

Abstract
The heart is an omnivore organ that requires constant energy production to match its functional demands. In the adult heart, ATP production occurs mainly through mitochondrial fatty acid and glucose oxidation. The heart must constantly adapt its energy production in response to changes in substrate supply and work demands across diverse physiologic and pathophysiologic conditions. The cardiac myocyte maintains a high level of mitochondrial ATP production through a complex transcriptional regulatory network that is orchestrated by the members of the peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) family. There is increasing evidence that during the development of cardiac hypertrophy and in the failing heart, the activity of this network, including PGC-1, is altered. This review summarizes our current understanding of the perturbations in the gene regulatory pathways that occur during the development of heart failure. An appreciation of the role this regulatory circuitry serves in the regulation of cardiac energy metabolism may unveil novel therapeutic targets aimed at the metabolic disturbances that presage heart failure.
Keywords: Heart failure, mitochondria, peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α), fatty acid oxidation (FAO)
1. Introduction
To meet the workload demands of a constant pump, the mammalian heart requires an incredibly high capacity for energy production. The ability of the heart to store energy, however, is limited. Therefore, the capacity to produce ATP in the heart must be tightly matched with work demands and fuel delivery. Much of this regulation occurs through a complex network of transcription factors that respond to changes in substrate (fatty acids and glucose) delivery, oxygen availability, and a myriad of physiological conditions.
The mitochondrion serves as the principal energy-producing organelle in the heart and accounts for up to 40% of the cardiac myocyte volume. Under normal conditions, 60–80% of ATP production in the heart is derived from mitochondrial fatty acid β-oxidation. The remainder is from carbohydrate (glucose and lactate) and to a lesser degree, ketone body oxidation. However, the heart is extremely metabolically flexible, capable of changing fuel source depending on substrate availability or hormonal milieu (e.g. insulin levels). Structural changes of the heart including physiological and pathological forms of cardiac hypertrophic growth are also associated with changes in fuel selection and energy-producing capacity. In the setting of heart failure, perturbations in cardiac energy metabolism have numerous consequences including a reduction in capacity and efficiency of mitochondrial respiration and ATP production. It is now recognized that the failing heart is “energy starved”. This review focuses on the gene regulatory pathways controlling mitochondrial energy production in the cardiac myocyte with an emphasis on the dysregulation that occurs during development of heart failure.
2. Changes in mitochondrial energy transduction and ATP generation
2.1 Energy production
The concept of the “energy starved” failing heart has been extensively reviewed elsewhere [1] and will not be addressed in detail here. It is well known that levels of the main storage buffer for ATP, phosphocreatine (PCr), fall during the development of pathologic cardiac hypertrophy and during the progression to heart failure. Phosphocreatine serves as a shuttle molecule to deliver high-energy phosphate from ATP produced in the mitochondria to ATP consumed at the myofibrillar structure. The PCr:ATP ratio has been shown to be a powerful predictor of mortality in patients with dilated cardiomyopathy [2]. A lower total [PCr] in the cell may reflect reduced mitochondrial respiration and ATP flux. When the [PCr] falls, ADP, AMP and Pi concentrations rise and activate glycolytic pathways through increased glucose transport and utilization. Although initially increased to meet the deficiency in energy production, the increased glycolytic rate is insufficient to meet the energy demands of the failing heart. It is now recognized that the energetic remodeling in the failing human heart leads to an approximate 30% decrease in [ATP] [2]. Of particular interest are the signaling events and mechanisms that lead to decreased energy production in the failing heart as well as potential therapeutic strategies to improve the energy starved heart.
2.2 Energy transduction pathways
There have been a number of studies aimed at assessing the expression of the components of energy-producing pathways in the failing heart [3, 4]. Similar to the expression of many structural and contractile proteins, there is a switch to the “fetal gene program” evident in metabolic enzyme gene expression in cardiac hypertrophy and failure [5]. Among the most evident changes is a decrease in the expression of genes encoding fatty acid β-oxidation enzymes that begins during the development of cardiac hypertrophy. The rates of FAO have also been shown to be lower in experimental and human heart failure [6, 7]. Coordinate changes in FAO enzyme levels have now been confirmed by a number of studies using proteomic approaches to measure changes in mitochondrial protein expression. For example, a recent mitochondrial comparative proteomic study in a rat pressure-overload model revealed downregulation of multiple FAO enzymes [7]. This was associated with increased expression of glucose oxidation enzymes in the mitochondria. Interestingly, upregulation of cardiac mitochondrial FAO enzyme expression has been observed in a number of diabetic models consistent with an activation of fatty acid oxidation rates [8–10]. This likely reflects the high influx of free fatty acids and activating ligands for this pathway (discussed more below). Cardiac hypertrophy and failure are also associated with changes in multiple components of the electron transport chain (ETC). However, even within the same model and study, divergent responses in different ETC components have been observed, underscoring the complex regulatory network that controls cardiac mitochondrial energy production [7, 11].
Interestingly, exercise training can produce cardiac hypertrophy that is not characterized by the classic pathologic fetal gene signature or dysfunction [12]. This so-called “physiologic hypertrophy” is accompanied by increases in capacity for mitochondrial respiration and ATP synthesis [13]. In addition, some studies have also demonstrated an increase in FAO rates and enzyme expression in physiological forms of cardiac hypertrophy [14, 15]. These observations raise the possibility that the metabolic effects of exercise training may be of some benefit in the setting of heart failure. Indeed, both animal and clinical studies have demonstrated a benefit from exercise training in both cardiac function and quality of life [16, 17].
Finally, advances in clinical imaging have enabled measurements of myocardial glucose and fatty acid metabolism in humans with heart failure. The use of positron emission tomography (PET) with 11C-palmitate has demonstrated that a decrease in myocardial FAO is an independent predictor of left ventricular hypertrophy (LVH) in hypertension [18]. Furthermore, LVH is associated with a decrease in both fatty acid metabolism and myocardial efficiency [19]. These studies correlate well with the molecular changes that have been observed in FAO enzyme expression during cardiac hypertrophy.
3. Nuclear receptors: Key transcriptional regulators of cardiac fuel and energy metabolism
A network of transcription factors respond to changes in substrate availability, as well as growth and stress stimuli, to ensure that the capacity for fuel burning keeps up with demands. Changes in the levels and activities of these factors play a key role in the perturbations in energy production pathways known to occur during development of heart failure. In addition, nuclear receptors, as ligand-activated proteins (discussed further below), have been actively pursued as therapeutic targets for metabolic and cardiovascular disease. The nuclear receptor family of transcription factors is now known to regulate virtually all aspects of cardiac mitochondrial energy production.
3.1 Peroxisome proliferator-activated receptors (PPARs)
Peroxisome proliferator-activated receptors (PPARs) are members of the extended nuclear hormone receptor family [20, 21]. All 3 isoforms, PPARα, PPARβ/δ and PPARγ, heterodimerize with the retinoid X receptor (RXR) and bind a specific DNA sequence or PPAR response element (PPRE) to activate transcription. PPARα is highly expressed in the myocardium and participates in the regulation of fatty acid transport and mitochondrial and peroxisomal fatty acid oxidation pathways [22]. PPARα is able to respond to changes in substrate availability through its ligand binding domain. Recent studies relevant to the liver suggest that long-chain fatty acids and phospholipids can serve as endogenous ligands [23]. In the heart, a recent study suggests that adipose triglyceride lipase (ATGL) generates biologically active lipid species that serve as PPARα activating ligands [24]. In support of this, ATGL knockout mice display a profound reduction of cardiac FAO gene expression along with multiple mitochondrial derangements. This finding unveils a potential mechanism in matching cardiac myocyte fatty acid storage and utilization.
The activity of PPARα signaling has been implicated in the known and divergent substrate shifts in different pathophysiological scenarios known to lead to heart failure (Figure 1). Fatty acid utilization is known to be downregulated coincident with an increase in glucose utilization in pressure overload-induced cardiac hypertrophy. In the insulin resistant and diabetic heart the opposite substrate shifts occur (Figure 1). The roles of PPARα in controlling cardiac fuel metabolism in health and disease have been defined, in part, by genetically-engineered gain-of-function and loss-of function mouse models. Mice that overexpress PPARα exclusively in the heart (MHC-PPARα) exhibit increased fatty acid oxidation associated with a decrease in glucose utilization resulting in an accumulation of triglyceride and a diabetic-like phenotype [25, 26]. These animals exhibit left ventricular hypertrophy and cardiac dysfunction that can be inhibited by deletion of CD36, a cellular fatty acid import protein [27, 28]. High circulating levels of free fatty acids present in obesity and metabolic disease may activate cardiac PPARα and contribute to the observed high FAO rates in this condition (see Figure 2). In addition, several groups have also reported an induction of PPARα levels in the diabetic mouse heart [29–31]. In contrast, mice lacking PPARα demonstrate a decrease in fatty acid oxidation and increased glucose utilization [32–34]. Interestingly, and in contradistinction to the diabetic heart, PPARα expression is decreased in human and animal models of pressure overload-induced cardiac hypertrophy and in heart failure [5, 35–37]. This is in agreement with the observed metabolic shift from fatty acid oxidation to glucose metabolism that occurs during the transition to cardiac hypertrophy and heart failure [38–40].
Figure 1.
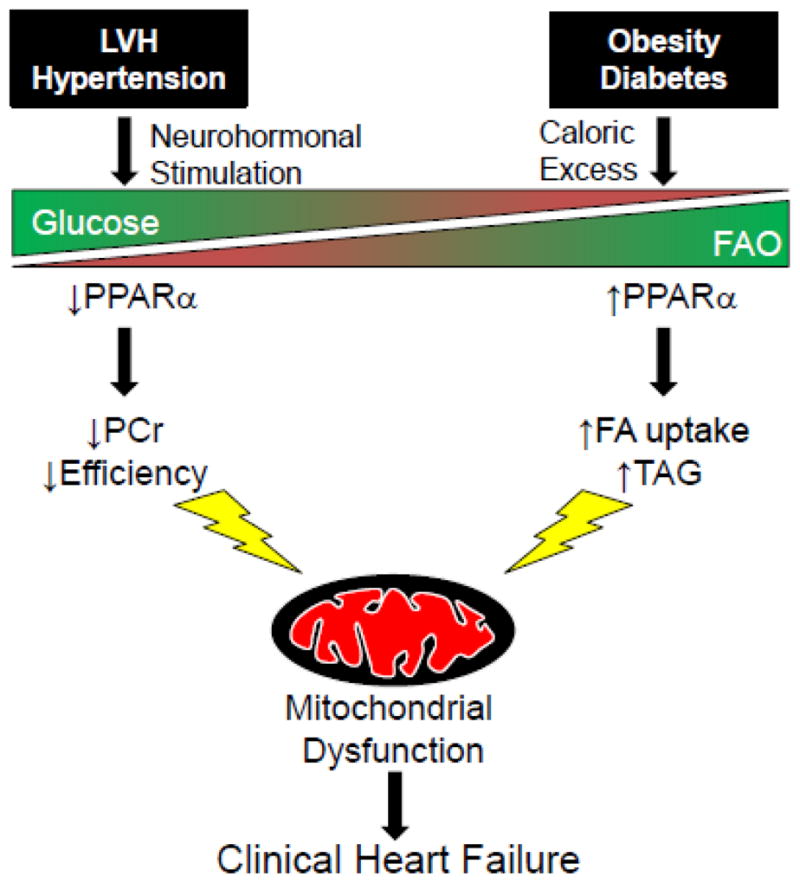
Divergent substrate utilization in the hypertensive, hypertrophic and insulin resistant, diabetic hearts. The specific etiology of heart disease promotes different substrate utilization in the cardiac myocyte. Cardiac hypertrophy frequently associated with hypertension and ischemia promotes glucose utilization while obesity and diabetes favors increased fatty acid oxidation (FAO). PPARα activity is central to the control of FAO and its modulation in hypertrophy (down) or diabetes (up) contributes to the observed changes in fuel source. However, both substrate switches eventually lead to mitochondrial dysfunction contributing to the progression of clinical heart failure. LVH, left ventricular hypertrophy; PPARα, peroxisome proliferator- activated receptor alpha; PCr, phosphocreatine; TAG, triacylglycerol.
Figure 2.

Mechanistic model of myocardial lipid accumulation relevant to obesity-related forms of heart failure that occur in the diabetic heart. There is an influx of fatty acids into the cardiac myocyte in the setting of obesity and insulin resistance. Excess intracellular fatty acids are stored as triglycerides or can act as activating ligands for PPARα. At least in the early stages of insulin resistance, activation of PPARα results in increased mitochondrial fatty acid β-oxidation. However, PPARα also activates fatty acid import pathways, including CD36, to begin a vicious cycle of increased import and storage of fatty acids. Eventually, PGC-1 levels fall and mitochondrial and cardiac dysfunction ensues. DGAT1, diacylglycerol acyltransferase 1; SCD1, stearoyl-CoA desaturase 1; TAG, triacylglycerol; PPARα, peroxisome proliferator- activated receptor alpha; PGC-1, PPARγ coactivator-1
PPARβ/δ regulates expression of a set of overlapping targets with PPARα including FAO enzymes. However, in contrast to PPARα, PPARβ/δ also activates glucose utilization in the heart [41]. Mice expressing PPARβ/δ in the heart are also relatively resistant to diet-induced cardiac myocyte lipid accumulation and lipotoxic cardiomyopathy. Consistent with these observations, cardiac-specific deletion of the PPARβ/δ gene in mice results in a decrease in fatty acid and glucose oxidation rates concomitant with a decrease in the expression of genes in both pathways [42].
The role of cardiac PPARγ is less well understood. Although it is expressed at much lower levels in heart compared to either PPARα or PPARβ/δ, deleterious effects of cardiac PPARγ-deficiency in mice have been reported by independent groups [43, 44]. Cardiac overexpression of PPARγ also leads to cardiac dysfunction with lipid accumulation and mitochondrial abnormalities [45]. Therefore, it would seem that a critical balance in the level of PPARγ activity is necessary for proper cardiac function.
3.2 Estrogen-related receptor (ERR)
Also belonging to the nuclear receptor superfamily, the estrogen-related receptor (ERR) family is comprised of three members, ERRα, ERRβ and ERRγ [46–48]. Endogenous ligands for ERRs have not been identified and crystallography studies suggest the ligand-binding pocket of ERRα may be too small for any to exist [49]. ERRα and ERRγ are enriched in tissues with high oxidative metabolic rates such as the heart.
ERRα controls the expression of many genes involved in energy metabolism pathways including cellular fatty acid transport, mitochondrial and peroxisomal fatty acid oxidation and mitochondrial respiration [50]. ERRα also activates expression of PPARα providing an additional layer of cross-regulation [50]. Genome-wide association studies have determined that ERRα and ERRγ likely act as nonobligatory heterodimers and target a common set of promoters [51]. ERRα is required for the adaptive response to hemodynamic stress as loss-of-function results in decompensated heart failure when mice are subjected to left ventricular pressure overload. This is associated with myocardial phosphocreatine depletion and reduced maximal ATP synthesis [52]. Furthermore, expression of ERR target genes including those involved in glucose metabolism, FAO and the ETC are significantly downregulated in human heart failure samples suggesting that interruption of normal ERR function contributes to the pathophysiology of heart failure.
A critical role for ERRγ in the control of cardiac energy metabolism has also been demonstrated in loss-of-function studies. ERRγ knockout mice display a downregulation of several ERR targets in the heart, exhibit cardiomyopathy, and die immediately following birth. The early postnatal death in these animals probably results from the inability to transition from glucose to fatty acid utilization following birth [53].
4. PPARγ coactivator 1 (PGC-1)
The discovery of peroxisome proliferator-activated receptor (PPAR)γ coactivator-1α (PGC-1α) provided the first clue as to how the complex transcriptional regulatory circuit controlling cardiac metabolism was orchestrated in accordance with energy demands. PGC-1α was first cloned in brown adipose tissue as a PPARγ interacting protein [54]. The PGC-1 family also contains two other members, PGC-1β and PGC-1-related coactivator (PRC) [55, 56]. PGC-1α is an inducible transcriptional coregulator activated by stimuli that increase demands of mitochondrial flux such as cold exposure and exercise [57–59]. PGC-1α expression in the heart is induced during heart development concomitant with the mitochondrial burst that occurs just before birth [60]. An increase in PGC-1α cardiac expression is also observed following acute and chronic exercise training which may be dependent, in part, upon insulin growth factor 1 (IGF1) signaling [61]. Overexpression studies confirmed that PGC-1α directly interacts and activates the targets of PPARα [62], PPARβ/δ [63], ERRα and ERRγ [64–66]. These results suggest that PGC-1 integrates physiologic signals and developmental cues to regulate virtually all aspects of mitochondrial function (Figure 3).
Figure 3.
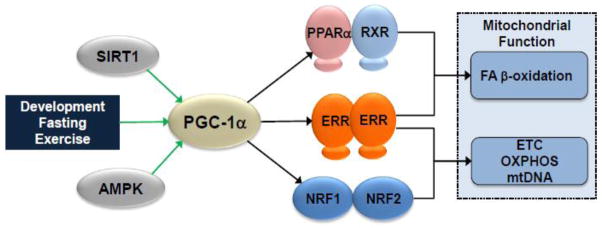
PGC-1α control of cardiac mitochondrial function. PGC-1α integrates signals from physiologic stimuli as well as upstream modulators such as sirtuins (SIRT) and AMPK. Direct interaction with multiple transcription factors drives expression of proteins involved in all aspects of mitochondrial function. SIRT1, sirtuin 1; AMPK, AMP-activated protein kinase; PGC-1α, PPARγ coactivator-1 alpha; PPARα, peroxisome proliferator-activated receptor alpha; RXR, retinoid X receptor; ERR, estrogen-related receptor; NRF, nuclear respiratory factor; ETC, electron transport chain; OXPHOS, oxidative phosphorylation.
Somewhat surprisingly, myocardial mitochondrial volume and ventricular function is not diminished in PGC-1α knockout animals [67, 68]. The relatively mild phenotype is related to functional redundancies between PGC-1α and PGC-1β. Indeed, disruption of both the PGC-1α and PGC-1β genes prevents perinatal mitochondrial biogenesis in heart causing cardiomyopathy and death shortly after birth [69]. Given its critical role in mitochondrial function, a logical question is whether dysregulation of PGC-1 is involved in the pathogenesis of the “energy-starved” phenotype of the failing heart. The changes in fuel selection and mitochondrial function in the hypertrophied and failing heart are likely consequences of dysregulation of the PGC-1 circuit. Consistent with this notion, the expression of PGC-1α is downregulated in both animal models and human heart failure samples [5, 70, 71]. In addition, PGC-1α and PGC-1β deficient mice develop heart failure following pressure overload [72, 73]. Taken together, these data suggest that deactivation of PGC-1 and accompanying metabolic derangements contribute to the development of heart failure. Furthermore, the activity of PGC-1 may be altered independently of its expression by post-translational modifications including phosphorylation and acetylation. PGC-1α activity can be increased through direct phosphorylation by AMPK [74]. Further regulation of PGC-1α activity is controlled by opposing actions of GCN5 (acetylation) [75] and SIRT1 (deacetylation) [76]. Changes in intracellular energy production and nutrient availability known to occur during heart failure impact the activity of both AMPK and SIRT1. However, the role of alterations in the upstream signaling pathways and consequence of these changes on PGC-1 activity during the development of heart failure have not been fully explored in animal models or humans.
5. Other relevant transcription factors
5.1 MEF2
The myocyte enhancer factor-2 transcription factor has been shown to regulate many growth and remodeling genes during cardiac hypertrophy [77]. A link between MEF2A and cardiac metabolism was first shown in MEF2A knockout mice which displayed decreased mitochondrial content and perinatal lethality [78]. Subsequently, MEF2 was shown to directly activate transcription of PGC-1α [79]. Cardiac MEF2 activity is tightly controlled by class II histone deacetylases including HDAC4 and HDAC5. Class II HDACs have been shown to be signal-responsive inhibitors of MEF2 and pathologic cardiac growth [80, 81]. In this context, dysregulation of MEF2 activity could contribute to perturbations in cardiac energy production during hypertrophy and failure.
5.2 NRF-1
Nuclear respiratory factor-1 (NRF-1) was originally identified through its binding of the cytochrome c promoter [82]. Subsequent characterization of its binding site demonstrated that NRF-1 regulates the expression of different genes encoding mitochondrial electron transport chain complex subunits [83]. Moreover, NRF-1 induces the expression of the mitochondrial transcription factor A (Tfam), that plays a role in mitochondrial DNA transcription, stabilization and maintenance [84, 85]. NRF-1 knockouts are embryonically lethal at day 6.5 and show impaired mitochondrial membrane potential as well as a decrease in the mitochondrial DNA content [86]. Interestingly, NRF-1 also induces MEF2 expression to coordinate expression of mitochondrial respiratory chain subunits [87]. Along with direct regulation of PGC-1α by MEF2A, this network provides a positive feedback loop through NRF-1, MEF2A, and PGC-1α to control mitochondrial function and content in the cardiac myocyte.
6. Implications for the development of new therapeutic approaches targeting cardiac energy metabolism
The rates of heart failure are rising as the population continues to age. There is a clear unmet medical need for new therapeutic approaches for this problem, particularly aimed at the early stages. Indeed, the prognosis for heart failure patients on current therapies remains dismal, with a 5-year mortality rate of around 50% [88]. In addition, a significant proportion of heart failure involves preserved ejection fraction (HFpEF) or so-called diastolic heart failure, for which there is no evidence-based medical treatment that improves mortality in these patients [89]. The traditional emphasis on heart failure with decreased ejection fraction and lack of reliable animal models has also limited our understanding of the metabolic derangements that occur in HFpEF. There is currently no heart failure therapeutic that directly targets metabolism or energy production.
6.1 Myocardial substrate utilization
To date, the most extensively studied and cited potential therapeutic intervention, particularly in the setting of ischemia, is to decrease mitochondrial FAO and force a shift to increased glucose utilization. This shift would lead to a more oxygen efficient state in terms of ATP production. It should be noted, however, that the utility of this approach, particularly in non-ischemic scenarios remains to be proven. Several strategies have been employed in preclinical studies including targeting free fatty acid uptake, fatty acid entry into the mitochondria or direct FAO inhibition [3]. Although certain agents have now been approved for the treatment of angina, none have progressed far for the treatment of chronic heart failure. However, some evidence suggests that partial FAO inhibitors, such as trimetazidine, have a positive effect on cardiac function and remodeling in addition to their anti-anginal activities [90]. Definitive clinical trials are needed to establish these initial findings in chronic heart failure.
Not all evidence supports the strategy to inhibit FAO. Cardiac overexpression of pyruvate dehydrogenase kinase 4 (PDK4) in transgenic mice leads to very high FAO rates and decreased glucose utilization [91]. Surprisingly, however, these mice have normal recovery following ischemia/reperfusion injury and some evidence for protecting against myocyte lipid accumulation due to a high fat diet, which would potentially be beneficial in diabetic forms of cardiac dysfunction. PGC-1α levels and target gene expression are also upregulated in these mice. These results underscore that a “one-size fits all” approach to modulating fuel utilization may not be optimal and that therapies targeting myocardial energy production will need to be tailored to the specific disease state. Moreover, strategies to increase fatty acid catabolism in heart must be matched with a corresponding increase in mitochondrial respiratory capacity. Lastly, one therapeutic approach may be well-suited for a diabetic patient with ectopic lipid accumulation and high FAO rates but ineffective or even detrimental to a patient with hypertension and a different metabolic profile.
6.2 PGC-1 as a therapeutic target to maintain or increase mitochondrial function
Given the importance of PGC-1 in the regulation of virtually all aspects of mitochondria, it is reasonable to hypothesize that strategies to increase PGC-1 levels and/or activity will increase energy production in the failing heart. However, this concept has not been definitively tested. The approach in pre-clinical models is complicated by the fact that cardiac overexpression of PGC-1α results in cardiomyopathy as a result of uncontrolled mitochondrial biogenesis [60]. To date, effective small molecule activators of PGC-1α have not been developed. However, cardiac overexpression of PGC-1β is cardioprotective in a lipopolysaccharide (LPS) challenge model [92]. Upstream modulators of PGC-1 may also serve as interventional points. Specific inhibition or activation of GCN5 and SIRT1, respectively, would be predicted to activate PGC-1. Modulation of each of these factors has been shown to increase PGC-1 activity [93, 94]. Definitive studies are needed to establish the benefit of PGC-1 activation in the setting of heart failure.
6.3 Nuclear receptor activation
Ligands for specific nuclear receptors, particularly the PPARs, have been developed and, in some cases, serve to increase the interaction with PGC-1. Data with PPARα ligands in experimental heart failure models has been mixed [95]. Clinically, PPARα ligands have a very favorable safety profile; however, they have not been specifically tested in a heart failure population. Moreover, PPARα ligands would theoretically be restricted to those patients with reduced myocardial FAO. PPARα activation may actually participate in the development of lipotoxic cardiomyopathy in the setting of obesity and diabetes. In addition, the extra-cardiac effects of PPARα activation must be considered.
As discussed above, ERRs also interact with PGC-1 to drive virtually all aspects of mitochondrial biogenesis. In contrast to the PPARs, the relatively small ligand binding domain has hindered the development of ERR activating ligands. ERRγ would appear to offer the best target in this regard.
6.3 Lipid storage pathways
Perturbations in lipid handling and storage in the cardiac myocyte may also impact energy production in the heart, especially in the cardiac dysfunction associated with obesity and diabetes. Mice deficient in stearoyl-CoA desaturase (SCD1), the rate-limiting enzyme in the biosynthesis of monounsaturated fatty acids, has been shown to decrease cardiac lipid accumulation and improve cardiac function on the leptin-deficient ob/ob background [96]. Loss of SCD1 also decreased PPARα levels and β-oxidation rates observed in ob/ob mice. Clinical development of SCD1 inhibitors, however, has been problematic due to unwanted side effects. Similar effects have been observed with deletion of diacylglycerol acyltransferase 1 (DGAT1), which catalyzes the final step in triglyceride synthesis. Cardiac triglyceride accumulation is reduced and increases in PPARα, β-oxidation, and fatty acid import genes are inhibited when DGAT1 knockout mice are placed on a high-fat diet [97]. A remaining question is the fate of the unstored fatty acids in the absence of DGAT1 activity. However, at least one study suggests that these are shuttled to the mitochondrial β-oxidation pathway [98]. Taken together, these data suggest that inhibition of cardiac lipid accumulation could serve as a promising therapeutic target in the development of lipotoxic cardiomyopathy.
7. Concluding Remarks
The healthy heart has an amazing capacity to shift substrate utilization preferences in response to pathophysiologic stimuli to meet its energy needs. This fuel utilization flexibility becomes constrained during the development of heart failure. In addition to changes in structural and contractile proteins, myocardial energy metabolism is remodeled during cardiac hypertrophy and failure. Importantly, different forms of cardiac disease, such as hypertension/ischemia versus obesity/diabetes are characterized by distinct and directionally different alterations in cardiac fuel metabolism. These changes are driven, in part, by a complex transcriptional regulatory network that responds to numerous inputs including nutrient availability and increased workload. There is a prime opportunity and medical need to develop therapeutics that directly address the underlying metabolic derangements that occur early during the development of heart failure. This could, in essence, lead to a more precise or personalized approach to this problem, and at an early stage. In the long-term, such therapies could, perhaps, be tailored to the etiology of heart failure and the accompanying metabolic derangements.
Highlights.
Cardiac hypertrophy and failure are associated with changes in energy substrate utilization
ERR and PPAR play key roles in the regulation of cardiac fuel metabolism
PGC-1 integrates multiple stimuli to control cardiac myocyte energy metabolism
Acknowledgments
This work was supported by NIH grants (RO1 DK045416, RO1 HL058493, RO1 HL101189 [D.P.K.]) and The Swiss National Science Foundation (PBLAP3 136880 [G.A]). We thank Lorenzo Thomas for help with manuscript preparation and Teresa Leone for critical reading of the manuscript.
Footnotes
Disclosures
D.P.K. serves on Scientific Advisory Boards for Johnson & Johnson, Pfizer, and Eli Lilly & Company.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ingwall JS. On the hypothesis that the failing heart is energy starved: lessons learned from the metabolism of ATP and creatine. Current hypertension reports. 2006;8:457–464. doi: 10.1007/s11906-006-0023-x. [DOI] [PubMed] [Google Scholar]
- 2.Neubauer S, Horn M, Cramer M, Harre K, Newell JB, Peters W, Pabst T, Ertl G, Hahn D, Ingwall JS, Kochsiek K. Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation. 1997;96:2190–2196. doi: 10.1161/01.cir.96.7.2190. [DOI] [PubMed] [Google Scholar]
- 3.Jaswal JS, Keung W, Wang W, Ussher JR, Lopaschuk GD. Targeting fatty acid and carbohydrate oxidation--a novel therapeutic intervention in the ischemic and failing heart. Biochimica et biophysica acta. 2011;1813:1333–1350. doi: 10.1016/j.bbamcr.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 4.Ardehali H, Sabbah HN, Burke MA, Sarma S, Liu PP, Cleland JG, Maggioni A, Fonarow GC, Abel ED, Campia U, Gheorghiade M. Targeting myocardial substrate metabolism in heart failure: potential for new therapies. European journal of heart failure. 2012;14:120–129. doi: 10.1093/eurjhf/hfr173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sack MN, Rader TA, Park S, Bastin J, McCune SA, Kelly DP. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation. 1996;94:2837–2842. doi: 10.1161/01.cir.94.11.2837. [DOI] [PubMed] [Google Scholar]
- 6.Taegtmeyer H, Golfman L, Sharma S, Razeghi P, van Arsdall M. Linking gene expression to function: metabolic flexibility in the normal and diseased heart. Annals of the New York Academy of Sciences. 2004;1015:202–213. doi: 10.1196/annals.1302.017. [DOI] [PubMed] [Google Scholar]
- 7.Bugger H, Schwarzer M, Chen D, Schrepper A, Amorim PA, Schoepe M, Nguyen TD, Mohr FW, Khalimonchuk O, Weimer BC, Doenst T. Proteomic remodelling of mitochondrial oxidative pathways in pressure overload-induced heart failure. Cardiovascular research. 2010;85:376–384. doi: 10.1093/cvr/cvp344. [DOI] [PubMed] [Google Scholar]
- 8.Hamblin M, Friedman DB, Hill S, Caprioli RM, Smith HM, Hill MF. Alterations in the diabetic myocardial proteome coupled with increased myocardial oxidative stress underlies diabetic cardiomyopathy. Journal of molecular and cellular cardiology. 2007;42:884–895. doi: 10.1016/j.yjmcc.2006.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jullig M, Hickey AJ, Middleditch MJ, Crossman DJ, Lee SC, Cooper GJ. Characterization of proteomic changes in cardiac mitochondria in streptozotocin-diabetic rats using iTRAQ isobaric tags. Proteomics Clinical applications. 2007;1:565–576. doi: 10.1002/prca.200600831. [DOI] [PubMed] [Google Scholar]
- 10.Bugger H, Chen D, Riehle C, Soto J, Theobald HA, Hu XX, Ganesan B, Weimer BC, Abel ED. Tissue-specific remodeling of the mitochondrial proteome in type 1 diabetic akita mice. Diabetes. 2009;58:1986–1997. doi: 10.2337/db09-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jullig M, Hickey AJ, Chai CC, Skea GL, Middleditch MJ, Costa S, Choong SY, Philips AR, Cooper GJ. Is the failing heart out of fuel or a worn engine running rich? A study of mitochondria in old spontaneously hypertensive rats. Proteomics. 2008;8:2556–2572. doi: 10.1002/pmic.200700977. [DOI] [PubMed] [Google Scholar]
- 12.Abel ED, Doenst T. Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovascular research. 2011;90:234–242. doi: 10.1093/cvr/cvr015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Neill BT, Kim J, Wende AR, Theobald HA, Tuinei J, Buchanan J, Guo A, Zaha VG, Davis DK, Schell JC, Boudina S, Wayment B, Litwin SE, Shioi T, Izumo S, Birnbaum MJ, Abel ED. A conserved role for phosphatidylinositol 3-kinase but not Akt signaling in mitochondrial adaptations that accompany physiological cardiac hypertrophy. Cell Metab. 2007;6:294–306. doi: 10.1016/j.cmet.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burelle Y, Wambolt RB, Grist M, Parsons HL, Chow JC, Antler C, Bonen A, Keller A, Dunaway GA, Popov KM, Hochachka PW, Allard MF. Regular exercise is associated with a protective metabolic phenotype in the rat heart. Am J Physiol Heart Circ Physiol. 2004;287:H1055–1063. doi: 10.1152/ajpheart.00925.2003. [DOI] [PubMed] [Google Scholar]
- 15.Strom CC, Aplin M, Ploug T, Christoffersen TE, Langfort J, Viese M, Galbo H, Haunso S, Sheikh SP. Expression profiling reveals differences in metabolic gene expression between exercise-induced cardiac effects and maladaptive cardiac hypertrophy. The FEBS journal. 2005;272:2684–2695. doi: 10.1111/j.1742-4658.2005.04684.x. [DOI] [PubMed] [Google Scholar]
- 16.McMullen JR, Amirahmadi F, Woodcock EA, Schinke-Braun M, Bouwman RD, Hewitt KA, Mollica JP, Zhang L, Zhang Y, Shioi T, Buerger A, Izumo S, Jay PY, Jennings GL. Protective effects of exercise and phosphoinositide 3-kinase(p110alpha) signaling in dilated and hypertrophic cardiomyopathy. Proc Natl Acad Sci U S A. 2007;104:612–617. doi: 10.1073/pnas.0606663104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Downing J, Balady GJ. The role of exercise training in heart failure. Journal of the American College of Cardiology. 2011;58:561–569. doi: 10.1016/j.jacc.2011.04.020. [DOI] [PubMed] [Google Scholar]
- 18.de las Fuentes L, Herrero P, Peterson LR, Kelly DP, Gropler RJ, Davila-Roman VG. Myocardial fatty acid metabolism: independent predictor of left ventricular mass in hypertensive heart disease. Hypertension. 2003;41:83–87. doi: 10.1161/01.hyp.0000047668.48494.39. [DOI] [PubMed] [Google Scholar]
- 19.de las Fuentes L, Soto PF, Cupps BP, Pasque MK, Herrero P, Gropler RJ, Waggoner AD, Davila-Roman VG. Hypertensive left ventricular hypertrophy is associated with abnormal myocardial fatty acid metabolism and myocardial efficiency. Journal of nuclear cardiology: official publication of the American Society of Nuclear Cardiology. 2006;13:369–377. doi: 10.1016/j.nuclcard.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 20.Dreyer C, Krey G, Keller H, Givel F, Helftenbein G, Wahli W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell. 1992;68:879–887. doi: 10.1016/0092-8674(92)90031-7. [DOI] [PubMed] [Google Scholar]
- 21.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 22.Gilde AJ, van der Lee KA, Willemsen PH, Chinetti G, van der Leij FR, van der Vusse GJ, Staels B, van Bilsen M. Peroxisome proliferator-activated receptor (PPAR) alpha and PPAR beta/delta, but not PPAR gamma, modulate the expression of genes involved in cardiac lipid metabolism. Circ Res. 2003;92:518–524. doi: 10.1161/01.RES.0000060700.55247.7C. [DOI] [PubMed] [Google Scholar]
- 23.Chakravarthy MV, Lodhi IJ, Yin L, Malapaka RR, Xu HE, Turk J, Semenkovich CF. Identification of a physiologically relevant endogenous ligand for PPAR alpha in liver. Cell. 2009;138:476–488. doi: 10.1016/j.cell.2009.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haemmerle G, Moustafa T, Woelkart G, Buttner S, Schmidt A, van de Weijer T, Hesselink M, Jaeger D, Kienesberger PC, Zierler K, Schreiber R, Eichmann T, Kolb D, Kotzbeck P, Schweiger M, Kumari M, Eder S, Schoiswohl G, Wongsiriroj N, Pollak NM, Radner FP, Preiss-Landl K, Kolbe T, Rulicke T, Pieske B, Trauner M, Lass A, Zimmermann R, Hoefler G, Cinti S, Kershaw EE, Schrauwen P, Madeo F, Mayer B, Zechner R. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1. Nat Med. 2011;17:1076–1085. doi: 10.1038/nm.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP. The cardiac phenotype induced by PPAR alpha overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109:121–130. doi: 10.1172/JCI14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park SY, Cho YR, Finck BN, Kim HJ, Higashimori T, Hong EG, Lee MK, Danton C, Deshmukh S, Cline GW, Wu JJ, Bennett AM, Rothermel B, Kalinowski A, Russell KS, Kim YB, Kelly DP, Kim JK. Cardiac-specific overexpression of peroxisome proliferator-activated receptor-alpha causes insulin resistance in heart and liver. Diabetes. 2005;54:2514–2524. doi: 10.2337/diabetes.54.9.2514. [DOI] [PubMed] [Google Scholar]
- 27.Finck BN, Han X, Courtois M, Aimond F, Nerbonne JM, Kovacs A, Gross RW, Kelly DP. A critical role for PPAR alpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proc Natl Acad Sci U S A. 2003;100:1226–1231. doi: 10.1073/pnas.0336724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang J, Sambandam N, Han X, Gross RW, Courtois M, Kovacs A, Febbraio M, Finck BN, Kelly DP. CD36 deficiency rescues lipotoxic cardiomyopathy. Circ Res. 2007;100:1208–1217. doi: 10.1161/01.RES.0000264104.25265.b6. [DOI] [PubMed] [Google Scholar]
- 29.Bernal-Mizrachi C, Weng S, Feng C, Finck BN, Knutsen RH, Leone TC, Coleman T, Mecham RP, Kelly DP, Semenkovich CF. Dexamethasone induction of hypertension and diabetes is PPAR-alpha dependent in LDL receptor-null mice. Nat Med. 2003;9:1069–1075. doi: 10.1038/nm898. [DOI] [PubMed] [Google Scholar]
- 30.Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Jeong Yun U, Cooksey RC, Litwin SE, Abel ED. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology. 2005;146:5341–5349. doi: 10.1210/en.2005-0938. [DOI] [PubMed] [Google Scholar]
- 31.Duncan JG, Fong JL, Medeiros DM, Finck BN, Kelly DP. Insulin-resistant heart exhibits a mitochondrial biogenic response driven by the peroxisome proliferator-activated receptor-alpha/PGC-1 alpha gene regulatory pathway. Circulation. 2007;115:909–917. doi: 10.1161/CIRCULATIONAHA.106.662296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Campbell FM, Kozak R, Wagner A, Altarejos JY, Dyck JR, Belke DD, Severson DL, Kelly DP, Lopaschuk GD. A role for peroxisome proliferator-activated receptor alpha (PPAR alpha) in the control of cardiac malonyl-CoA levels: reduced fatty acid oxidation rates and increased glucose oxidation rates in the hearts of mice lacking PPAR alpha are associated with higher concentrations of malonyl-CoA and reduced expression of malonyl-CoA decarboxylase. J Biol Chem. 2002;277:4098–4103. doi: 10.1074/jbc.M106054200. [DOI] [PubMed] [Google Scholar]
- 33.Leone TC, Weinheimer CJ, Kelly DP. A critical role for the peroxisome proliferator-activated receptor alpha (PPAR alpha) in the cellular fasting response: The PPAR alpha-null mouse as a model of fatty acid oxidation disorders. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:7473–7478. doi: 10.1073/pnas.96.13.7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Panagia M, Gibbons GF, Radda GK, Clarke K. PPAR-alpha activation required for decreased glucose uptake and increased susceptibility to injury during ischemia. Am J Physiol Heart Circ Physiol. 2005;288:H2677–2683. doi: 10.1152/ajpheart.00200.2004. [DOI] [PubMed] [Google Scholar]
- 35.Dewald O, Sharma S, Adrogue J, Salazar R, Duerr GD, Crapo JD, Entman ML, Taegtmeyer H. Downregulation of peroxisome proliferator-activated receptor-alpha gene expression in a mouse model of ischemic cardiomyopathy is dependent on reactive oxygen species and prevents lipotoxicity. Circulation. 2005;112:407–415. doi: 10.1161/CIRCULATIONAHA.105.536318. [DOI] [PubMed] [Google Scholar]
- 36.Goikoetxea MJ, Beaumont J, Gonzalez A, Lopez B, Querejeta R, Larman M, Diez J. Altered cardiac expression of peroxisome proliferator-activated receptor-isoforms in patients with hypertensive heart disease. Cardiovascular research. 2006;69:899–907. doi: 10.1016/j.cardiores.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 37.Karbowska J, Kochan Z, Smolenski RT. Peroxisome proliferator-activated receptor alpha is downregulated in the failing human heart. Cellular & molecular biology letters. 2003;8:49–53. [PubMed] [Google Scholar]
- 38.Bishop SP, Altschuld RA. Increased glycolytic metabolism in cardiac hypertrophy and congestive failure. The American journal of physiology. 1970;218:153–159. doi: 10.1152/ajplegacy.1970.218.1.153. [DOI] [PubMed] [Google Scholar]
- 39.Nascimben L, Ingwall JS, Lorell BH, Pinz I, Schultz V, Tornheim K, Tian R. Mechanisms for increased glycolysis in the hypertophied rat heart. Hypertension. 2004;44:662–667. doi: 10.1161/01.HYP.0000144292.69599.0c. [DOI] [PubMed] [Google Scholar]
- 40.Osorio JC, Stanley WC, Linke A, Castellari M, Diep QN, Panchal AR, Hintze TH, Lopaschuk GD, Recchia RA. Impaired myocardial fatty acid oxidation and reduced protein expression of retinoid X receptor-alpha in pacing-induced heart failure. Circulation. 2002;106:606–612. doi: 10.1161/01.cir.0000023531.22727.c1. [DOI] [PubMed] [Google Scholar]
- 41.Burkart EM, Sambandam N, Han X, Gross RW, Courtois M, Gierasch CM, Shoghi K, Welch MJ, Kelly DP. Nuclear receptors PPARbeta/delta and PPARalpha direct distinct metabolic regulatory programs in the mouse heart. J Clin Invest. 2007;117:3930–3939. doi: 10.1172/JCI32578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang P, Liu J, Li Y, Wu S, Luo J, Yang H, Subbiah R, Chatham J, Zhelyabovska O, Yang Q. Peroxisome proliferator-activated receptor {delta} is an essential transcriptional regulator for mitochondrial protection and biogenesis in adult heart. Circ Res. 2010;106:911–919. doi: 10.1161/CIRCRESAHA.109.206185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duan SZ, Ivashchenko CY, Russell MW, Milstone DS, Mortensen RM. Cardiomyocyte-specific knockout and agonist of peroxisome proliferator-activated receptor-gamma both induce cardiac hypertrophy in mice. Circ Res. 2005;97:372–379. doi: 10.1161/01.RES.0000179226.34112.6d. [DOI] [PubMed] [Google Scholar]
- 44.Ding G, Fu M, Qin Q, Lewis W, Kim HW, Fukai T, Bacanamwo M, Chen YE, Schneider MD, Mangelsdorf DJ, Evans RM, Yang Q. Cardiac peroxisome proliferator-activated receptor gamma is essential in protecting cardiomyocytes from oxidative damage. Cardiovascular research. 2007;76:269–279. doi: 10.1016/j.cardiores.2007.06.027. [DOI] [PubMed] [Google Scholar]
- 45.Son NH, Park TS, Yamashita H, Yokoyama M, Huggins LA, Okajima K, Homma S, Szabolcs MJ, Huang LS, Goldberg IJ. Cardiomyocyte expression of PPARgamma leads to cardiac dysfunction in mice. J Clin Invest. 2007;117:2791–2801. doi: 10.1172/JCI30335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Giguere V, Yang N, Segui P, Evans RM. Identification of a new class of steroid hormone receptors. Nature. 1988;331:91–94. doi: 10.1038/331091a0. [DOI] [PubMed] [Google Scholar]
- 47.Heard DJ, Norby PL, Holloway J, Vissing H. Human ERR gamma, a third member of the estrogen receptor-related receptor (ERR) subfamily of orphan nuclear receptors: tissue-specific isoforms are expressed during development and in the adult. Mol Endocrinol. 2000;14:382–392. doi: 10.1210/mend.14.3.0431. [DOI] [PubMed] [Google Scholar]
- 48.Hong H, Yang L, Stallcup MR. Hormone-independent transcriptional activation and coactivator binding by novel orphan nuclear receptor ERR3. Journal of Biological Chemistry. 1999;274:22618–22626. doi: 10.1074/jbc.274.32.22618. [DOI] [PubMed] [Google Scholar]
- 49.Kallen J, Schlaeppi JM, Bitsch F, Filipuzzi I, Schilb A, Riou V, Graham A, Strauss A, Geiser M, Fournier B. Evidence for ligand-independent transcriptional activation of the human estrogen-related receptor alpha (ERR alpha): crystal structure of ERR alpha ligand binding domain in complex with peroxisome proliferator-activated receptor coactivator-1 alpha. J Biol Chem. 2004;279:49330–49337. doi: 10.1074/jbc.M407999200. [DOI] [PubMed] [Google Scholar]
- 50.Huss JM, Torra IP, Staels B, Giguere V, Kelly DP. Estrogen-related receptor alpha directs peroxisome proliferator-activated receptor alpha signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol Cell Biol. 2004;24:9079–9091. doi: 10.1128/MCB.24.20.9079-9091.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dufour CR, Wilson BJ, Huss JM, Kelly DP, Alaynick WA, Downes M, Evans RM, Blanchette M, Giguere V. Genome-wide orchestration of cardiac functions by the orphan nuclear receptors ERR alpha and gamma. Cell Metab. 2007;5:345–356. doi: 10.1016/j.cmet.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 52.Huss JM, Imahashi K, Dufour CR, Weinheimer CJ, Courtois M, Kovacs A, Giguere V, Murphy E, Kelly DP. The nuclear receptor ERR alpha is required for the bioenergetic and functional adaptation to cardiac pressure overload. Cell Metab. 2007;6:25–37. doi: 10.1016/j.cmet.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 53.Alaynick WA, Kondo RP, Xie W, He W, Dufour CR, Downes M, Jonker JW, Giles W, Naviaux RK, Giguere V, Evans RM. ERR gamma directs and maintains the transition to oxidative metabolism in the postnatal heart. Cell Metabolism. 2007;6:13–24. doi: 10.1016/j.cmet.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 54.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 55.Lin J, Puigserver P, Donovan J, Tarr P, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1 alpha), a novel PGC-1-related transcription coactivator associated with host cell factor. Journal of Biological Chemistry. 2002;277:1645–1648. doi: 10.1074/jbc.C100631200. [DOI] [PubMed] [Google Scholar]
- 56.Andersson U, Scarpulla RC. PGC-1-related coactivator, a novel, serum-inducible coactivator of nuclear respiratory factor-1-dependent transcription in mammalian cells. Molecular and Cellular Biology. 2001;21:3738–3749. doi: 10.1128/MCB.21.11.3738-3749.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baar K, Wende AR, Jones TE, Marison M, Nolte LA, Chen M, Kelly DP, Holloszy JO. Adaptations of skeletal muscle to exercise: Rapid increase in the transcriptional coactivator PGC-1. FASEB Journal. 2002;16:1879–1886. doi: 10.1096/fj.02-0367com. [DOI] [PubMed] [Google Scholar]
- 58.Terada S, Goto M, Kato M, Kawanaka K, Shimokawa T, Tabata I. Effects of low-intensity prolonged exercise on PGC-1 mRNA expression in rat epitrochlearis muscle. Biochemical and biophysical research communications. 2002;296:350–354. doi: 10.1016/s0006-291x(02)00881-1. [DOI] [PubMed] [Google Scholar]
- 59.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 60.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim J, Wende AR, Sena S, Theobald HA, Soto J, Sloan C, Wayment BE, Litwin SE, Holzenberger M, LeRoith D, Abel ED. Insulin-like growth factor I receptor signaling is required for exercise-induced cardiac hypertrophy. Mol Endocrinol. 2008;22:2531–2543. doi: 10.1210/me.2008-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol. 2000;20:1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H, Evans RM. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell. 2003;113:159–170. doi: 10.1016/s0092-8674(03)00269-1. [DOI] [PubMed] [Google Scholar]
- 64.Hentschke M, Susens U, Borgmeyer U. PGC-1 and PERC, coactivators of the estrogen receptor-related receptor gamma. Biochemical and biophysical research communications. 2002;299:872–879. doi: 10.1016/s0006-291x(02)02753-5. [DOI] [PubMed] [Google Scholar]
- 65.Huss JM, Kopp RP, Kelly DP. Peroxisome proliferator-activated receptor coactivator-1 alpha (PGC-1 alpha) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-alpha and -gamma. Identification of novel leucine-rich interaction motif within PGC-1alpha. J Biol Chem. 2002;277:40265–40274. doi: 10.1074/jbc.M206324200. [DOI] [PubMed] [Google Scholar]
- 66.Schreiber SN, Knutti D, Brogli K, Uhlmann T, Kralli A. The transcriptional coactivator PGC-1 regulates the expression and activity of the orphan nuclear receptor estrogen-related receptor alpha (ERRalpha) J Biol Chem. 2003;278:9013–9018. doi: 10.1074/jbc.M212923200. [DOI] [PubMed] [Google Scholar]
- 67.Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, Ahmad F, Matsui T, Chin S, Wu PH, Rybkin, Shelton JM, Manieri M, Cinti S, Schoen FJ, Bassel-Duby R, Rosenzweig A, Ingwall JS, Spiegelman BM. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259–271. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 68.Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, Courtois M, Wozniak DF, Sambandam N, Bernal-Mizrachi C, Chen Z, Holloszy JO, Medeiros DM, Schmidt RE, Saffitz JE, Abel ED, Semenkovich CF, Kelly DP. PGC-1 alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005;3:e101. doi: 10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lai L, Leone TC, Zechner C, Schaeffer PJ, Kelly SM, Flanagan DP, Medeiros DM, Kovacs A, Kelly DP. Transcriptional coactivators PGC-1 alpha and PGC-1 beta control overlapping programs required for perinatal maturation of the heart. Genes Dev. 2008;22:1948–1961. doi: 10.1101/gad.1661708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sihag S, Cresci S, Li AY, Sucharov CC, Lehman JJ. PGC-1 alpha and ERR alpha target gene downregulation is a signature of the failing human heart. Journal of molecular and cellular cardiology. 2009;46:201–212. doi: 10.1016/j.yjmcc.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garnier A, Zoll J, Fortin D, N’Guessan B, Lefebvre F, Geny B, Mettauer B, Veksler V, Ventura-Clapier R. Control by circulating factors of mitochondrial function and transcription cascade in heart failure: a role for endothelin-1 and angiotensin II. Circulation Heart failure. 2009;2:342–350. doi: 10.1161/CIRCHEARTFAILURE.108.812099. [DOI] [PubMed] [Google Scholar]
- 72.Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc Natl Acad Sci U S A. 2006;103:10086–10091. doi: 10.1073/pnas.0603615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Riehle C, Wende AR, Zaha VG, Pires KM, Wayment B, Olsen C, Bugger H, Buchanan J, Wang X, Moreira AB, Doenst T, Medina-Gomez G, Litwin SE, Lelliott CJ, Vidal-Puig A, Abel ED. PGC-1beta deficiency accelerates the transition to heart failure in pressure overload hypertrophy. Circ Res. 2011;109:783–793. doi: 10.1161/CIRCRESAHA.111.243964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1 alpha. Proc Natl Acad Sci U S A. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lerin C, Rodgers JT, Kalume DE, Kim SH, Pandey A, Puigserver P. GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1 alpha. Cell Metab. 2006;3:429–438. doi: 10.1016/j.cmet.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 76.Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. The EMBO journal. 2007;26:1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Olson EN, Backs J, McKinsey TA. Control of cardiac hypertrophy and heart failure by histone acetylation/deacetylation. Novartis Foundation symposium. 2006;274:3–12. discussion 13–19, 152–155, 272–156. [PubMed] [Google Scholar]
- 78.Naya FJ, Black BL, Wu H, Bassel-Duby R, Richardson JA, Hill JA, Olson EN. Mitochondrial deficiency and cardiac sudden death in mice lacking the MEF2A transcription factor. Nat Med. 2002;8:1303–1309. doi: 10.1038/nm789. [DOI] [PubMed] [Google Scholar]
- 79.Czubryt MP, McAnally J, Fishman GI, Olson EN. Regulation of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1 alpha) and mitochondrial function by MEF2 and HDAC5. Proc Natl Acad Sci U S A. 2003;100:1711–1716. doi: 10.1073/pnas.0337639100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chang S, McKinsey TA, Zhang CL, Richardson JA, Hill JA, Olson EN. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol Cell Biol. 2004;24:8467–8476. doi: 10.1128/MCB.24.19.8467-8476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, McKinsey TA. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol. 2004;24:8374–8385. doi: 10.1128/MCB.24.19.8374-8385.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Evans MJ, Scarpulla RC. Interaction of nuclear factors with multiple sites in the somatic cytochrome c promoter. Characterization of upstream NRF-1, ATF and intron Sp1 recognition sites. Journal of Biological Chemistry. 1989;264:14361–14368. [PubMed] [Google Scholar]
- 83.Virbasius CA, Virbasius JV, Scarpulla RC. NRF-1, an activator involved in nuclear-mitochondrial interactions, utilizes a new DNA-binding domain conserved in a family of developmental regulators. Genes & Development. 1993;7:2431–2445. doi: 10.1101/gad.7.12a.2431. [DOI] [PubMed] [Google Scholar]
- 84.Fisher RP, Lisowsky T, Parisi MA, Clayton DA. DNA wrapping and bending by a mitochondrial high mobility group-like transcriptional activator protein. Journal of Biological Chemistry. 1992;267:3358–3367. [PubMed] [Google Scholar]
- 85.Parisi MA, Xu B, Clayton DA. A human mitochondrial transcriptional activator can functionally replace a yeast mitochondrial HMG-box protein both in vivo and in vitro. Molecular and Cellular Biology. 1993;13:1951–1961. doi: 10.1128/mcb.13.3.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huo L, Scarpulla RC. Mitochondrial DNA instability and peri-implantation lethality associated with targeted disruption of nuclear respiratory factor 1 in mice. Molecular and Cellular Biology. 2001;21:644–654. doi: 10.1128/MCB.21.2.644-654.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ramachandran B, Yu G, Gulick T. Nuclear respiratory factor 1 controls myocyte enhancer factor 2A transcription to provide a mechanism for coordinate expression of respiratory chain subunits. J Biol Chem. 2008;283:11935–11946. doi: 10.1074/jbc.M707389200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Roger VL. The heart failure epidemic. International journal of environmental research and public health. 2010;7:1807–1830. doi: 10.3390/ijerph7041807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. The New England journal of medicine. 2006;355:251–259. doi: 10.1056/NEJMoa052256. [DOI] [PubMed] [Google Scholar]
- 90.Zhang L, Lu Y, Jiang H, Zhang L, Sun A, Zou Y, Ge J. Additional use of trimetazidine in patients with chronic heart failure: a meta-analysis. Journal of the American College of Cardiology. 2012;59:913–922. doi: 10.1016/j.jacc.2011.11.027. [DOI] [PubMed] [Google Scholar]
- 91.Chambers KT, Leone TC, Sambandam N, Kovacs A, Wagg CS, Lopaschuk GD, Finck BN, Kelly DP. Chronic inhibition of pyruvate dehydrogenase in heart triggers an adaptive metabolic response. J Biol Chem. 2011;286:11155–11162. doi: 10.1074/jbc.M110.217349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schilling J, Lai L, Sambandam N, Dey CE, Leone TC, Kelly DP. Toll-like receptor-mediated inflammatory signaling reprograms cardiac energy metabolism by repressing peroxisome proliferator-activated receptor gamma coactivator-1 signaling. Circulation Heart failure. 2011;4:474–482. doi: 10.1161/CIRCHEARTFAILURE.110.959833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Price NL, Gomes AP, Ling AJ, Duarte FV, Martin-Montalvo A, North BJ, Agarwal B, Ye L, Ramadori G, Teodoro JS, Hubbard BP, Varela AT, Davis JG, Varamini B, Hafner A, Moaddel R, Rolo AP, Coppari R, Palmeira CM, de Cabo R, Baur JA, Sinclair DA. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012;15:675–690. doi: 10.1016/j.cmet.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Coste A, Louet JF, Lagouge M, Lerin C, Antal MC, Meziane H, Schoonjans K, Puigserver P, O’Malley BW, Auwerx J. The genetic ablation of SRC-3 protects against obesity and improves insulin sensitivity by reducing the acetylation of PGC-1 alpha. Proc Natl Acad Sci U S A. 2008;105:17187–17192. doi: 10.1073/pnas.0808207105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sarma S, Ardehali H, Gheorghiade M. Enhancing the metabolic substrate: PPAR-alpha agonists in heart failure. Heart failure reviews. 2012;17:35–43. doi: 10.1007/s10741-010-9208-0. [DOI] [PubMed] [Google Scholar]
- 96.Dobrzyn P, Dobrzyn A, Miyazaki M, Ntambi JM. Loss of stearoyl-CoA desaturase 1 rescues cardiac function in obese leptin-deficient mice. Journal of lipid research. 2010;51:2202–2210. doi: 10.1194/jlr.M003780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu L, Yu S, Khan RS, Ables GP, Bharadwaj KG, Hu Y, Huggins LA, Eriksson JW, Buckett LK, Turnbull AV, Ginsberg HN, Blaner WS, Huang LS, Goldberg IJ. DGAT1 deficiency decreases PPAR expression and does not lead to lipotoxicity in cardiac and skeletal muscle. Journal of lipid research. 2011;52:732–744. doi: 10.1194/jlr.M011395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yamamoto T, Yamaguchi H, Miki H, Kitamura S, Nakada Y, Aicher TD, Pratt SA, Kato K. A novel coenzyme A:diacylglycerol acyltransferase 1 inhibitor stimulates lipid metabolism in muscle and lowers weight in animal models of obesity. European journal of pharmacology. 2011;650:663–672. doi: 10.1016/j.ejphar.2010.10.040. [DOI] [PubMed] [Google Scholar]