

Abstract
Preterm birth in the United States is now 12%. Multiple genes, gene networks, variants have been associated with this disease. Using a custom database for preterm birth (dbPTB) with a refined set of genes extensively curated from literature and biological databases, we analyzed a GWAS of preterm birth for complete genotype data on nearly 2000 preterm and term mothers. We used both the curated genes and a genome-wide approach to carry out a pathway-based analysis. There were 19 significant pathways, which withstood FDR correction for multiple testing that were identified using both the curated genes and the genome-wide approach. The analysis based on the curated genes was more significant than genome-wide in 15 out of 19 pathways. This approach demonstrates the use of a validated set of genes, in the analysis of otherwise unsuccessful GWAS data, to identify gene-gene interactions in a way that enhances statistical power and discovery.
Keywords: Preterm birth, Pathway analysis, GWAS
1. Introduction
Genome-wide association studies (GWAS) enable investigation of the genetic associations underlying complex diseases without a priori hypotheses [1, 2]. Advances in high-throughput genotyping, sequencing technology and developments in computational power have enhanced the feasibility of large case-controlled studies and reduced costs [3]. Since they have the potential of identifying novel genetic variants, GWAS have become a popular approach to the investigation of complex diseases. By the second quarter of 2011, there were 1449 reports in the Catalog of Published Genome-Wide Association Studies (http://www.genome.gov/gwastudies/) covering hundreds of associations of common genetic variants with complex traits [4]. These reports have provided valuable insights into the genetic architecture of disease, including inflammatory bowel disease, macular degeneration, and obesity [5–7]. Nonetheless, GWAS for complex diseases have had only a measured success. While, there have been many loci identified and replicated in GWAS, many studies have failed to identify significant associations. Likewise, the genetic markers that have been identified through the GWAS approach are rarely functional variants in the diseases with which they are associated. In addition, most common variants that are identified by GWAS are responsible for only a small portion of the genetic variation and thus there remains a large amount of “missing heritability” [8, 9]. If the “common disease common variant hypothesis” underlying the GWAS approach does not explain the genetic contributions to complex diseases than what does [8, 10]? It is likely that rare variants and/or genetic interactions, epistasis, underlie a significant portion of the ‘missing heritability’ not revealed by conventional GWAS analyses [11–13]. It is also likely that complex mechanisms and higher orders of gene-gene interactions underlie the pathogenesis of many (most) complex diseases and lead to variations/alterations of the phenotype [14–17]. Identification of the multiple genes contributing to disease pathogenesis may help in understanding the effects on phenotype and in the search for missing heritability [18]. Nonetheless, the GWAS-based interrogation of large numbers of anonymous single nucleotide polymorphisms (SNP) severely limits power, thus weakening our computational ability to examine combinatorial gene-gene interactions [19–21].
We are interested in the genetic contribution(s) to preterm birth. Preterm birth is an important, poorly understood clinical problem [22–25]. The incidence of preterm birth (PTB) in the United States is now 12%, or 1 in 8 women [26]. It creates enormous clinical, economic and psychological burdens. The pathogenesis has remained elusive. Clinical tests and interventions to identify the patients at risks for preterm birth have relied heavily on assessment of common pathways associated with labor, such as myometrial contractility, cervical ripening, and decidual/membrane activation [27]. Interventions to prevent preterm birth are aimed at these common pathways. However, most of these interventions have proven ineffective. Multiple genes, gene networks, variants have been associated with preterm birth, however, single genes and pathways and simple patterns of inheritance are inadequate to explain the pathogenesis of the majority of preterm births [24]. The pathogenesis of PTB may be better understood if the analysis incorporated a more complex model that entails a host of genes [25] and with environmental triggers overlaying the genetics as well.
We have developed an approach for identifying a parsimonious set of genes for the study of preterm birth validated by a priori biological information. We used a semantic data mining and natural language processing approach to extract all published articles related to preterm birth [28]. Then, the genes identified from public databases and archives of expression arrays were aggregated with the gene set curated from the literature. Lastly, pathway analysis was used to impute genes from pathways identified during curation. The curated articles and collected genetic information form a unique resource for investigators interested in preterm birth, the Database for Preterm Birth (dbPTB), publicly-accessible at http://ptbdb.cs.brown.edu/dbPTBv1.php. Recently, results from a genome-wide study of preterm birth, “GENEVA,” became available in dbGAP [29]. The dataset includes phenotypic information and complete genotype data on nearly 2000 mothers, ranging from 20 to 42 weeks gestation. Since It has been demonstrated that that the genetic risk of preterm birth segregates heavily to the maternal genome, we have concentrated our analysis only on maternal genotype information [30]. Using the curated genes from dbPTB, we have analyzed the GENEVA data set from mothers only. The results of the refined curated genes were further analyzed by gene set enrichment analysis.
2. Results
2.1. SNP basic association analysis
We applied standard case/control allelic testing in Plink v1.07 to analyze the association of individual SNPs with preterm birth. In the first analysis, we only used SNPs that belonged to the curated dbPTB genes [28]. We included SNPs within the genomic region encompassing each gene as well as SNPs within 5kb upstream or downstream. Of the 617 genes identified in dbPTB, 551 were mapped onto the Illumina 660 quad platform encompassing 9077 tag SNPs. In the second analysis, we ran a genome-wide comparison using all of the SNPs on the Illumina platform (n= 560,768 SNPs). Preterm women were divided into three gestational age categories: less than 30 weeks gestation (n= 92), less than 34 weeks (n= 446), and less than 37 weeks gestation (n= 884) and compared to women who delivered greater than or equal to 38 weeks gestation (n=960). A Manhattan plot showing the results for all three preterm gestational age groups across the dbPTB SNPs is shown in Figure 1. As can be seen from this representative plot of chromosome 1, there were multiple regions where associations were seen for all three patient groups. Similar results were seen for the other chromosomes. While several regions demonstrated −log P values greater than 2.5 (p<0.0032), no significant single variants were identified in the dbPTB set of curated genes that withstood Bonferroni correction for multiple comparisons (p< 5.5×10−6). The lowest P-value was 1.45 10−4 for the SNP (rs5742637) which belongs to insulin-like growth factor 1 gene (IGF1). In the genome-wide analysis only a single variant reached the Bonferroni-corrected significance threshold (p<8.9×10−8). The P-value for this SNP (rs12682166) was 4.99 10−8. This SNP did not map within any known gene nor were there any genes within 5kb upstream or downstream from this variant.
Figure 1. Manhattan plots of three preterm gestational age groups.
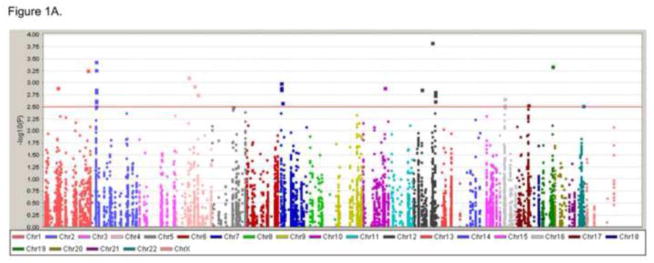
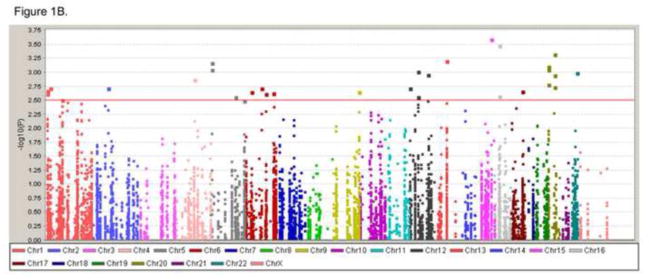
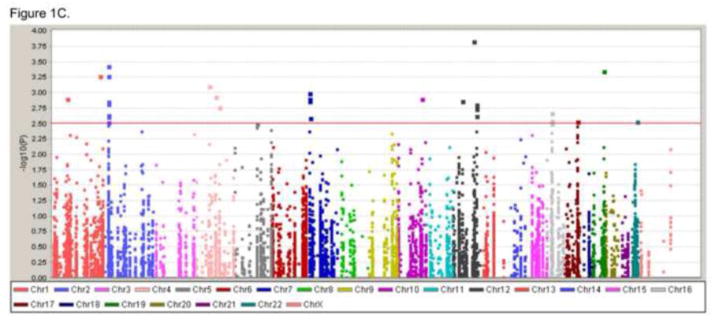
We analyzed the single SNP association with genes from dbPTB by comparing the controls (38 gestation weeks and high) and three gestational groups (37, 34, 30 gestation weeks or less). Manhattan plots showing the results for three preterm gestational age groups. The Y-axis indicates the −log P value for each SNP, X-axis shows the genomic positions. A) Figure represents the comparison of groups for 38 gestation weeks or high vs. 30 gestational weeks or less. B) Figure represents the comparison of groups for 38 gestation weeks or high vs. 34 gestational weeks or less. C) Figure represents the comparison of groups for 38 gestation weeks or high vs. 37 gestation weeks or less.
2.2. GSEA analysis of dbPTB genes
Gene set enrichment analysis was carried out using the 9077 SNPs within the 515 curated dbPTB genes. We compared the cases less than 30 weeks gestation with the controls described in the previous section. For the GSEA analysis we selected the following analytical options: tag SNPS with the 5kb upstream and downstream of each gene [31]; gene sets were selected, including “canonical pathways, GO biological process, GO molecular function, GO cellular component;” lastly, we used gene set size ranges from 5 to 200. For comparison, we performed GSEA on the genome-wide SNP data (n= 560,768 SNPs). All parameters were kept the same when running the genome wide analysis.
From the dbPTB-based pathway analysis, we identified a total of 30 pathways with high confidence values (false discovery rate correction for multiple comparison, FDR, <0.05), see supplemental Table 1. From the whole genome based pathway analysis 39 pathways with high confidence were identified. When we compared the analyses, there were 37 shared pathways. A view illustrating the pathway analysis results for both analyses is shown in Figure 2 and in Table 1. The Vertical axis represents the −log P values for each of the pathways. The statistical values for the pathways identified by dbPTB are shown in dark blue and for the whole genome analysis are shown in light blue. Each of the pathways is shown along the horizontal axis. The threshold value of −log P= 1.3 corresponds to an FDR p value less than 0.05 which has already been adjusted for multiple comparisons. There were nineteen shared pathways that reached significance by either analysis. The dbPTB analysis showed greater significance in 15 out of the 19 shared significant pathways (Figure 2). There were 13 significant pathways that were only identified using the dbPTB curated set of genes and 33 significant pathways that were only identified in the whole genome analysis. Remarkably, most of the pathways involving inflammation were either shared or identified only by the dbPTB-based pathway analysis (Supplemental Table 1). Prominent among the results from the genome-wide analysis were metabolomic pathways including phospholipase A2 activity, amino acid derivative, biosynthetic processes, plus the pathway involving the trans-Golgi network, enzyme inhibitor pathway, lipase activity and nitrogen compound biosynthetic processes and carboxyl esterase activity, hypotaurine metabolism (Supplemental Table 2). A summary comparison of the results of the pathway analyses from both dbPTB and the genome-wide data is shown in supplemental Table 3 and supplemental Figure 1.
Figure 2. Comparison of the pathway analysis results from dbPTB genes and from genome wide analysis.
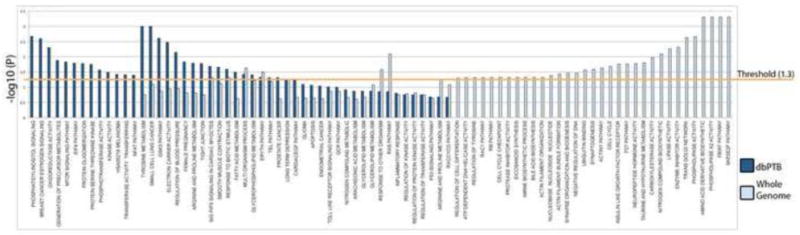
The Vertical axis represents the −log P values for each the pathways. The statistical values for dbPTB are shown in dark blue bars and for the whole genome analysis are shown in light blue bars. Each of the pathways are shown along the horizontal axis. The threshold value of −log P= 1.3 corresponds to an FDR p value less than 0.05 which has already been corrected for the multiple comparisons.
Table 1. The significant pathways from dbPTB in comparison to the analysis from the whole genome.
Table shows the gene set FDR values of the significant pathways from dbPTB in comparison to the analysis from the whole genome. The threshold of FDR < 0.25 denotes the confidence of ‘possible’ or ‘hypothesis’, while the threshold of FDR < 0.05 is regarded as ‘high confidence’ or ‘with statistical significance’.
| GENE SETS | Gene Set FDR* for dbPTB | Gene Set FDR* for Whole Genome |
|---|---|---|
| ARGININE AND PROLINE METABOLISM | 0.01625 | 0.15443571 |
| CARDIACEGF PATHWAY | 0.059756756 | 0.20725532 |
| ELECTRON CARRIER ACTIVITY | 0.003333333 | 0.114043474 |
| ERYTH PATHWAY | 0.04771875 | 0.032863636 |
| FEMALE PREGNANCY | 0.015 | 0.15270229 |
| GCR PATHWAY | 0.10168333 | 0.13590197 |
| GLYCEROLIPID METABOLISM | 0.13772368 | 0.20964974 |
| GSK3 PATHWAY | 0.0025 | 0.136 |
| FATTY ACID METABOLISM | 0.032652173 | 0.23582296 |
| ARGININE AND PROLINE METABOLISM | 0.20881133 | 0.054365855 |
| TYROSINE METABOLISM | 0.001 | 0.17828049 |
| GLYCEROLIPID METABOLISM | 0.13772368 | 0.08272603 |
| GLYCEROPHOSPHOLIPID METABOLISM | 0.039499998 | 0.059211537 |
| ARACHIDONIC ACID METABOLISM | 0.13734783 | 0.24749328 |
| P53 SIGNALING PATHWAY | 0.20801903 | 0.22548293 |
| APOPTOSIS | 0.087419994 | 0.22546601 |
| TIGHT JUNCTION | 0.016727272 | 0.17913496 |
| TOLL LIKE RECEPTOR SIGNALING PATHWAY | 0.098 | 0.1351 |
| LONG TERM DEPRESSION | 0.059526317 | 0.055510636 |
| ENDOMETRIAL CANCER | 0.092660375 | 0.2393868 |
| GLIOMA | 0.082 | 0.22672947 |
| PROSTATE CANCER | 0.04909091 | 0.24307871 |
| SMALL CELL LUNG CANCER | 0.001 | 0.08212857 |
| INFLAMMATORY RESPONSE | 0.15717977 | 0.17977108 |
| MULTI ORGANISM PROCESS | 0.03808 | 0.023111112 |
| NITROGEN COMPOUND METABOLIC PROCESS | 0.12135385 | 0.2158209 |
| RACCYCD PATHWAY | 0.21092594 | 0.08395946 |
| RAS PATHWAY | 0.14021127 | 0.0081 |
| REGULATION OF BLOOD PRESSURE | 0.00725 | 0.11133333 |
| REGULATION OF KINASE ACTIVITY | 0.1786701 | 0.16406667 |
| REGULATION OF PROTEIN KINASE ACTIVITY | 0.1786701 | 0.15386029 |
| REGULATION OF TRANSFERASE ACTIVITY | 0.1786701 | 0.1796467 |
| RESPONSE TO BIOTIC STIMULUS | 0.025818182 | 0.048911765 |
| RESPONSE TO OTHER ORGANISM | 0.1394875 | 0.026736842 |
| SIG PIP3 SIGNALING IN CARDIAC MYOCTES | 0.020499999 | 0.048689652 |
| SMOOTH MUSCLE CONTRACTION | 0.022105262 | 0.07357576 |
| TEL PATHWAY | 0.049032256 | 0.065125 |
3. Discussion
Although there have been some successes, GWAS based approaches have failed to provide comprehensive explanations for the genetic basis of many complex diseases [32]. There are many challenges in identification the causative genes. As noted above for complex diseases, gene-gene interactions are a far more likely model as complex molecular networks and metabolic pathways are involved in polygenic diseases [31, 33–35]. For our approach, we took into consideration the a priori biological information about genes involved in preterm birth from the published literature and from available expression arrays. In addition to these initial steps we included pathway analysis to impute additional genes likely to be involved from pathways identified during curation. Combining these three sources powered the curated gene set for our disease of interest, preterm birth [28]. We increased our power by focusing on a smaller number of comparisons, none of the identified single gene variants reached statistical significance. By employing pathway based permutation testing we identified important genes and their variants in this important disorder. Moreover, by using a more parsimonious, curated set of genes or variants with demonstrated biological significance, we greatly enhance our statistical power. This was most evident in the statistical validation of pathways involved in inflammation. Those pathways were not evident in the genome wide analysis but were SOLEY identified using the curated set of genes for permutation testing.
Since a portion of the ‘missing heritability’ is likely explained by gene-gene interactions, we employed a pathway-based approach to analyze the results from a large GWAS on preterm birth [13]. Our pathway-based approach used the SNPs selected for the dbPTB set of genes and whole genome from GENEVA data. In order to enhance our likelihood of success by selecting the most “extreme phenotype”, we restricted our analysis to comparison of controls which delivered at 38 weeks gestation or higher to patients who delivered at 30 weeks gestation or lower.
In order to generate the “p-values” needed for the pathway analysis, we first carried out single variant analysis using both dbPTB curated genes and whole genome data. As already noted, we did not find significant single variant associated with any known genes using either the dbPTB curated gene set or the genome-wide data. By comparison, the pathway based approached yielded some rich and significant results which replicate the findings from other studies [24]. Among the ranked list of SNPs in the dbPTB curated gene analysis, the best SNP (rs5742637) mapped onto the IGF1 gene. IGF1 was identified in the dbPTB gene set from a single manuscript which sought candidate genes associated with coagulation and inflammatory pathways in preterm birth [24]. In that report, 1536 SNPs in 130 candidate genes were interrogated and IGF1 was one of the significant findings. In the pathway analysis, there were a total of 3 significant pathways which included IGF1. These included the erythrocyte differentiation pathway, prostate cancer and PIP3 signaling in cardiac myocytes. These overlap with the pathways with which IGF1 has been more broadly been associated and are listed in the preterm birth database and includes hypertrophic cardiomyopathy, the mTOR signaling pathway and prostate cancer. Also of note, the IGF1R was identified in the preterm birth database as associated with preterm birth. This was the result of a large linkage analysis done in the Finnish population [36]. In the latter report, the association of IGF1R with preterm birth was verified by haplotype analysis in a larger, independent group of patients [36]. It is likely that the failure to identify IGF1R in both our curated gene study and the pathway analysis is due to the omission of these tag SNPs on our genotyping platform. Nonetheless, the importance of this pathway is suggested by our results and others’ [36]. IGF1 and its signaling pathway were included in previous candidate analyses because of their participation in the decidua-chorioamniotic, and systemic inflammation signaling pathways which involve the PI3kinase and mTOR signaling pathways [36], both of which were prominent in our results.
The pathways identified in our analysis are not independent but instead show a rich network of connectivity. This can best be seen graphically Figure 3. Gephi was used to make the network maps [37]. In this figure, the blue nodes represent the shared pathways and orange nodes that pathways only identified by dbPTB. Likewise the genes forming the connectivity are displayed. AKT1 was the most connected gene, being identified as contributing to a significant role in preterm birth in 15 pathways. This is shown in Figure 4. A listing of the other, highly connected dbPTB genes is provided in supplemental Table 4. An alternative way to view the strength of the pathway analysis is not to look solely at the gene contributing the most pathways, but to indentify which pathways had the most genes contributing to their significance and which genes were these. These results are shown in Figures 5A and 5B. Breast cancer estrogen signaling and oxidoreductase activity pathways each had 10 different genes contributed significantly to their involvement in preterm birth. The latter two pathways were seen solely in the dbPTB analysis.
Figure 3. Network map of significant pathways for dbPTB genes.

The blue nodes represent the shared pathways and orange nodes the pathways only identified by dbPTB. Likewise the genes (green symbols) forming the connectivity are displayed.
Figure 4. The most connected gene.
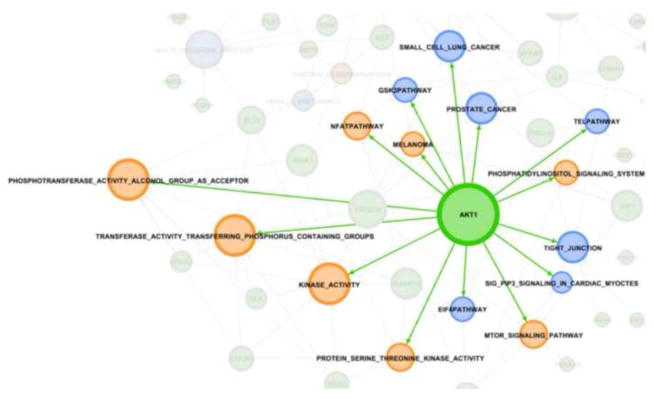
AKT1 was the most connected gene, being identified as contributing to a significant role in preterm birth in 15 pathways.
Figure 5. The most connected pathways.


Breast cancer estrogen signaling and oxidoreductase activity pathways each had 10 different genes contributed significantly to their involvement in preterm birth. 5A. Breast cancer estrogen signaling. 5B. Oxidoreductase activity.
While our approach to pathway analysis was hypothesis-free, inspection of the genes which showed a significant relationship to preterm birth reveals that several of the traditional mechanisms for preterm birth were highly represented. This includes inflammation and metabolomic disorders. Infection and inflammation have been strongly linked to preterm birth [25]. Genes involved in inflammatory mechanisms that emerged from the pathway analysis include: IL6, TGFβ2, NOS1, NFKB1, AKT1, IRAK1, TLR3, TLR7, TP53, IFNG, and AR2. These are all important genes, receptors and signaling elements in inflammation. Many of the associations of these inflammatory genes with preterm birth emerged from their involvement in other, related pathways including GSK3 signaling, small lung cell cancers, organism processes in response to biotic stimulus and PI3K signaling, protein serine3 kinase activity and the NFAT pathway, Figure 3. These genes were not identified in the only other published candidate and pathway-based interrogation although, in the latter study, pathways associated with inflammation were identified including JAK-STAT signaling, MAP kinase signaling, T cell receptor signaling and the Toll-like receptor signaling pathway [24].
Metabolomics has recently been identified as an emerging technology that may provide clues to the pathogenesis of preterm birth that were not previously apparent [38, 39]. In a recent report, gas chromatography-mass spectrometry was used to profile low molecular weight compounds in amniotic fluid of patients delivering with preterm birth with and without inter-amniotic inflammation [39]. A classification profile was developed which subsequently allowed correct classification of patients with preterm birth. We identified several pathways involved in metabolomics that may provide a clue to the genetic architecture underlying the role of metabolomics in preterm birth. These include electron carrier activity, arginine and proline metabolism, the signaling pathways involved in GSK3 and PI3K, tyrosine metabolism, response to biotic stimuli, the oxido-reductase pathway, Figure 5B, protein oligomerization and serine threonine kinase activity. Of the genes associated with these pathways, the most prominent were NOS1 (which is also involved in inflammation), protein kinase C-alpha and ALK associated with both phosphotransferase activity alcohol group as acceptor and kinase activity and transferase activity transferring phosphorus containing groups. The strength of our approach can be seen through the inclusion of NOS1. NOS1 was not identified during the literature curation process or during the aggregation of genes from transcriptome-wide arrays. NOS1 was included in the dbPTB curated genes through the pathway imputation process. Remarkable now is the inclusion of NOS1 in multiple pathways through the GSEA analysis. NOS1 contributed prominently to the significance of the smooth muscle contraction pathway, the oxidoreductase activity pathway, arginine and proline metabolism, small lung cell cancer. In similar fashion, AKT1 was included in the dbPTB set of curated genes through pathway imputation. What is likewise remarkable is that AKT1 was the most frequently identified gene whose variants contributed to significance in the pathway analysis, Figure 4. AKT1 contributed to identification of signaling pathways as diverse as GSK3 signaling tight junctions, prostate cancer, small lung cell cancer, PI3K signaling in cardiac myocytes, telomerase pathways. AKT1 was also prominent in the pathways that were shared between the dbPTB analysis and the whole genome-based pathway analysis including PI3K signaling, HSA41/50 motor signaling, eIF4 pathway, protein serine3 kinase activity, phosphotransferase activity to alcohol groups, general kinase activity, melanoma, transferase activity transferring phosphorus-containing groups, the NFAT pathway.
We are especially interested in comparing the pathway results from the dbPTB curated genes and the pathway results from the genome-wide analysis. As noted above, while there were 37 shared pathways, there were 33 significant pathways that were only identified in the whole genome-based pathway analysis. Prominent among these were metabolomic pathways including phospholipase A2 activity, amino acid derivative, biosynthetic processes, plus the pathway involving the trans-Golgi network, enzyme inhibitor pathway, lipase activity and nitrogen compound biosynthetic processes and carboxyl esterase activity, hypotaurine metabolism. In contrast, most of the pathways involving inflammation were either shared or identified only by the dbPTB-based pathway analysis. This demonstrates the strength of our hybrid approach to identification of relevant genes and pathways in complex diseases. There have been other efforts to collate information on preterm birth. PTBGene is a publicly available database which stores published information on genetic associations with preterm birth [40]. The database currently includes 84 genes with 189 polymorphisms. Using meta analyses these investigators reported 5 significant variants. Four of them were maternal and one was in the newborn.
In summary, we used a bioinformatically-driven strategy to identify a parsimonious set of genes associated with preterm birth. By aggregating genes from literature curation, publically-available databases (most often from transcriptome-wide analysis) and then using pathway-based imputation, we identified 617 genes for which there was a priori biological evidence for involvement in preterm birth. The tag SNPs associated with these genes were then used in traditional candidate gene association testing using data from the GENEVA genome-wide association study. While we increased our power by focusing on a smaller number of comparisons, none of the identified single gene variance reached statistical significance. We did, however, corroborate the best of those curated genes, IGF1 in the pathway-based analyses in both the dbPTB pathway analysis and the genome-wide analysis. The database for preterm birth was built to support analysis of gene/gene interactions. It is clear using extremely large sets of SNPs that it’s computationally expensive to carry out even pairwise comparisons of genes. Moreover, the knowledge-based association of genetic variation with disease dictates that all variants are not interacting with each other. Rather, gene/gene interactions occur on the basis of known biological information. This body of information has been built into robust databases including KEGG, Biocarta, DAVID, GO and Ingenuity [41]. Although pathway-based analysis methods help us in understanding and evaluating GWAS data, improvements are forthcoming. Better summary statistics will help to evaluate the results more robustly as described in Wang et al [41]. Likewise, gene level p-values which usually depend on SNP association test are limited by the number and preference of SNPs on the arrays. Another limitation is to identify which SNP is the best representative of a given gene by not only considering the best association p-values but also considering combined effects of SNPs in linkage disequilibrium [33].
The gene set enrichment approach allowed us to interrogate the known biological associations annotated by several of these databases. By permutation testing, we compared the association of the single nucleotide polymorphisms tagging the genes in our dataset and their association in cases and controls. Even given the anticipated improvements in pathway-based methods, the results were extraordinary. We identified a large number of significant pathways in which biologically relevant curated genes and their associated variants showed significant segregation between the preterm birth and full term births. Moreover, the curated genes from the dbPTB dataset gave much stronger associations than the genome wide analysis in all but a few of these pathways. These results provide important confirmation of the role of genetic architecture in the risk of preterm birth. They also provide important mechanistic insights and curated genes which are suitable for future genetic association testing or ideal targets for more thorough evaluation including targeted re-sequencing. We recognize that, due to the lack of a replication dataset, this study should be considered hypothesis generating and that these results will need to be replicated in an appropriate dataset.
4. Materials and methods
4.1. dbPTB; The Database for Preterm Birth
We identified 186 genes using the literature-based curation, 215 genes from publically-available databases and an additional 216 genes from the pathway-based interpolation [28]. These 617 genes represent a robust set of genes for which there is good prior biological evidence for involvement in preterm birth [28].
4.2. The Gene Environment Association Studies initiative (GENEVA) Data
We analyzed the single nucleotide polymorphism (SNP) genotyping data from a prospective cohort study in Denmark. The data were derived from the Gene Environment Association Studies initiative (GENEVA) funded by the trans-NIH Genes, Environment, and Health Initiative (GEI) [42]. The data from GENEVA consist of approximately 4000 Danish women and children and includes phenotype and genotype information from a genome-wide case/control study using approximately 1000 preterm mother-child pairs. There is also data from 1000 control mother-child pairs where the child was born greater than or equal to 38 weeks’ gestation. All data were deposited into the Database for Genotypes and Phenotypes (dbGaP) [29]. Genome wide SNP genotyping was performed using Illumina Human 660W-Quad_v1_A (n=560,768 SNPs) at the Center for Inherited Disease Research, Baltimore, MD. As reported in the data set release, genotypes were not reported for any SNP which had a call rate less than 85% or which had more than 1 replicate error as defined with the HapMap control samples.
4.3. SNP association testing in PLINK
We ran basic SNP association tests in PLINK to obtain individual marker P-values [43]. The basic association test is based on comparing allele frequencies between cases and controls. PLINK is a free, open-source whole genome association analysis toolset which performs a range of basic, large-scale analyses [43]. The SNP-association analyses were conducted in PLINK using only curated-genes from dbPTB as well as using all the SNPs from the genome-wide analysis. For these analyses, the study “controls” consisted of the 960 mothers who had delivered at 38 weeks gestation or higher. For comparison we carried out the same curated gene analysis using three different patient groups from the GENEVA study. We analyzed the single SNP association with PTB by comparing the controls with the 884 patients delivering less than 37 weeks, the 446 patients delivering less than 34 weeks, and the 92 patients delivering less than 30 weeks.
4.4. Gene set enrichment analysis
In recent years, gene set enrichment analysis (GSEA, [33]) has become increasingly popular to support analysis of gene-gene interactions and to help in understanding the individual contribution(s) of biological pathways to genetic architecture. GSEA employs a new way of considering the SNPs in GWAS [33]. Instead of analyzing single SNPs individually, GSEA tests disease association with genetic variants in functionally related genes by analyzing the genes that belong to the same pathway that may represent the possible SNP or gene association with complex diseases [33]. We first performed GSEA on GENEVA GWAS data, using the SNP P-values from the dbPTB curated genes with i-GSEA4GWAS. The i-GSEA4GWAS web server implements i-GSEA to explore GWAS data efficiently. “i-GSEA” is an enhanced application and extension of GSEA. The program runs the analysis in three steps. First, it maps the variants to the genes, each gene is represented by −log (P-value) of closely spaced SNPs in a gene. Second, i-GSEA is performed to identify the pathways correlated to traits based on the distribution of enrichment scores generated by permutation. FDR is calculated and used to correct for multiple testing. The threshold of FDR < 0.25 denotes the confidence of ‘possible’ or ‘hypothesis’, while the threshold of FDR < 0.05 is regarded as ‘high confidence’ or ‘with statistical significance’. Finally, program lists significant pathways, the genes within those pathways which contributed to the significant associations along with the SNPs that contributed to the association.
Supplementary Material
Highlights.
We use both the curated genes and a genome-wide approach for pathway analysis.
The analysis based on the curated genes was more significant than genome-wide.
We identify gene-gene interactions that enhance statistical power and discovery.
We report significant pathway-based associations for preterm birth.
The results suggest several, important mechanistic implications for preterm birth.
Acknowledgments
AU built the original curation database and web based interfaces, participated in weekly curation meetings, had direct involvement in all genetic analysis, wrote and edited the paper.
ATD provided oversight and guidance on the initial genetic approaches, contributed to single variant analysis, gave feedback and guidance on GSEA analysis, edited the paper.
SI aided in identification of TAG SNPs, provided alternative semantic data mining results to cross checking the original data, participated in monthly discussions of the evolving data and analyses, contributor to the original funding, edited the paper.
JFP developed the initial concepts, participated in the weekly curation meetings, had direct involvement in all genetic analyses, wrote and edited the paper.
This work was supported by the National Foundation March of Dimes Prematurity Initiative # 21-FY08-563, and National Institutes of Health Grants NIH-5T35HL094308-02 and NIH-NCRR P20 RR018728. GWAS data for preterm birth was analyzed from the Genome-Wide Association Studies of Prematurity and Its Complications, dbGaP study accession numbered “phs000103.v1.p1”. This research was conducted using computational resources and services at the Center for Computation and Visualization, Brown University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Feero WG, Guttmacher AE, Collins FS. Genomic medicine--an updated primer. N Engl J Med. 2010;362:2001–2011. doi: 10.1056/NEJMra0907175. [DOI] [PubMed] [Google Scholar]
- 2.Collins FS, Green ED, Guttmacher AE, Guyer MS. A vision for the future of genomics research. Nature. 2003;422:835–847. doi: 10.1038/nature01626. [DOI] [PubMed] [Google Scholar]
- 3.Iles MM. Genome-wide association studies. Methods Mol Biol. 2011;713:89–103. doi: 10.1007/978-1-60327-416-6_7. [DOI] [PubMed] [Google Scholar]
- 4.Hindorff LA, MacArthur J, Wise A, Junkins HA, Hall PN, Klemm AK, Manolio TA. A Catalog of Published Genome-Wide Association Studies. 2012. [Google Scholar]
- 5.Mathew CG. New links to the pathogenesis of Crohn disease provided by genome-wide association scans. Nat Rev Genet. 2008;9:9–14. doi: 10.1038/nrg2203. [DOI] [PubMed] [Google Scholar]
- 6.Dewan A, Liu M, Hartman S, Zhang SS, Liu DT, Zhao C, Tam PO, Chan WM, Lam DS, Snyder M, Barnstable C, Pang CP, Hoh J. HTRA1 promoter polymorphism in wet age-related macular degeneration. Science. 2006;314:989–992. doi: 10.1126/science.1133807. [DOI] [PubMed] [Google Scholar]
- 7.Glessner JT, Bradfield JP, Wang K, Takahashi N, Zhang H, Sleiman PM, Mentch FD, Kim CE, Hou C, Thomas KA, Garris ML, Deliard S, Frackelton EC, Otieno FG, Zhao J, Chiavacci RM, Li M, Buxbaum JD, Berkowitz RI, Hakonarson H, Grant SF. A genome-wide study reveals copy number variants exclusive to childhood obesity cases. Am J Hum Genet. 2010;87:661–666. doi: 10.1016/j.ajhg.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldstein DB. Common genetic variation and human traits. N Engl J Med. 2009;360:1696–1698. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- 9.Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet. 2010;11:415–425. doi: 10.1038/nrg2779. [DOI] [PubMed] [Google Scholar]
- 10.Hirschhorn JN. Genomewide association studies--illuminating biologic pathways. N Engl J Med. 2009;360:1699–1701. doi: 10.1056/NEJMp0808934. [DOI] [PubMed] [Google Scholar]
- 11.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TFC, McCarroll SA, Visscher PM. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cowper-Sallari R, Cole MD, Karagas MR, Lupien M, Moore JH. Layers of epistasis: genome-wide regulatory networks and network approaches to genome-wide association studies. Wiley Interdiscip Rev Syst Biol Med. 2011;3:513–526. doi: 10.1002/wsbm.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eichler EE, Flint J, Gibson G, Kong A, Leal SM, Moore JH, Nadeau JH. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11:446–450. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu T, Sinnott-Armstrong NA, Kiralis JW, Andrew AS, Karagas MR, Moore JH. Characterizing genetic interactions in human disease association studies using statistical epistasis networks. BMC Bioinformatics. 2011;12:364. doi: 10.1186/1471-2105-12-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Askland K, Read C, Moore J. Pathways-based analyses of whole-genome association study data in bipolar disorder reveal genes mediating ion channel activity and synaptic neurotransmission. Human Genetics. 2008;125:63–79. doi: 10.1007/s00439-008-0600-y. [DOI] [PubMed] [Google Scholar]
- 16.Lesnick TG, Papapetropoulos S, Mash DC, Ffrench-Mullen J, Shehadeh L, de Andrade M, Henley JR, Rocca WA, Ahlskog JE, Maraganore DM. A genomic pathway approach to a complex disease: axon guidance and Parkinson disease. PLoS Genet. 2007;3:e98. doi: 10.1371/journal.pgen.0030098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lesnick TG, Sorenson EJ, Ahlskog JE, Henley JR, Shehadeh L, Papapetropoulos S, Maraganore DM. Beyond Parkinson disease: amyotrophic lateral sclerosis and the axon guidance pathway. PLoS ONE. 2008;3:e1449. doi: 10.1371/journal.pone.0001449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gibson G. Hints of hidden heritability in GWAS. Nat Genet. 2010;42:558–560. doi: 10.1038/ng0710-558. [DOI] [PubMed] [Google Scholar]
- 19.Gui J, Andrew AS, Andrews P, Nelson HM, Kelsey KT, Karagas MR, Moore JH. A Robust Multifactor Dimensionality Reduction Method for Detecting Gene-Gene Interactions with Application to the Genetic Analysis of Bladder Cancer Susceptibility. Ann Hum Genet. 2010 doi: 10.1111/j.1469-1809.2010.00624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cordell HJ. Detecting gene-gene interactions that underlie human diseases. Nat Rev Genet. 2009;10:392–404. doi: 10.1038/nrg2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moore JH. Detecting, characterizing, and interpreting nonlinear gene-gene interactions using multifactor dimensionality reduction. Adv Genet. 2010;72:101–116. doi: 10.1016/B978-0-12-380862-2.00005-9. [DOI] [PubMed] [Google Scholar]
- 22.Adams KM, Eschenbach DA. The genetic contribution towards preterm delivery. Semin Fetal Neonatal Med. 2004;9:445–452. doi: 10.1016/j.siny.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 23.Crider KS, Whitehead N, Buus RM. Genetic variation associated with preterm birth: a HuGE review. Genet Med. 2005;7:593–604. doi: 10.1097/01.gim.0000187223.69947.db. [DOI] [PubMed] [Google Scholar]
- 24.Menon R, Fortunato SJ, Thorsen P, Williams S. Genetic associations in preterm birth: a primer of marker selection, study design, and data analysis. J Soc Gynecol Investig. 2006;13:531–541. doi: 10.1016/j.jsgi.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 25.Muglia LJ, Katz M. The enigma of spontaneous preterm birth. N Engl J Med. 2010;362:529–535. doi: 10.1056/NEJMra0904308. [DOI] [PubMed] [Google Scholar]
- 26.Weinberg CR, Shi M. The genetics of preterm birth: using what we know to design better association studies. Am J Epidemiol. 2009;170:1373–1381. doi: 10.1093/aje/kwp325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weiner CP, Mason CW, Dong Y, Buhimschi IA, Swaan PW, Buhimschi CS. Human effector/initiator gene sets that regulate myometrial contractility during term and preterm labor. Am J Obstet Gynecol. 2010;202:474 e471–420. doi: 10.1016/j.ajog.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uzun A, Laliberte A, Parker J, Andrew C, Winterrowd E, Sharma S, Istrail S, Padbury JF. dbPTB: a database for preterm birth. Database (Oxford) 2012;2012:bar069. doi: 10.1093/database/bar069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mailman MD, Feolo M, Jin Y, Kimura M, Tryka K, Bagoutdinov R, Hao L, Kiang A, Paschall J, Phan L, Popova N, Pretel S, Ziyabari L, Lee M, Shao Y, Wang ZY, Sirotkin K, Ward M, Kholodov M, Zbicz K, Beck J, Kimelman M, Shevelev S, Preuss D, Yaschenko E, Graeff A, Ostell J, Sherry ST. The NCBI dbGaP database of genotypes and phenotypes. Nat Genet. 2007;39:1181–1186. doi: 10.1038/ng1007-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boyd HA, Poulsen G, Wohlfahrt J, Murray JC, Feenstra B, Melbye M. Maternal Contributions to Preterm Delivery. American Journal of Epidemiology. 2009;170:1358–1364. doi: 10.1093/aje/kwp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 32.McClellan J, King M-C. Genetic Heterogeneity in Human Disease. Cell. 2010;141:210–217. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 33.Wang K, Li M, Hakonarson H. Analysing biological pathways in genome-wide association studies. Nat Rev Genet. 2010;11:843–854. doi: 10.1038/nrg2884. [DOI] [PubMed] [Google Scholar]
- 34.Zhang K, Cui S, Chang S, Zhang L, Wang J. i-GSEA4GWAS: a web server for identification of pathways/gene sets associated with traits by applying an improved gene set enrichment analysis to genome-wide association study. Nucleic Acids Res. 2010;38:W90–95. doi: 10.1093/nar/gkq324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schadt EE. Molecular networks as sensors and drivers of common human diseases. Nature. 2009;461:218–223. doi: 10.1038/nature08454. [DOI] [PubMed] [Google Scholar]
- 36.Haataja R, Karjalainen MK, Luukkonen A, Teramo K, Puttonen H, Ojaniemi M, Varilo T, Chaudhari BP, Plunkett J, Murray JC, McCarroll SA, Peltonen L, Muglia LJ, Palotie A, Hallman M. Mapping a new spontaneous preterm birth susceptibility gene, IGF1R, using linkage, haplotype sharing, and association analysis. PLoS Genet. 2011;7:e1001293. doi: 10.1371/journal.pgen.1001293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bastian HSM, Jacomy M. Gephi: an open source software for exploring and manipulating networks. International AAAI Conference on Weblogs and Social Media; 2009. [Google Scholar]
- 38.Romero R, Espinoza J, Gotsch F, Kusanovic JP, Friel LA, Erez O, Mazaki-Tovi S, Than NG, Hassan S, Tromp G. The use of high-dimensional biology (genomics, transcriptomics, proteomics, and metabolomics) to understand the preterm parturition syndrome. BJOG. 2006;113(Suppl 3):118–135. doi: 10.1111/j.1471-0528.2006.01150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gracie S, Pennell C, Ekman-Ordeberg G, Lye S, McManaman J, Williams S, Palmer L, Kelley M, Menon R, Gravett M. An integrated systems biology approach to the study of preterm birth using “-omic” technology--a guideline for research. BMC Pregnancy Childbirth. 2011;11:71. doi: 10.1186/1471-2393-11-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dolan SM, Hollegaard MV, Merialdi M, Betran AP, Allen T, Abelow C, Nace J, Lin BK, Khoury MJ, Ioannidis JP, Bagade S, Zheng X, Dubin RA, Bertram L, Velez Edwards DR, Menon R. Synopsis of preterm birth genetic association studies: the preterm birth genetics knowledge base (PTBGene) Public Health Genomics. 2010;13:514–523. doi: 10.1159/000294202. [DOI] [PubMed] [Google Scholar]
- 41.Ramanan VK, Shen L, Moore JH, Saykin AJ. Pathway analysis of genomic data: concepts, methods and prospects for future development. Trends Genet. 2012 doi: 10.1016/j.tig.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.GENEVA (Gene-Environment Association Studies) 2012. [Google Scholar]
- 43.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.