

Abstract
Dopamine cell loss and increased iron in the substantia nigra (SN) characterize Parkinson’s disease (PD), with cerebellar involvement increasingly recognized, particularly in motor compensation and levodopa-induced-dyskinesia (LID) development. Because the red nucleus (RN) mediates cerebellar circuitry, we hypothesized that RN iron changes may reflect cerebellum-related compensation, and/or the intrinsic capacity for LID development. We acquired high resolution MRI images from 23 Controls and 38 PD subjects [12 with (PD+DYS) and 26 without (PD−DYS) LID history]. Iron content was estimated from bilateral RN and SN transverse relaxation rates (R2*). PD subjects overall displayed higher R2* values in both the SN and RN. RN R2* values correlated with off-drug Unified Parkinson’s Disease Rating Scale-motor scores, but not disease duration or drug dosage. RN R2* values were significantly higher in PD+DYS subjects compared to Controls and PD−DYS; Controls and PD−DYS did not differ. The association of higher RN iron content with PD-related dyskinesia suggests increased iron content is involved in, or reflects, greater cerebellar compensatory capacity and thus increased likelihood of LID development.
Keywords: Parkinson’s disease (PD), red nucleus, transverse relaxation rate (R2*), magnetic resonance imaging (MRI)
1. INTRODUCTION
Parkinson’s disease (PD) is characterized by loss of dopaminergic neurons in the substantia nigra (SN) of the basal ganglia (BG). Levodopa corrects the primary motor dysfunction, but with continuing use and disease progression is associated with levodopa-induced dyskinesia (LID). The classic striato-thalamo-cortical model posits that dopamine deficiency leads to primary motor dysfunction via excessive inhibition of the thalamus resulting in reduced excitatory thalamo-cortical output. Functional somatotopy after pallidotomy, however, suggests PD primary motor symptoms have a different anatomic substrate from LID (Kishore et al., 2000) that cannot be explained purely by traditional striato-thalamo-cortical models.
In recent years, we (Lewis et al., 2011; Lewis et al., 2007; Sen et al., 2009) and others (Cerasa et al., 2006; Yu et al., 2007) have suggested cerebellar pathways may be important in the pathophysiology of PD, particularly in compensating for BG dysfunction with increased functional activity. Increased cerebellar function may contribute to the development of LID in PD (Brusa et al., 2011; Koch et al., 2009) as shown by increased cerebellar activity during PD progression (Sen et al., 2009) and by more LID in PD subjects with younger onset age (Jankovic, 2005), possibly due to more robust compensatory mechanisms (Fuente-Fernandez et al., 2011). In addition, di- and trisynaptic connections between the striatum and cerebellum have been demonstrated (Bostan et al., 2010; Hoshi et al., 2005), and in the context of our previously proposed model of motor control (Lewis et al., 2007), these data lead to a testable hypothetical model for motor control (Figure 1, Panel A, gray arrows) in which motor tasks are processed through combined activity of cortico-striato-cortical and cortico-cerebello-cortical circuits. The model also suggests that the subcortical striato-cerebellar and cerebello-striatal pathways that may modulate function in a parallel fashion [the implications of PD (Figure 1, panels B&C) are addressed in the Discussion].
Figure 1.

Model of motor function. Panel A (normal subjects): Motor tasks are accomplished through combined activity in striato- and cerebello-thalamo-cortical circuits (gray arrows) with modulation from striato-cerebellar and cerebello-striatal subcortical pathways (solid lines). Panels B & C (see Discussion): Model of motor dysfunction in PD with and without dyskinesia. In PD+DYS, decreased input from dysfunctional striatal circuits (dotted lines) leads to markedly increased activity in cerebellar circuits that set the stage for LID development (Panel B). In PD−DYS patients, the slightly increased recruitment of cerebellar circuits is not dysfunctional enough to result in predisposition for development of LID (Panel C).
The red nucleus (RN) receives significant somatotopically-organized input from ipsilateral motor cortex and contralateral cerebellum (Habasand Cabanis, 2007), thus providing a pivotal intersection between primary and cerebellar motor pathways (Birdand Shaw, 1978; Lapresleand Hamida, 1970). Animal studies suggest that RN output provides important compensation after corticospinal lesions (Belhaj-Saifand Cheney, 2000; Kanagaland Muir, 2009). In PD, the RN may increase its function to fulfill cerebellar compensation in the presence of BG dysfunction (see Figure 1, Panel B), although the exact mechanism is not known.
Iron is a cofactor for the synthesis and degradation of several neurotransmitters (Beard et al., 1993), and occurs in high concentration in many subcortical nuclei with high metabolic demands (e.g., SN, globus pallidus). In the face of oxidative stress challenges, there is both an induction of ferritin intracelluarly (possibly as a “sink” for toxic iron) and an up-regulation of heme oxygenase (increasing iron sequestration into glia). These changes, however, may promote iron “trapping” or “overload” (Zukor et al., 2009). We thus hypothesized that RN iron content may be increased in PD, particularly in patients with higher cerebellar compensation (i.e., higher metabolic demand) in the presence of oxidative stress; these subjects would be hypothesized to have increased likelihood of developing LID.
Iron is paramagnetic and causes a strong reduction in T2* relaxation time. Several MRI studies have demonstrated that the SN transverse relaxation rate (R2*=1/T2*) is correlated with iron concentration in vivo (Gelman et al., 1999), is increased in PD (Sofic et al., 1988), and discriminates PDs from Controls (Du et al., 2011). The RN lies near the SN, is similarly iron-rich (Drayer et al., 1986), and is recognized easily in midbrain MRI images (see Figure 2). This is the first MRI study focused on the RN to test the hypothesis that RN iron may mark the intrinsic capacity of PD subjects to develop LID.
Figure 2.
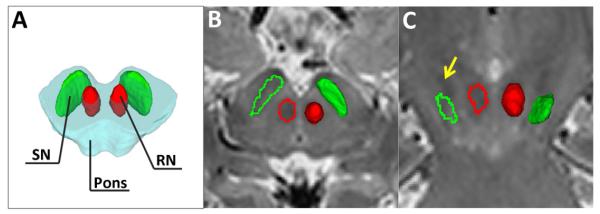
Relative location of the substantia nigra (SN) and red nucleus (RN) on a schematic of the midbrain (A). A typical MRI image illustrates the location of the SN (green) and RN (red) in T2-weighted images (B) and the co-registered regions on the R2* (C) map in an axial section.
2. METHODS
2.1. Subjects
Thirty-eight PD [12 with (PD+DYS) and 26 without (PD−DYS) history of dyskinesia] and 23 control subjects (Controls) were recruited from patients and their companions presenting to a tertiary movement disorders clinic (Table 1). PD diagnosis was confirmed by a movement disorder specialist (XH) according to published criteria (Calne et al., 1992), and PD medications optimized prior to enrollment in the study. Disease duration from time of diagnosis and history of dyskinesia were obtained from subject history. All 12 PD+DYS subjects were medically optimized such that dyskinesia was either minimal or totally absent at the time of study enrollment. Thus, the brain MRI results are unlikely to be influenced by abnormal movements per se.
Table 1.
Overall characteristics of study subjects
| Controls | Parkinson’s disease subjects | P values | |||||
|---|---|---|---|---|---|---|---|
| ALL | PD−DYS | PD+DYS |
Control vs.
PD |
PD−DYS
vs. PD+DYS |
Control,
PD−DYS, PD+DYS |
||
| No. (female, male) | 23 (17, 12) | 38 (17, 23) | 26 (12, 14) | 12 (5, 7) | 1.00a | 1.00a | 1.00b |
| Age, years (mean ± SD) | 59.9±7.0 | 60.6±8.0 | 59.7±8.9 | 62.6±5.4 | 0.483c | 1.00c | 1.00d |
| UPDRS-III motor score (mean ± SD) | NA | 23.8±15.4 | 21.2±15.1 | 29.5±15.3 | NA | 0.132c | NA |
| Disease duration, years (mean ± SD) | NA | 4.4±4.7 | 2.5±2.4 | 8.5±5.9 | NA | 0.005c | NA |
| Hohen & Yahr Stage (I/II/III) | NA | 13/21/4 | 11/12/3 | 2/9/1 | NA | 0.455c | NA |
| Levodopa-equivalent dosage (LEDD, mg) | NA | 535±400 | 429±310 | 767.5±484 | NA | 0.042c | NA |
| Total levodopa dose (tLD, mg) | NA | 385±367 | 288±313 | 596±401 | NA | 0.031c | NA |
p-value is calculated from Fisher’s exact test comparing Controls and all PD subjects or PD−DYS to PD+DYS.
p-value is calculated from Fisher’s exact test comparing all combinations of Controls, PD−DYS, and PD+DYS.
p-value is calculated from two-sample Student’s t-test with unequal variance comparing Controls and all PD subjects or PD−DYS to PD+DYS.
Analysis of variance comparing Controls, PD−DYS, and PD+DYS subjects.
NA = Not applicable
Unified Parkinson’s Disease Rating Scale part III-motor scores (UPDRS-III; Goetz et al., 2008) were obtained for each PD subject after withholding all PD medication overnight (~12 h). Levodopa-equivalent daily dose (LEDD) was estimated (Tomlinson et al., 2010), and total levodopa dose (tLD) calculated by summing the amount of levodopa taken per day. All subjects were free of major acute medical issues such as liver, kidney or thyroid abnormalities, or deficiencies of B12 or folate. A medical history on all subjects, noting any previous or current conditions, was obtained. Each brain MRI (vide infra) was inspected and reviewed by a board-certified neurologist, and deemed to be free of any cerebral white matter or ischemic changes. All subjects gave written informed consent, and the study was reviewed and approved by the Penn State Hershey Institutional Review Board.
2.2. MRI data acquisition
All subjects were scanned using a 3.0 Tesla MR Scanner (Trio, Siemens Magnetom, Erlangen, Germany) and high-resolution T2-weighted and multi-gradient-echo T2*-weighted images were collected. T2-weighted images were acquired using a fast-spin-echo sequence with TR/TE=2500/316, FoV=256 mm × 256 mm, matrix=256 × 256, slice thickness=1 mm (with no gap), and slice number=176. A multi-gradient-echo sequence was used to estimate the proton transverse relaxation rate, R2* (R2*=1/T2*). Six echoes with a TE ranging from 7 to 47 ms and an interval of 8 ms were acquired with TR=54 ms, flip angle=20°, FoV=256 mm × 256 mm, matrix=256 × 256, slice thickness=1 mm (with no gap), slice number=64. The middle slice of the T2* images was placed on the line between the anterior and posterior commissures.
2.3. Image processing and analysis
2.3.1. ROI definition
Bilateral SN and RN were delineated manually on multi-slice high resolution T2-weighted images using ITK-SNAP [www.itksnap.org (Yushkevich et al., 2006)]. A schematic of the location and shape of the SN and RN are depicted in Figure 2A. The SN was defined as a hypointensity band between the RN and cerebral peduncle in T2-weighted axial sections according to a previously published method (Du et al., 2011) as illustrated in Figure 2B (rostral segment) and 2C (caudal segment). To avoid including the subthalamic nucleus, the SN segmentation was started at the level of the RN showing the largest radius or one slice lower, depending on which slice the tail of subthalamic nucleus disappeared. A total of 6-8 slices (6-8 mm height) were used. The RN was defined as a hypointensity circle posterior-medial to the SN in axial sections (Figure 2B). A total of 4-6 slices (4-6 mm height) were used for the RN. The SN and RN regions were drawn independently by two raters (MK and NP) blinded to subject group designation and the inter-rater reliability was at an acceptable level, with the Pearson’s correlation coefficients of estimated R2* values being 0.960 for the RN and 0.703 for the SN.
2.3.2. R2* values
Multi-gradient-echo images were used to estimate the R2* maps by using a voxel-wise linear least-squares fit to a mono-exponential function with free baseline using an in-house MATLAB (The MathWorks, Inc., Natick, MA) tool. An affine registration process implemented in 3D Slicer (www.slicer.org) then was used to map the SN and RN regions to the R2* maps in order to reduce motions artifacts between T2-weighted and multi-gradient-echo sequences by co-registering T2-weighted images to an averaged T2*-weighted image of the six echoes. R2* values in the SN and RN of each subject were calculated using trimmed means (from the 5%-95% percentile) to reduce variability introduced by manual segmentation and imperfect registration processes. This process entailed plotting the histogram of R2* values for each subject and then ‘trimming’ the bottom and top of the distribution. The trimmed mean represents a more robust central estimator of the measurement, and is based on the assumption that the top and bottom values represent extreme measures (potential noise). For each subject, the trimmed mean R2* value from the left and right SN or RN then was combined and averaged, resulting in one R2* value for each structure.
2.4. Statistical Analysis
Demographic data were compared using two-sample t-tests and Fisher’s exact tests as appropriate. Regional R2* values were compared between PD and Controls or among Controls and PD subgroups by analyses of covariance with adjustments for age and gender. For some analyses, further adjustments for UPDRS-III scores, disease duration, LEDD, or tLD also were made. The relationship between UPDRS-III scores, disease duration, LEDD, tLD, and R2* values was explored by Pearson’s correlation analysis in the overall PD group and subgroups. Due to the exploratory nature of these correlational analyses, correction for multiple comparisons was not implemented. Logistic regression and receiver operating characteristic (ROC) curves compared the sensitivity and specificity separately for SN R2*values, RN R2* values, and disease duration and for their combinations. The c-statistic, representing the area under the ROC curve, was calculated as a measure of discrimination. All statistical analyses were performed using SAS 9.2 (SAS Institute Inc., Cary, NC, USA).
3. RESULTS
3.1. Subject Characteristics
There were no significant differences among PD, PD subgroups, and Controls in age or gender, although there were more males in the PD group and more females in the Controls, reflecting PD being more common in males and many Controls being spouses of the PD subjects. Clinically, most PD subjects were relatively early in disease course (Table 1). The PD+DYS and PD−DYS subgroups demonstrated no significant difference in age or gender. As expected, (Hauser et al., 2006; Jankovic, 2005), PD+DYS subjects had significantly higher tLD, LEDD, and longer disease duration compared to PD−DYS. The two PD groups were remarkably similar in UPDRS-III scores and Hoehn & Yahr staging.
3.2. Comparison of R2* Values in SN and RN between groups
R2* values in the SN and RN of PD subjects were significantly higher compared to Controls (Figure 3A). In the PD subgroups, PD−DYS subjects had significantly increased R2* in SN (p=0.036), but not RN (p=0.180), compared to Controls (Figure 3B). PD+DYS subjects, however, demonstrated significantly increased R2* values in both the SN (p=0.0001) and RN (p=0.002) compared to Controls. Moreover, both SN (p=0.023) and RN (p=0.011) R2* values in PD+DYS subjects were significantly higher compared to PD−DYS subjects (Figure 3B). Whereas the significant difference in RN (p=0.025) and SN (p=0.047) R2* values between the two PD subgroups was maintained following adjustment for UPDRS-III scores, only the RN R2* values remained significant after adjusting further for disease duration (SN p=0.106, RN p=0.003), LEDD (SN p=0.117, RN p=0.014) or tLD (SN p=0.084, RN p=0.009).
Figure 3.
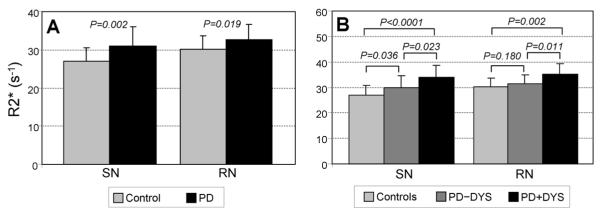
Mean SN and RN R2* values (± SD) for Control and PD subjects. (3A) In both the SN and RN, PD subjects had higher R2* values compared to control subjects. (3B) In both the SN and RN, PD subjects with a history of dyskinesia (PD+DYS) had higher R2* values compared to both control subjects and PD subjects without a history of dyskinesia (PD−DYS). PD−DYS subjects showed significantly higher R2* values in the SN compared to Controls, but this comparison did not reach significance in the RN.
3.3. Clinical correlations of R2* Values in SN and RN
In the overall PD group, SN R2* values were correlated with UPDRS-III scores, disease duration, tLD, and LEDD values, whereas RN R2* values were associated significantly only with UPDRS-III scores (Table 2). None of the SN R2* values were associated with clinical measures in either PD subgroup. The association between UPDRS-III scores and R2* values in the RN, however, was found in the PD−DYS group (r=0.422, p=0.033), but absent in the PD+DYS group (r=0.04, p=0.907; Table 2).
Table 2.
Pearson’s correlation coefficients and p-values (in parentheses) between clinical scores and SN and RN R2* values in the overall PD group and PD subjects with (PD+DYS) and without (PD−DYS) history of dyskinesia.
| Clinical measurement | Overall PD group | PD−DYS | PD+DYS |
|---|---|---|---|
| SN | |||
| Total UPDRS-III |
0.364
(0.025) |
0.336 (0.093) |
0.217 (0.498) |
| tLD |
0.346
(0.035) |
0.228 (0.262) |
0.244 (0.445) |
| LEDD |
0.448
(0.005) |
0.202 (0.322) |
0.190 (0.554) |
| Disease duration |
0.357
(0.028) |
0.275 (0.174) |
0.484 (0.111) |
| RN | |||
| Total UPDRS-III |
0.356
(0.028) |
0.422
(0.032) |
0.038 (0.907) |
| tLD | 0.068 (0.684) |
0.122 (0.554) |
−0.458 (0.134) |
| LEDD | 0.159 (0.341) |
0.195 (0.339) |
−0.498 (0.099) |
| Disease duration | 0.114 (0.495) |
.240 (0.237) |
−0.304 (0.337) |
Bold and italic highlight statistically significant results with p<0.05.
tLD = levodopa dosage; LEDD = levodopa equivalent daily dosage
3.4. Logistic regression analysis to discriminate PD+DYS and PD−DYS subjects
Since UPDRS-III scores were not significantly different between the two PD subgroups, they were not included in the regression analyses. Although LEDD and tLD were different between the subgroups, regression analyses indicated these factors either alone or in combination with disease duration and/or the imaging measures did not contribute to the discrimination of the two subgroups (data not shown). As a result, the remaining regression analyses included disease duration, SN R2* values, and RN R2* values alone or in combination. There was significant discrimination between PD+DYS and PD−DYS subjects using the individual factors of disease duration, SN R2* values, or RN R2* values alone (Table 3). The combination of SN and RN R2* values improved the discrimination between the PD subgroups compared to either factor alone, although it was not better than disease duration alone. Combining SN R2* values and disease duration slightly increased the discrimination between the PD subgroups (Table 3, Figure 4A). The combination of RN R2* values and disease duration, however, yielded the greatest discrimination (Table 3, Figure 4B).
Table 3.
Logistic regression results for predicting PD dyskinesia status from SN R2* values, RN R2* values, or disease duration alone or in combination.
| Parameter (s) used in the Model |
Odds ratio (95% confidence interval) |
Model p-value |
C-statistic | |
|---|---|---|---|---|
| Disease duration | 0.686 (0.525, 0.897) | 0.006 | 0.825 | |
| SN R2* | 0.844 (0.725, 0.983) | 0.029 | 0.731 | |
| RN R2* | 0.774 (0.619, 0.970) | 0.026 | 0.766 | |
| SN R2* + RN R2* |
SN R2* | 0.893 (0.754, 1.058) | 0.014 | 0.811 |
| RN R2* | 0.819 (0.642, 1.046) | |||
| Disease duration + SN R2* |
Disease duration | 0.706 (0.539, 0.924) | <0.001 | 0.846 |
| SN R2* | 0.888 (0.739, 1.067) | |||
| Disease duration + RN R2* |
Disease duration | 0.624 (0.438, 0.888) | <0.001 | 0.917 |
| RN R2* | 0.713 (0.541, 0.940) |
Figure 4.

ROC curves for discriminating between PD subjects with and without history of dyskinesia. Panel A represents the model that tests disease duration and SN R2* values alone or in combination. Panel B depicts the model that tests disease duration and RN R2* values alone or in combination.
4. DISCUSSION
It is well-established that the SN (Jellinger, 1991) has increased iron and R2* values in PD (Du et al., 2011; Gorell et al., 1995; Martin et al., 2008; Sofic et al., 1988), suggesting that iron accumulation plays an etiological (Lavaute et al., 2001) and/or compensatory (Rivera-Mancia et al., 2010) role. Prior studies reported increased SN iron in PD was correlated with motor scores (Martin et al., 2008), and we found that increased SN iron in PD (Figure 3A) was associated with clinical measures (Table 2). These data support the notion that SN iron may be a good marker for PD progression since it is correlated with all clinical measurements, including disease duration, UPDRS-III scores, tLD, and LEDD. The key finding of this study is, however, the R2* changes in the RN.
4.1. The red nucleus and PD
Despite lying adjacent to the SN in the midbrain and being similarly rich in iron (Drayer et al., 1986), few studies have interrogated iron RN levels in PD subjects; the available data are equivocal or non-significant (Table 4). The current study is the first to demonstrate significantly increased RN iron in PD that appears to be due to increased iron content in PD+DYS subjects (Figure 3). The inclusion of PD+DYS patients in imaging studies is challenging because motion can affect the quality of MR images. In the current study, all PD+DYS subjects were medically optimized and had minimal movement making MRI scans better tolerated, and enabling us to delineate midbrain structures with confidence. It is interesting to note that if only PD−DYS subjects had been studied, RN R2* differences between PD and controls would not have been statistically different (Figure 3B), similar to previous reports (Table 4). RN iron was associated significantly with UPDRS-III scores, but not with disease duration, tLD, or LEDD in PD subjects as a whole. The association remained only in the PD−DYS group, suggesting these patients may be in a different (pre-clinical) state as it relates to developing LID.
Table 4.
Prior studies reporting RN iron values.
| Study | Subjects | R2* values (p value) |
|---|---|---|
| (Vymazal et al., 1999) | 23 PD, 18 Controls | ↑ (>0.05) |
| (Martin et al., 2008) | 26 PD, 13 Controls | ↑ (>0.05) |
| (Peran et al., 2010) | 30 PD, 22 Controls | ↑ (>0.05) |
| (Zhang et al., 2010) | 40 PD, 26 Controls | ↓ (0.272) |
| (Jin et al., 2011) | 45 PD, 45 Controls | ↑ (0.247) |
↑: increased; ↓: decreased; PD: Parkinson’s disease
4.2. The role of the cerebellum in dyskinesia
Iron in the RN differed between the PD subgroups, and provides the best predictor for occurrence of dyskinesia when combined with disease duration. This supports the idea that RN R2* may be a marker for the propensity to develop dyskinesia, whether in an etiological or compensatory sense. It further supports the notion that structures outside traditional striato-thalamo-cortical pathways, specifically cerebello-thalamo-cortical structures, may play a role in LID development. Consistent with this hypothesis, Sen et al. (2009) reported increased cerebellar activity with PD progression. Recent studies have demonstrated cerebellar involvement in somatosensory integration (Bastian, 2006; Wolpert et al., 1998), and Liu et al. (2001) reported decreased drug-induced dyskinesia after turning off visual guidance. These results suggest that decreased somatosensory integration may diminish cerebellar pathway compensation resulting in less dyskinesia. Additionally, decreased glucose utilization in the cerebellum following continuous theta burst stimulation mirrors decreased dyskinesia scores in PD (Brusa et al., 2011).
Strick and colleagues (Bostan et al., 2010; Hoshi et al., 2005) demonstrated that the striatum and cerebellum may communicate and/or influence motor circuitry function at the subcortical level through di- and trisynaptic connections. Although previous studies indicate that several brain structures like the striatum and subthalamic nucleus play a role in LID (Fabbrini et al., 2007), we (Lewis et al., 2011; Lewis et al., 2007; Sen et al., 2009) and others (Cerasa et al., 2006; Yu et al., 2007) have suggested cerebellar pathways also may be important in PD pathophysiology, particularly in compensating for BG dysfunction. The RN may provide another point of functional intersection between striato- and cerebello-thalamo-cortical motor pathways. Increased RN iron may reflect structural changes associated with cerebello-thalamo-cortical compensation in PD. Additional research is needed to determine the interplay of the two motor circuits and their role in dyskinesia, but these data may be placed in the context of the proposed model of striato- and cerebello-thalamo-cortical function in either normal (Figure 1, Panel A) or dysfunctional motor control (Figure 1, Panels B and C) that extends the ideas proposed by Strick and coworkers (Bostan et al., 2010; Hoshi et al., 2005).
As stated, motor tasks normally are processed through combined activity of cortico-striato-cortical and cortico-cerebello-cortical circuits (Figure 1, Panel A, gray arrows), modulated by subcortical striato-cerebellar and cerebello-striatal pathways in a parallel fashion. When striatal dysfunction leads to diminished input to thalamo-cortical and striato-cerebellar structures in PD+DYS, markedly increased activity in cerebellar pathways (through the RN and thalamus) sets the stage for LID development (Figure 1, Panel B). In PD−DYS, there may be slightly increased activity in cerebellar circuits, but insufficiently dysfunctional to predispose patients to LID (Figure 1, Panel C).
4.3. The meaning of the RN R2* signal
The exact biochemical substrate for the observed R2* increase is unknown. Although R2* most likely reflects iron content, the current study could not assess the state of the iron. Nevertheless, there are a number of possibilities for the observed increased RN R2*. For example, the RN contains both transferrin and ferritin, whose iron binding capacities may be increased with higher metabolism (Beard, 2003). It also is likely that there may be increased iron sequestration in RN glia from up-regulation of heme oxygenase due to increased metabolic demand in conjunction with oxidative stress (Zukor et al., 2009). In addition, the RN contains neuromelanin (Nietoand Nieto, 1986) and the increased iron may reflect neuromelanin over-saturation. Additional pathological work focused on PD subjects demonstrating a history of dyskinesia can help determine the form of iron and the mechanism behind increased R2*.
Iron has been linked etiologically to cell loss in several neurodegenerative disorders, including dopamine cell loss in the SN in PD (Jellinger, 1991; Sofic et al., 1988). This link is strengthened by recent work demonstrating markedly increased SN iron in tau-knockout mice having significant loss of dopaminergic neurons (Lei et al., 2012). Iron in the remaining SN cells also may be compensatory to the primary cell damage (Rivera-Mancia et al., 2010). The RN, however, is not a primary site of PD pathology, and previous studies (Braak et al., 2003; Jellinger, 1991) have not reported RN cell loss possibly because PD−DYS and PD+DYS patients were not distinguished. Pathological studies on patients with LID history may help resolve the role of iron; a lack of RN cell loss would argue against increased RN iron being linked etiologically to PD-related cell death. The fact that the observed iron changes in the RN were seen only in PD+DYS patients is consistent with this notion. It is possible, however, that longitudinal examination of non-dyskinetic subjects might show a delayed, but equivalent, increase in RN iron that does not correlate with LID induction. Such a result would argue against the hypothesis we favor (vide infra). The current study has other limitations. Although our sample size of 38 patients and 23 Controls is substantial compared to other neuroimaging-based PD studies, it is relatively small for a cross-sectional study. Moreover, there were more males in the PD group and more females in the Controls. This reflects that PD is more common in males (Baldereschi et al., 2000; Mayeux et al., 1992), and also that many Controls were spouses of the PD subjects. The different gender proportions were not significantly different, but although we corrected for gender differences in our statistical analyses, we cannot rule out a possible gender effect in our results. In addition, tLD or LEDD after medical optimization may not reflect lifelong PD drug exposure. Thus, we could not test adequately the hypothesis that RN R2* changes result from prior medication usage. It will be critical to test this vigorously in drug naïve, newly diagnosed PD patients in a prospective, longitudinal study.
4.4. Conclusions
The current data are consistent with the hypothesis that, as an integral part of cerebellar circuitry, the RN may mediate PD-related compensatory changes and the occurrence of dyskinesia. This may be reflected by higher RN iron content, as increased RN R2* values were most prominent in PD+DYS subjects. The increased iron content was not influenced by the use of levodopa or other antiparkinsonian drugs, and was detectable even though dyskinesia in PD+DYS subjects had been markedly attenuated by medication optimization. Although it is generally accepted that pulsatility of dopamine receptor occupation is a factor that initiates LID (Karlsborg et al., 2010; Nyholm, 2007; Olanow et al., 2006a; Olanow et al., 2006b), this is the first indication that intrinsic cerebellar circuitry may play a role in the causation of this debilitating aspect of PD treatment. These data require independent confirmation from imaging and pathological studies, but may lead to a new understanding of LID and potential interventions.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported in part by the National Institutes of Health (NS060722, NS082151 and ES019672 to XH), the Penn State Clinical & Translational Science Institute, Pennsylvania State University CTSA (UL-1RR033184), and the Pennsylvania Department of Health Tobacco Settlement Funds (C06 RR016499). All analyses, interpretations, and conclusions are those of the authors and not the research sponsors.
We would like to express our gratitude to the subjects who graciously volunteered to participate in this study. We acknowledge the MRI technical support of Mr. Jeffery Vesek and the assistance of study coordinator Ms. Eleanore Hernandez.
ABBREVIATIONS
- BG
basal ganglia
- LEDD
levodopa-equivalent daily dosage
- LID
levodopa-induced dyskinesia
- PD
Parkinson’s disease
- PD+DYS
subjects with Parkinson’s disease and history of LID
- PD−DYS−
subjects with Parkinson’s disease but no history of LID
- RN
red nucleus
- SN
substantia nigra
- tLD
total levodopa daily dosage
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURE STATEMENT
The authors report no conflicts of interest related to this study.
REFERENCES
- Baldereschi M, Di Carlo A, Rocca WA, Vanni P, Maggi S, Perissinotto E, Grigoletto F, Amaducci L, Inzitari D, ILSA Working Group. Italian Longitudinal Study on Aging Parkinson’s disease and parkinsonism in a longitudinal study: two-fold higher incidence in men. Neurology. 2000;55:1358–1363. doi: 10.1212/wnl.55.9.1358. [DOI] [PubMed] [Google Scholar]
- Bastian AJ. Learning to predict the future: the cerebellum adapts feedforward movement control. Curr.Opin.Neurobiol. 2006;16:645–649. doi: 10.1016/j.conb.2006.08.016. [DOI] [PubMed] [Google Scholar]
- Beard J. Iron deficiency alters brain development and functioning. J Nutr. 2003;133:1468S–1472S. doi: 10.1093/jn/133.5.1468S. [DOI] [PubMed] [Google Scholar]
- Beard JL, Connor JR, Jones BC. Iron in the brain. Nutr.Rev. 1993;51:157–170. doi: 10.1111/j.1753-4887.1993.tb03096.x. [DOI] [PubMed] [Google Scholar]
- Belhaj-Saif A, Cheney PD. Plasticity in the distribution of the red nucleus output to forearm muscles after unilateral lesions of the pyramidal tract. J Neurophysiol. 2000;83:3147–3153. doi: 10.1152/jn.2000.83.5.3147. [DOI] [PubMed] [Google Scholar]
- Bird TD, Shaw CM. Progressive myoclonus and epilepsy with dentatorubral degeneration: a clinicopathological study of the Ramsay Hunt syndrome. J Neurol.Neurosurg.Psychiatry. 1978;41:140–149. doi: 10.1136/jnnp.41.2.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostan AC, Dum RP, Strick PL. The basal ganglia communicate with the cerebellum. Proc.Natl.Acad.Sci.U.S.A. 2010;107:8452–8456. doi: 10.1073/pnas.1000496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol.Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Brusa L, Ceravolo R, Kiferle L, Monteleone F, Iani C, Schillaci O, Stanzione P, Koch G. Metabolic changes induced by theta burst stimulation of the cerebellum in dyskinetic Parkinson’s disease patients. Parkinsonism.Relat Disord. 2011 doi: 10.1016/j.parkreldis.2011.08.019. [DOI] [PubMed] [Google Scholar]
- Calne DB, Snow BJ, Lee C. Criteria for diagnosing Parkinson’s disease. Ann.Neurol. 1992;32(Suppl):S125–S127. doi: 10.1002/ana.410320721. [DOI] [PubMed] [Google Scholar]
- Cerasa A, Hagberg GE, Peppe A, Bianciardi M, Gioia MC, Costa A, Castriota-Scanderbeg A, Caltagirone C, Sabatini U. Functional changes in the activity of cerebellum and frontostriatal regions during externally and internally timed movement in Parkinson’s disease. Brain Res.Bull. 2006;71:259–269. doi: 10.1016/j.brainresbull.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Drayer B, Burger P, Darwin R, Riederer S, Herfkens R, Johnson GA. MRI of brain iron. AJR Am.J Roentgenol. 1986;147:103–110. doi: 10.2214/ajr.147.1.103. [DOI] [PubMed] [Google Scholar]
- Du G, Lewis MM, Styner M, Shaffer ML, Sen S, Yang QX, Huang X. Combined R2* and Diffusion Tensor Imaging Changes in the Substantia Nigra in Parkinson’s Disease. Mov Disord. 2011 doi: 10.1002/mds.23643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbrini G, Brotchie JM, Grandas F, Nomoto M, Goetz CG. Levodopa-induced dyskinesias. Mov Disord. 2007;22:1379–1389. doi: 10.1002/mds.21475. [DOI] [PubMed] [Google Scholar]
- Fuente-Fernandez R, Schulzer M, Kuramoto L, Cragg J, Ramachandiran N, Au WL, Mak E, McKenzie J, McCormick S, Sossi V, Ruth TJ, Lee CS, Calne DB, Stoessl AJ. Age-specific progression of nigrostriatal dysfunction in Parkinson’s disease. Ann.Neurol. 2011;69:803–810. doi: 10.1002/ana.22284. [DOI] [PubMed] [Google Scholar]
- Gelman N, Gorell JM, Barker PB, Savage RM, Spickler EM, Windham JP, Knight RA. MR imaging of human brain at 3.0 T: preliminary report on transverse relaxation rates and relation to estimated iron content. Radiology. 1999;210:759–767. doi: 10.1148/radiology.210.3.r99fe41759. [DOI] [PubMed] [Google Scholar]
- Goetz CG, Tilley BC, Shaftman SR, Stebbins GT, Fahn S, Martinez-Martin P, Poewe W, Sampaio C, Stern MB, Dodel R, Dubois B, Holloway R, Jankovic J, Kulisevsky J, Lang AE, Lees A, Leurgans S, LeWitt PA, Nyenhuis D, Olanow CW, Rascol O, Schrag A, Teresi JA, van Hilten JJ, LaPelle N. Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov Disord. 2008;23:2129–2170. doi: 10.1002/mds.22340. [DOI] [PubMed] [Google Scholar]
- Gorell JM, Ordidge RJ, Brown GG, Deniau JC, Buderer NM, Helpern JA. Increased iron-related MRI contrast in the substantia nigra in Parkinson’s disease. Neurology. 1995;45:1138–1143. doi: 10.1212/wnl.45.6.1138. [DOI] [PubMed] [Google Scholar]
- Habas C, Cabanis EA. Cortical projection to the human red nucleus: complementary results with probabilistic tractography at 3 T. Neuroradiology. 2007;49:777–784. doi: 10.1007/s00234-007-0260-y. [DOI] [PubMed] [Google Scholar]
- Hauser RA, McDermott MP, Messing S. Factors associated with the development of motor fluctuations and dyskinesias in Parkinson disease. Arch.Neurol. 2006;63:1756–1760. doi: 10.1001/archneur.63.12.1756. [DOI] [PubMed] [Google Scholar]
- Hoshi E, Tremblay L, Feger J, Carras PL, Strick PL. The cerebellum communicates with the basal ganglia. Nat.Neurosci. 2005;8:1491–1493. doi: 10.1038/nn1544. [DOI] [PubMed] [Google Scholar]
- Jankovic J. Motor fluctuations and dyskinesias in Parkinson’s disease: clinical manifestations. Mov Disord. 2005;20(Suppl 11):S11–S16. doi: 10.1002/mds.20458. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Pathology of Parkinson’s disease. Changes other than the nigrostriatal pathway. Mol.Chem.Neuropathol. 1991;14:153–197. doi: 10.1007/BF03159935. [DOI] [PubMed] [Google Scholar]
- Kanagal SG, Muir GD. Task-dependent compensation after pyramidal tract and dorsolateral spinal lesions in rats. Exp.Neurol. 2009;216:193–206. doi: 10.1016/j.expneurol.2008.11.028. [DOI] [PubMed] [Google Scholar]
- Karlsborg M, Korbo L, Regeur L, Glad A. Duodopa pump treatment in patients with advanced Parkinson’s disease. Dan.Med.Bull. 2010;57:A4155. [PubMed] [Google Scholar]
- Kishore A, Panikar D, Balakrishnan S, Joseph S, Sarma S. Evidence of functional somatotopy in GPi from results of pallidotomy. Brain. 2000;123(Pt 12):2491–2500. doi: 10.1093/brain/123.12.2491. [DOI] [PubMed] [Google Scholar]
- Koch G, Brusa L, Carrillo F, Lo GE, Torriero S, Oliveri M, Mir P, Caltagirone C, Stanzione P. Cerebellar magnetic stimulation decreases levodopa-induced dyskinesias in Parkinson disease. Neurology. 2009;73:113–119. doi: 10.1212/WNL.0b013e3181ad5387. [DOI] [PubMed] [Google Scholar]
- Lapresle J, Hamida MB. The dentato-olivary pathway. Somatotopic relationship between the dentate nucleus and the contralateral inferior olive. Arch.Neurol. 1970;22:135–143. doi: 10.1001/archneur.1970.00480200041004. [DOI] [PubMed] [Google Scholar]
- Lavaute T, Smith S, Cooperman S, Iwai K, Land W, Meyron-Holtz E, Drake SK, Miller G, Abu-Asab M, Tsokos M, Switzer R, III, Grinberg A, Love P, Tresser N, Rouault TA. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat.Genet. 2001;27:209–214. doi: 10.1038/84859. [DOI] [PubMed] [Google Scholar]
- Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK, Wong BX, Adlard PA, Cherny RA, Lam LQ, Roberts BR, Volitakis I, Egan GF, McLean CA, Cappai R, Duce JA, Bush AI. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat.Med. 2012 doi: 10.1038/nm.2613. [DOI] [PubMed] [Google Scholar]
- Lewis MM, Du G, Sen S, Kawaguchi A, Truong Y, Lee S, Mailman RB, Huang X. Differential involvement of striato- and cerebello-thalamo-cortical pathways in tremor- and akinetic/rigid-predominant Parkinson’s disease. Neuroscience. 2011;177:230–239. doi: 10.1016/j.neuroscience.2010.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis MM, Slagle CG, Smith AB, Truong Y, Bai P, McKeown MJ, Mailman RB, Belger A, Huang X. Task specific influences of Parkinson’s disease on the striato-thalamo-cortical and cerebello-thalamo-cortical motor circuitries. Neuroscience. 2007;147:224–235. doi: 10.1016/j.neuroscience.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Osterbauer R, Aziz TZ, Miall RC, Stein JF. Increased response to visual feedback of drug-induced dyskinetic movements in advanced Parkinson’s disease. Neurosci.Lett. 2001;304:25–28. doi: 10.1016/s0304-3940(01)01740-2. [DOI] [PubMed] [Google Scholar]
- Martin WR, Wieler M, Gee M. Midbrain iron content in early Parkinson disease: a potential biomarker of disease status. Neurology. 2008;70:1411–1417. doi: 10.1212/01.wnl.0000286384.31050.b5. [DOI] [PubMed] [Google Scholar]
- Mayeux R, Denaro J, Hemenegildo N, Marder K, Tang MX, Cote LJ, Stern Y. A population-based investigation of Parkinson’s disease with and without dementia. Relationship to age and gender. Arch.Neurol. 1992;49:492–497. doi: 10.1001/archneur.1992.00530290076015. [DOI] [PubMed] [Google Scholar]
- Nieto D, Nieto A. Neurochemical significance of the red nucleus. Bull.Clin Neurosci. 1986;51:89–93. [PubMed] [Google Scholar]
- Nyholm D. The rationale for continuous dopaminergic stimulation in advanced Parkinson’s disease. Parkinsonism.Relat Disord. 2007;13(Suppl):S13–S17. doi: 10.1016/j.parkreldis.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Obeso JA, Stocchi F. Continuous dopamine-receptor treatment of Parkinson’s disease: scientific rationale and clinical implications. Lancet Neurol. 2006a;5:677–687. doi: 10.1016/S1474-4422(06)70521-X. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Obeso JA, Stocchi F. Drug insight: Continuous dopaminergic stimulation in the treatment of Parkinson’s disease. Nat.Clin.Pract.Neurol. 2006b;2:382–392. doi: 10.1038/ncpneuro0222. [DOI] [PubMed] [Google Scholar]
- Rivera-Mancia S, Perez-Neri I, Rios C, Tristan-Lopez L, Rivera-Espinosa L, Montes S. The transition metals copper and iron in neurodegenerative diseases. Chem.Biol.Interact. 2010;186:184–199. doi: 10.1016/j.cbi.2010.04.010. [DOI] [PubMed] [Google Scholar]
- Sen S, Kawaguchi A, Truong Y, Lewis MM, Huang X. Dynamic changes in cerebello-thalamo-cortical motor circuitry during progression of Parkinson’s disease. Neuroscience. 2009 doi: 10.1016/j.neuroscience.2009.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofic E, Riederer P, Heinsen H, Beckmann H, Reynolds GP, Hebenstreit G, Youdim MB. Increased iron (III) and total iron content in post mortem substantia nigra of parkinsonian brain. J.Neural Transm. 1988;74:199–205. doi: 10.1007/BF01244786. [DOI] [PubMed] [Google Scholar]
- Tomlinson CL, Stowe R, Patel S, Rick C, Gray R, Clarke CE. Systematic review of levodopa dose equivalency reporting in Parkinson’s disease. Mov Disord. 2010;25:2649–2653. doi: 10.1002/mds.23429. [DOI] [PubMed] [Google Scholar]
- Wolpert DM, Miall RC, Kawato M. Internal models in the cerebellum. Trends Cogn Sci. 1998;2:338–347. doi: 10.1016/s1364-6613(98)01221-2. [DOI] [PubMed] [Google Scholar]
- Yu H, Sternad D, Corcos DM, Vaillancourt DE. Role of hyperactive cerebellum and motor cortex in Parkinson’s disease. Neuroimage. 2007;35:222–233. doi: 10.1016/j.neuroimage.2006.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yushkevich PA, Piven J, Hazlett HC, Smith RG, Ho S, Gee JC, Gerig G. User-guided 3D active contour segmentation of anatomical structures: significantly improved efficiency and reliability. Neuroimage. 2006;31:1116–1128. doi: 10.1016/j.neuroimage.2006.01.015. [DOI] [PubMed] [Google Scholar]
- Zukor H, Song W, Liberman A, Mui J, Vali H, Fillebeen C, Pantopoulos K, Wu TD, Guerquin-Kern JL, Schipper HM. HO-1-mediated macroautophagy: a mechanism for unregulated iron deposition in aging and degenerating neural tissues. J Neurochem. 2009;109:776–791. doi: 10.1111/j.1471-4159.2009.06007.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.