. Curr Opin Organ Transplant. 2009;14:674–82. doi: 10.1097/MOT.0b013e328332a489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Braun M, Ramracheya R, Bengtsson M, Zhang Q, Karanauskaite J, Partridge C, et al. Voltage-gated ion channels in human pancreatic beta-cells: electrophysiological characterization and role in insulin secretion. Diabetes. 2008;57:1618–28. doi: 10.2337/db07-0991. [DOI] [PubMed] [Google Scholar]
- [4].Braun M, Ramracheya R, Amisten S, Bengtsson M, Moritoh Y, Zhang Q, et al. Somatostatin release, electrical activity, membrane currents and exocytosis in human pancreatic delta cells. Diabetologia. 2009 doi: 10.1007/s00125-009-1382-z. [DOI] [PubMed] [Google Scholar]
- [5].Braun M, Ramracheya R, Bengtsson M, Clark A, Walker J, Johnson P, et al. GABA is an autocrine excitatory transmitter in human pancreatic {beta}-cells. Diabetes. 2010 doi: 10.2337/db09-0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hanna S, Pigeau G, Galvanovskis J, Clark A, Rorsman P, MacDonald P. Kiss-and-run exocytosis and fusion pores of secretory vesicles in human beta-cells. Pflugers Arch. 2009;457:1343–50. doi: 10.1007/s00424-008-0588-0. [DOI] [PubMed] [Google Scholar]
- [7].Fiaschi-Taesch NM, Salim F, Kleinberger J, Troxell R, Cozar-Castellano I, Selk K, et al. Induction of human beta-cell proliferation and engraftment using a single G1/S regulatory molecule, cdk6. Diabetes. 2010;59:1926–36. doi: 10.2337/db09-1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Brissova M, Fowler M, Nicholson W, Chu A, Hirshberg B, Harlan D, et al. Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J Histochem Cytochem. 2005;53:1087–97. doi: 10.1369/jhc.5C6684.2005. [DOI] [PubMed] [Google Scholar]
- [9].Bosco D, Armanet M, Morel P, Niclauss N, Sgroi A, Muller Y, et al. Unique arrangement of alpha- and beta-cells in human islets of Langerhans. Diabetes. 2010;59:1202–10. doi: 10.2337/db09-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rodriguez-Diaz R, Abdulreda MH, Formoso AL, Gans I, Ricordi C, Berggren PO, et al. Innervation patterns of autonomic axons in the human endocrine pancreas. Cell Metab. 2011;14:45–54. doi: 10.1016/j.cmet.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cabrera O, Berman D, Kenyon N, Ricordi C, Berggrern P, Caicedo A. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:2334–9. doi: 10.1073/pnas.0510790103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bonner-Weir S, O’Brien TD. Islets in type 2 diabetes: in honor of Dr. Robert C. Turner. Diabetes. 2008;57:2899–904. doi: 10.2337/db07-1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Erlandsen SL, Hegre OD, Parsons JA, McEvoy RC, Elde RP. Pancreatic islet cell hormones distribution of cell types in the islet and evidence for the presence of somatostatin and gastrin within the D cell. J Histochem Cytochem. 1976;24:883–97. doi: 10.1177/24.7.60437. [DOI] [PubMed] [Google Scholar]
- [14].Otonkoski T, Banerjee M, Korsgren O, Thornell LE, Virtanen I. Unique basement membrane structure of human pancreatic islets: implications for beta-cell growth and differentiation. Diabetes Obes Metab. 2008;10(Suppl 4):119–27. doi: 10.1111/j.1463-1326.2008.00955.x. [DOI] [PubMed] [Google Scholar]
- [15].Henquin J, Dufrane D, Nenquin M. Nutrient control of insulin secretion in isolated normal human islets. Diabetes. 2006;55:3470–7. doi: 10.2337/db06-0868. [DOI] [PubMed] [Google Scholar]
- [16].Pisania A, Weir GC, O’Neil JJ, Omer A, Tchipashvili V, Lei J, et al. Quantitative analysis of cell composition and purity of human pancreatic islet preparations. Lab Invest. United States. 2010:1661–75. doi: 10.1038/labinvest.2010.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Steiner DJ, Kim A, Miller K, Hara M. Pancreatic islet plasticity: interspecies comparison of islet architecture and composition. Islets. 2010;2:135–45. doi: 10.4161/isl.2.3.11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jain R, Lammert E. Cell-cell interactions in the endocrine pancreas. Diabetes Obes Metab. 2009;11(Suppl 4):159–67. doi: 10.1111/j.1463-1326.2009.01102.x. [DOI] [PubMed] [Google Scholar]
- [19].Wojtusciszyn A, Armanet M, Morel P, Berney T, Bosco D. Insulin secretion from human beta cells is heterogeneous and dependent on cell-to-cell contacts. Diabetologia. 2008;51:1843–52. doi: 10.1007/s00125-008-1103-z. [DOI] [PubMed] [Google Scholar]
- [20].Stagner JI, Samols E. The vascular order of islet cellular perfusion in the human pancreas. Diabetes. 1992;41:93–7. doi: 10.2337/diab.41.1.93. [DOI] [PubMed] [Google Scholar]
- [21].Samols E, Stagner JI. Islet somatostatin--microvascular, paracrine, and pulsatile regulation. Metabolism. 1990;39:55–60. doi: 10.1016/0026-0495(90)90212-u. [DOI] [PubMed] [Google Scholar]
- [22].Henquin J. Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia. 2009;52:739–51. doi: 10.1007/s00125-009-1314-y. [DOI] [PubMed] [Google Scholar]
- [23].Ramracheya R, Ward C, Shigeto M, Walker JN, Amisten S, Zhang Q, et al. Membrane potential-dependent inactivation of voltage-gated ion channels in alpha-cells inhibits glucagon secretion from human islets. Diabetes. 2010;59:2198–208. doi: 10.2337/db09-1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gromada J, Franklin I, Wollheim C. Alpha-cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocr Rev. 2007;28:84–116. doi: 10.1210/er.2006-0007. [DOI] [PubMed] [Google Scholar]
- [25].Braun M, Ramracheya R, Bengtsson M, Zhang Q, Karanauskaite J, Partridge C, et al. Voltage-gated ion channels in human pancreatic beta-cells: electrophysiological characterization and role in insulin secretion. Diabetes. 2008;57:1618–28. doi: 10.2337/db07-0991. [DOI] [PubMed] [Google Scholar]
- [26].Wang Z, Thurmond DC. Mechanisms of biphasic insulin-granule exocytosis - roles of the cytoskeleton, small GTPases and SNARE proteins. J Cell Sci. 2009;122:893–903. doi: 10.1242/jcs.034355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jewell JL, Oh E, Thurmond DC. Exocytosis mechanisms underlying insulin release and glucose uptake: conserved roles for Munc18c and syntaxin 4. Am J Physiol Regul Integr Comp Physiol. 2010;298:R517–31. doi: 10.1152/ajpregu.00597.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Neuroscience. 4th ed Sinauer Associates, Inc; 2008. [Google Scholar]
- [29].Weihe E, Tao-Cheng J, Schäfer M, Erickson J, Eiden L. Visualization of the vesicular acetylcholine transporter in cholinergic nerve terminals and its targeting to a specific population of small synaptic vesicles. Proc Natl Acad Sci U S A. 1996;93:3547–52. doi: 10.1073/pnas.93.8.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Conn PM, Goodman HM, Kostyo JL. The endocrine system. Published for the American Physiological Society by Oxford University Press; New York: 1998. [Google Scholar]
- [31].Taborsky GJ, Smith PH, Porte D. Differential effects of somatostatin analogues on alpha- and beta-cells of the pancreas. Am J Physiol. 1979;236:E123–8. doi: 10.1152/ajpendo.1979.236.2.E123. [DOI] [PubMed] [Google Scholar]
- [32].Bertrand G, Gross R, Puech R, Loubatières-Mariani M, Bockaert J. Glutamate stimulates glucagon secretion via an excitatory amino acid receptor of the AMPA subtype in rat pancreas. Eur J Pharmacol. 1993;237:45–50. doi: 10.1016/0014-2999(93)90091-u. [DOI] [PubMed] [Google Scholar]
- [33].Hayashi M, Yamada H, Uehara S, Morimoto R, Muroyama A, Yatsushiro S, et al. Secretory granule-mediated co-secretion of L-glutamate and glucagon triggers glutamatergic signal transmission in islets of Langerhans. J Biol Chem. 2003;278:1966–74. doi: 10.1074/jbc.M206758200. [DOI] [PubMed] [Google Scholar]
- [34].Hayashi M, Otsuka M, Morimoto R, Hirota S, Yatsushiro S, Takeda J, et al. Differentiation-associated Na+-dependent inorganic phosphate cotransporter (DNPI) is a vesicular glutamate transporter in endocrine glutamatergic systems. J Biol Chem. 2001;276:43400–6. doi: 10.1074/jbc.M106244200. [DOI] [PubMed] [Google Scholar]
- [35].Cabrera O, Jacques-Silva M, Speier S, Yang S, Kohler M, Fachado A, et al. Glutamate is a positive autocrine signal for glucagon release. Cell Metabolism. 2008;7:545–54. doi: 10.1016/j.cmet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Feldmann N, del Rio RM, Gjinovci A, Tamarit-Rodriguez J, Wollheim CB, Wiederkehr A. Reduction of plasma membrane glutamate transport potentiates insulin but not glucagon secretion in pancreatic islet cells. Mol Cell Endocrinol. 2011;338:46–57. doi: 10.1016/j.mce.2011.02.019. [DOI] [PubMed] [Google Scholar]
- [37].Moriyama Y, Hayashi M. Glutamate-mediated signaling in the islets of Langerhans: a thread entangled. Trends Pharmacol Sci. 2003;24:511–7. doi: 10.1016/j.tips.2003.08.002. [DOI] [PubMed] [Google Scholar]
- [38].Cho JH, Chen L, Kim MH, Chow RH, Hille B, Koh DS. Characteristics and functions of {alpha}-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptors expressed in mouse pancreatic {alpha}-cells. Endocrinology. 2010;151:1541–50. doi: 10.1210/en.2009-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Uehara S, Muroyama A, Echigo N, Morimoto R, Otsuka M, Yatsushiro S, et al. Metabotropic glutamate receptor type 4 is involved in autoinhibitory cascade for glucagon secretion by alpha-cells of islet of Langerhans. Diabetes. 2004;53:998–1006. doi: 10.2337/diabetes.53.4.998. [DOI] [PubMed] [Google Scholar]
- [40].Bertrand G, Puech R, Loubatieres-Mariani M, Bockaert J. Glutamate stimulates insulin secretion and improves glucose tolerance in rats. Am J Physiol. 1995;269:E551–6. doi: 10.1152/ajpendo.1995.269.3.E551. [DOI] [PubMed] [Google Scholar]
- [41].Storto M, Capobianco L, Battaglia G, Molinaro G, Gradini R, Riozzi B, et al. Insulin secretion is controlled by mGlu5 metabotropic glutamate receptors. Mol Pharmacol. 2006;69:1234–41. doi: 10.1124/mol.105.018390. [DOI] [PubMed] [Google Scholar]
- [42].Muroyama A, Uehara S, Yatsushiro S, Echigo N, Morimoto R, Morita M, et al. A novel variant of ionotropic glutamate receptor regulates somatostatin secretion from delta-cells of islets of Langerhans. Diabetes. 2004;53:1743–53. doi: 10.2337/diabetes.53.7.1743. [DOI] [PubMed] [Google Scholar]
- [43].Burnstock G. Pathophysiology and therapeutic potential of purinergic signaling. Pharmacol Rev. 2006;58:58–86. doi: 10.1124/pr.58.1.5. [DOI] [PubMed] [Google Scholar]
- [44].Detimary P, Jonas J, Henquin J. Stable and diffusible pools of nucleotides in pancreatic islet cells. Endocrinology. 1996;137:4671–6. doi: 10.1210/endo.137.11.8895332. [DOI] [PubMed] [Google Scholar]
- [45].Hazama A, Hayashi S, Okada Y. Cell surface measurements of ATP release from single pancreatic beta cells using a novel biosensor technique. Pflugers Arch. 1998;437:31–5. doi: 10.1007/s004240050742. [DOI] [PubMed] [Google Scholar]
- [46].Leitner J, Sussman K, Vatter A, Schneider F. Adenine nucleotides in the secretory granule fraction of rat islets. Endocrinology. 1975;96:662–77. doi: 10.1210/endo-96-3-662. [DOI] [PubMed] [Google Scholar]
- [47].MacDonald PE, Braun M, Galvanovskis J, Rorsman P. Release of small transmitters through kiss-and-run fusion pores in rat pancreatic beta cells. Cell Metab. 2006;4:283–90. doi: 10.1016/j.cmet.2006.08.011. [DOI] [PubMed] [Google Scholar]
- [48].Obermüller S, Lindqvist A, Karanauskaite J, Galvanovskis J, Rorsman P, Barg S. Selective nucleotide-release from dense-core granules in insulin-secreting cells. J Cell Sci. 2005;118:4271–82. doi: 10.1242/jcs.02549. [DOI] [PubMed] [Google Scholar]
- [49].Jacques-Silva M, Correa-Medina M, Cabrera O, Rodriguez-Diaz R, Makeeva N, Fachado A, et al. ATP-gated P2X(3) receptors constitute a positive autocrine signal for insulin release in the human pancreatic beta cell. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:6465–70. doi: 10.1073/pnas.0908935107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Lavoie EG, Fausther M, Kauffenstein G, Kukulski F, Künzli BM, Friess H, et al. Identification of the ectonucleotidases expressed in mouse, rat, and human Langerhans islets: potential role of NTPDase3 in insulin secretion. Am J Physiol Endocrinol Metab. 2010;299:E647–56. doi: 10.1152/ajpendo.00126.2010. [DOI] [PubMed] [Google Scholar]
- [51].Léon C, Freund M, Latchoumanin O, Farret A, Petit P, Cazenave J, et al. The P2Y(1) receptor is involved in the maintenance of glucose homeostasis and in insulin secretion in mice. Purinergic Signal. 2005;1:145–51. doi: 10.1007/s11302-005-6209-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Petit P, Hillaire-Buys D, Manteghetti M, Debrus S, Chapal J, Loubatières-Mariani M. Evidence for two different types of P2 receptors stimulating insulin secretion from pancreatic B cell. Br J Pharmacol. 1998;125:1368–74. doi: 10.1038/sj.bjp.0702214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Poulsen C, Bokvist K, Olsen H, Høy M, Capito K, Gilon P, et al. Multiple sites of purinergic control of insulin secretion in mouse pancreatic beta-cells. Diabetes. 1999;48:2171–81. doi: 10.2337/diabetes.48.11.2171. [DOI] [PubMed] [Google Scholar]
- [54].Petit P, Manteghetti M, Puech R, Loubatieres-Mariani M. ATP and phosphate-modified adenine nucleotide analogues. Effects on insulin secretion and calcium uptake. Biochem Pharmacol. 1987;36:377–80. doi: 10.1016/0006-2952(87)90297-8. [DOI] [PubMed] [Google Scholar]
- [55].Salehi A, Eliasson L, Ma X, Rorsman P, Håkanson R, Lundquist I. Secretory and electrophysiological characteristics of insulin cells from gastrectomized mice: evidence for the existence of insulinotropic agents in the stomach. Regul Pept. 2007;139:31–8. doi: 10.1016/j.regpep.2006.10.001. [DOI] [PubMed] [Google Scholar]
- [56].Richards-Williams C, Contreras J, Berecek K, Schwiebert E. Extracellular ATP and zinc are co-secreted with insulin and activate multiple P2X purinergic receptor channels expressed by islet beta-cells to potentiate insulin secretion. Purinergic Signal. 2008;4:393–405. doi: 10.1007/s11302-008-9126-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Fernandez-Alvarez J, Hillaire-Buys D, Loubatières-Mariani M, Gomis R, Petit P. P2 receptor agonists stimulate insulin release from human pancreatic islets. Pancreas. 2001;22:69–71. doi: 10.1097/00006676-200101000-00012. [DOI] [PubMed] [Google Scholar]
- [58].Silva A, Rodrigues R, Tomé A, Cunha R, Misler S, Rosário L, et al. Electrophysiological and immunocytochemical evidence for P2X purinergic receptors in pancreatic beta cells. Pancreas. 2008;36:279–83. doi: 10.1097/MPA.0b013e31815a8473. [DOI] [PubMed] [Google Scholar]
- [59].Burnstock G. Purinergic regulation of vascular tone and remodelling. Auton Autacoid Pharmacol. 2009;29:63–72. doi: 10.1111/j.1474-8673.2009.00435.x. [DOI] [PubMed] [Google Scholar]
- [60].Gilon P, Henquin J. Mechanisms and physiological significance of the cholinergic control of pancreatic beta-cell function. Endocr Rev. 2001;22:565–604. doi: 10.1210/edrv.22.5.0440. [DOI] [PubMed] [Google Scholar]
- [61].Rodriguez-Diaz R, Dando R, Jacques-Silva MC, Fachado A, Molina J, Abdulreda MH, et al. Alpha cells secrete acetylcholine as a non-neuronal paracrine signal priming beta cell function in humans. Nat Med. 2011;17:888–92. doi: 10.1038/nm.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Zawalich W, Zawalich K, Rasmussen H. Cholinergic agonists prime the beta-cell to glucose stimulation. Endocrinology. 1989;125:2400–6. doi: 10.1210/endo-125-5-2400. [DOI] [PubMed] [Google Scholar]
- [63].Lang DA, Matthews DR, Peto J, Turner RC. Cyclic oscillations of basal plasma glucose and insulin concentrations in human beings. N Engl J Med. 1979;301:1023–7. doi: 10.1056/NEJM197911083011903. [DOI] [PubMed] [Google Scholar]
- [64].Gautam D, Gavrilova O, Jeon J, Pack S, Jou W, Cui Y, et al. Beneficial metabolic effects of M3 muscarinic acetylcholine receptor deficiency. Cell Metab. 2006;4:363–75. doi: 10.1016/j.cmet.2006.09.008. [DOI] [PubMed] [Google Scholar]
- [65].Rorsman P, Berggren PO, Bokvist K, Ericson H, Möhler H, Ostenson CG, et al. Glucose-inhibition of glucagon secretion involves activation of GABAA-receptor chloride channels. Nature. 1989;341:233–6. doi: 10.1038/341233a0. [DOI] [PubMed] [Google Scholar]
- [66].Soltani N, Qiu H, Aleksic M, Glinka Y, Zhao F, Liu R, et al. GABA exerts protective and regenerative effects on islet beta cells and reverses diabetes. Proc Natl Acad Sci U S A. 2011;108:11692–7. doi: 10.1073/pnas.1102715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Michalik M, Erecińska M. GABA in pancreatic islets: metabolism and function. Biochem Pharmacol. 1992;44:1–9. doi: 10.1016/0006-2952(92)90030-m. [DOI] [PubMed] [Google Scholar]
- [68].Kim J, Richter W, Aanstoot HJ, Shi Y, Fu Q, Rajotte R, et al. Differential expression of GAD65 and GAD67 in human, rat, and mouse pancreatic islets. Diabetes. 1993;42:1799–808. doi: 10.2337/diab.42.12.1799. [DOI] [PubMed] [Google Scholar]
- [69].Li L, Jiang J, Hagopian WA, Karlsen AE, Skelly M, Baskin DG, et al. Differential detection of rat islet and brain glutamic acid decarboxylase (GAD) isoforms with sequence-specific peptide antibodies. J Histochem Cytochem. 1995;43:53–9. doi: 10.1177/43.1.7822765. [DOI] [PubMed] [Google Scholar]
- [70].Petersen JS, Russel S, Marshall MO, Kofod H, Buschard K, Cambon N, et al. Differential expression of glutamic acid decarboxylase in rat and human islets. Diabetes. 1993;42:484–95. doi: 10.2337/diab.42.3.484. [DOI] [PubMed] [Google Scholar]
- [71].Chessler SD, Simonson WT, Sweet IR, Hammerle LP. Expression of the vesicular inhibitory amino acid transporter in pancreatic islet cells: distribution of the transporter within rat islets. Diabetes. 2002;51:1763–71. doi: 10.2337/diabetes.51.6.1763. [DOI] [PubMed] [Google Scholar]
- [72].Richerson GB, Wu Y. Dynamic equilibrium of neurotransmitter transporters: not just for reuptake anymore. J Neurophysiol. 2003;90:1363–74. doi: 10.1152/jn.00317.2003. [DOI] [PubMed] [Google Scholar]
- [73].Bonaventura MM, Catalano PN, Chamson-Reig A, Arany E, Hill D, Bettler B, et al. GABAB receptors and glucose homeostasis: evaluation in GABAB receptor knockout mice. Am J Physiol Endocrinol Metab. 2008;294:E157–67. doi: 10.1152/ajpendo.00615.2006. [DOI] [PubMed] [Google Scholar]
- [74].Wu Y, Wang W, Díez-Sampedro A, Richerson GB. Nonvesicular inhibitory neurotransmission via reversal of the GABA transporter GAT-1. Neuron. 2007;56:851–65. doi: 10.1016/j.neuron.2007.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Lernmark A, Bärmeier H, Dube S, Hagopian W, Karlsen A, Wassmuth R. Autoimmunity of diabetes. Endocrinol Metab Clin North Am. 1991;20:589–617. [PubMed] [Google Scholar]
- [76].Ohta Y, Kosaka Y, Kishimoto N, Wang J, Smith SB, Honig G, et al. Convergence of the insulin and serotonin programs in the pancreatic β-cell. Diabetes. 2011;60:3208–16. doi: 10.2337/db10-1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Rubí B, Ljubicic S, Pournourmohammadi S, Carobbio S, Armanet M, Bartley C, et al. Dopamine D2-like receptors are expressed in pancreatic beta cells and mediate inhibition of insulin secretion. J Biol Chem. 2005;280:36824–32. doi: 10.1074/jbc.M505560200. [DOI] [PubMed] [Google Scholar]
- [78].Arnerić SP, Chow SA, Long JP, Fischer LJ. Inhibition of insulin release from rat pancreatic islets by drugs that are analogues of dopamine. Diabetes. 1984;33:888–93. doi: 10.2337/diab.33.9.888. [DOI] [PubMed] [Google Scholar]
- [79].Rubí B, Maechler P. Minireview: new roles for peripheral dopamine on metabolic control and tumor growth: let’s seek the balance. Endocrinology. 2010;151:5570–81. doi: 10.1210/en.2010-0745. [DOI] [PubMed] [Google Scholar]
- [80].Bennet WM, Wang ZL, Jones PM, Wang RM, James RF, London NJ, et al. Presence of neuropeptide Y and its messenger ribonucleic acid in human islets: evidence for a possible paracrine role. J Clin Endocrinol Metab. 1996;81:2117–20. doi: 10.1210/jcem.81.6.8964837. [DOI] [PubMed] [Google Scholar]
- [81].Wang ZL, Bennet WM, Wang RM, Ghatei MA, Bloom SR. Evidence of a paracrine role of neuropeptide-Y in the regulation of insulin release from pancreatic islets of normal and dexamethasone-treated rats. Endocrinology. 1994;135:200–6. doi: 10.1210/endo.135.1.8013354. [DOI] [PubMed] [Google Scholar]
- [82].Whim MD. Pancreatic beta cells synthesize neuropeptide Y and can rapidly release peptide co-transmitters. PLoS One. 2011;6:e19478. doi: 10.1371/journal.pone.0019478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kim H, Toyofuku Y, Lynn FC, Chak E, Uchida T, Mizukami H, et al. Serotonin regulates pancreatic beta cell mass during pregnancy. Nat Med. 2010;16:804–8. doi: 10.1038/nm.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].MaassenVanDenBrink A, Centurión D, Villalón CM. Crosstalk of vascular 5-HT1 receptors with other receptors: clinical implications. Neuropharmacology. 2008;55:986–93. doi: 10.1016/j.neuropharm.2008.06.051. [DOI] [PubMed] [Google Scholar]
- [85].Speier S, Nyqvist D, Kohler M, Caicedo A, Leibiger I, Berggren P. Noninvasive high-resolution in vivo imaging of cell biology in the anterior chamber of the mouse eye. Nature Protocols. 2008;3:1278–86. doi: 10.1038/nprot.2008.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Speier S, Nyqvist D, Cabrera O, Yu J, Molano R, Pileggi A, et al. Noninvasive in vivo imaging of pancreatic islet cell biology. Nature Medicine. 2008;14:574–8. doi: 10.1038/nm1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Nguyen QT, Schroeder LF, Mank M, Muller A, Taylor P, Griesbeck O, et al. An in vivo biosensor for neurotransmitter release and in situ receptor activity. Nat Neurosci. 2010;13:127–32. doi: 10.1038/nn.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]