

Abstract
Objectives
Differences among cancer cells within a tumor are important in tumorigenesis and treatment resistance, yet no measure of intratumor heterogeneity is suitable for routine application. We developed a quantitative measure of intratumor genetic heterogeneity, based on differences among mutated loci in the mutant-allele fractions determined by next-generation sequencing (NGS) of tumor DNA. We then evaluated the application of this measure to head and neck squamous cell carcinoma (HNSCC).
Materials and Methods
We analyzed published electronically available NGS results for 74 HNSCC. For each tumor we calculated mutant-allele tumor heterogeneity (MATH) as the ratio of the width to the center of its distribution of mutant-allele fractions among tumor-specific mutated loci.
Results
Intratumor heterogeneity assessed by MATH was higher in 3 poor-outcome classes of HNSCC: tumors with disruptive mutations in the TP53 gene (versus wild-type TP53 or non-disruptive mutations), tumors negative versus positive for human papillomavirus (even when restricted to tumors having wild-type TP53), and HPV-negative tumors from smokers with more pack-years of cigarette exposure (with TP53 status taken into account).
Conclusion
The relation of this type of intratumor heterogeneity to HNSCC outcome classes supports its further evaluation as a prognostic biomarker. As NGS of tumor DNA becomes widespread in clinical research and practice, MATH should provide a simple, quantitative, and clinically practical biomarker to help evaluate relations of intratumor genetic heterogeneity to outcome in any type of cancer.
Keywords: head and neck cancer, intratumor heterogeneity, tumor biomarkers, next-generation DNA sequencing, somatic mutations, TP53, human papillomavirus, cigarette smoking
INTRODUCTION
Differences among cancer cells within a tumor are important in disease progression, metastasis, and treatment resistance, with heterogeneous tumors more likely to have developed a subpopulation that is therapy-resistant or metastasis-prone.1, 2 A measure of this intratumor heterogeneity might provide clinically significant information. Unfortunately, the techniques used to establish the importance of intratumor heterogeneity—such as examining intratumor distribution of pre-identified markers,3–5 extensive tumor dissection,3, 4, 6, 7 isolating and analyzing individual nuclei,3, 5, 8 and ultradeep sequencing of mutations9–are difficult to translate from research studies to the clinic.
We propose a way to use results of next-generation sequencing (NGS), expected to be applied soon in clinical oncology, to obtain a measure of intratumor genomic heterogeneity. Genomically distinct subpopulations of cells in a tumor lead to differences among mutated loci in terms of the fraction of sequence reads that show a mutant allele.9 The distribution of mutant-allele fractions among loci thus provides a straightforward measure of one type of intratumor heterogeneity, called mutant-allele tumor heterogeneity (MATH). MATH represents a consequence of multiple cell populations in a tumor, while avoiding the practical and theoretical difficulties of trying to identify and enumerate them directly. Using published NGS results on head and neck squamous cell carcinoma (HNSCC),10 we show that MATH is high in each of three poor-outcome classifications of HNSCC.
MATERIALS AND METHODS
Clinical data and NGS exome-sequencing results (approximately 1% of the genome, at 150-fold mean sequence coverage) for 74 HNSCC, and data on genomic copy-number alterations (CNA) for 55 of these, were imported from Supplementary Tables 6, 10 and 11 of Stransky et al.10 into R.11 Each tumor’s MATH value was calculated from the median absolute deviation (MAD) and the median of its mutant-allele fractions at tumor-specific mutated loci:
Calculation of MAD followed the default in R, with values scaled by a constant factor (1.4826) so that the expected MAD of a sample from a normal distribution equals the standard deviation.
Following Poeta et al.,12 we scored TP53 mutations whose predicted protein products were truncated or had altered charge or polarity of an amino-acid residue in the L2 or L3 binding domains as disruptive. Splice-site mutations were also scored as disruptive. HPV status was based on reported PCR results.10 Significance was taken at p < 0.05 in 2-sided tests.
RESULTS
Calculating MATH from NGS results
MATH is the ratio of the width to the center of the distribution of mutant-allele fractions among tumor-specific mutated loci. The basic idea is illustrated in Fig. 1A for an idealized situation with heterozygous loci, no CNA, and no normal tissue. A heterogeneous tumor will tend to have a wider distribution of mutant-allele fractions among loci, centered at a lower fraction, than a homogeneous tumor. The width of the distribution captures diversity among loci arising from different cell populations. Taking the ratio of the width to the center provides a first-order correction for the presence of normal tissue in the tumor, because the multiplicative factor correcting total cell numbers for normal-cell numbers appears in both the numerator and the denominator. Robust measures of the width and the center, the MAD and median, are used to minimize the influence of outlier loci (e.g., low-fraction loci with less precise values, and high-fraction loci common to most cell populations or with CNA favoring the mutant allele). Supplementary Text provides details on the relation of distribution width to patterns of mutation sharing among cell populations and CNA, the use of robust measures, and how the ratio of MAD to median corrects for normal tissue.
Fig. 1. Mutant-allele tumor heterogeneity.

A, Heterogeneity among mutant-allele fractions. Top, Three heterozygous loci (mutant alleles: lower-case a, b, c) in an idealized homogeneous tumor (left) or in a tumor having 2 populations (40/60 cell ratio, right), processed for sequencing. Middle, Mutant-allele fractions in DNA are 0.5 for all loci from the homogeneous tumor (left) or loci with shared mutations in the heterogeneous tumor (right), versus one-half of the cell fraction for population-specific mutations. Bottom, sampling in single-molecule sequencing smoothes the distributions, illustrated for 100 sequence reads at each of many such loci. The distribution of mutant-allele fractions from the heterogeneous tumor is wider (larger MAD) and has a lower median than for the homogeneous tumor. The ratio of the width to the center of the distribution (MATH = 100 * MAD/median) is thus larger for the heterogeneous tumor. B, Distributions of mutant-allele fractions among tumor-specific mutated loci for 3 HNSCC having similar numbers of mutated loci (top to bottom: 98, 102, 96 loci). Circles are mutant-allele fractions for individual loci. Curves are kernel density estimates from the R densityplot function (lattice package; default parameter values), with y-axes scaled so that the area under each curve equals 1. Each tumor’s MATH value is shown with a standard deviation (SD) estimated by bootstrap resampling; see text. C. Lack of relation between MATH and the number of tumor-specific mutations, a measure of overall mutation rate. Kendall rank correlation, p = 0.21. D, Relation of bootstrap-estimated SD of MATH determinations to the number of mutated loci among 74 HNSCC. r2 = 0.86.
MATH ranged from 19 to 55 (dimensionless units) among 74 HNSCC. Distributions of mutant-allele fractions and the corresponding MATH values are shown for 3 cases in Fig. 1B. CNA data available for a subset of 55 samples showed little influence of CNA on MATH. More than 90% of mutated loci had genomic copy numbers within 0.5 log2 units of normal, so that MATH values were similar whether calculated from all mutated loci (as in data presented here), only from loci having low CNA, or from mutant-allele fractions corrected for local CNA (Supplementary Text; Supplementary Figs. S1, S2; Supplementary Table S1).
MATH and mutation rate
Although intratumor heterogeneity and mutation rate are different concepts, it was possible that tumors with high mutation rates would simply have greater intratumor heterogeneity. This was not the case. MATH was not related to overall mutation rate, conventionally expressed as the number of mutated loci per MB of sequenced DNA,10 as illustrated by the 3 examples in Fig. 1B and by a plot of MATH versus the number of mutated loci (Fig. 1C). MATH thus represents a different aspect of tumor biology than the mutation rate itself.
As MATH is calculated from single-molecule DNA sequence reads, the precision of MATH values depends on sampling of loci and of mutant vs. reference alleles. We estimated the associated standard deviation (SD) of the MATH value for each tumor by bootstrap resampling from all sequence reads at mutated loci (median, 12,600 reads per tumor). At the median of 92 mutated loci per sample, the SD was 4 units; the SD decreased with the square root of the number of mutated loci (Fig. 1D). Thus the precision of determining MATH is better in samples with higher mutation rates, even though MATH values themselves are not related to mutation rate.
High intratumor heterogeneity in 3 poor-outcome classifications of HNSCC
Using MATH as a measure of intratumor heterogeneity, we examined the relationship of heterogeneity to 3 variables whose importance has been established clinically in HNSCC: disruptive TP53 mutations,12, 13 human papillomavirus (HPV) status,14, 15 and increasing exposure to cigarette smoke.16
First, we examined the relation of intratumor heterogeneity to mutations in the TP53 gene. The role of the p53 protein as guardian of the genome17 suggests a general hypothesis that TP53 mutations would lead to increased intratumor heterogeneity. Nevertheless, the multiple roles of p53 and the different functional consequences of different types of mutations18 mean that not all TP53 mutations might influence heterogeneity. In HNSCC, only a subset of TP53 mutations, called “disruptive,”12 is related to worse outcome, while patients with non-disruptive mutations have similar outcomes as those with wild-type TP53.12, 13 We thus tested the hypothesis that disruptive rather than non-disruptive TP53 mutations are associated with greater intratumor genetic heterogeneity in HNSCC.
Consistent with this hypothesis, disruptive mutations in TP53 were specifically related to higher MATH values. MATH was higher in HNSCC having disruptive TP53 mutations than in those with non-disruptive mutations (p = 0.038) or wild-type sequence (p = 0.008), but did not differ between tumors having non-disruptive mutations and wild-type TP53 (p = 0.93; Fig. 2).
Fig. 2. Relation of MATH to TP53 mutation status in HNSCC.
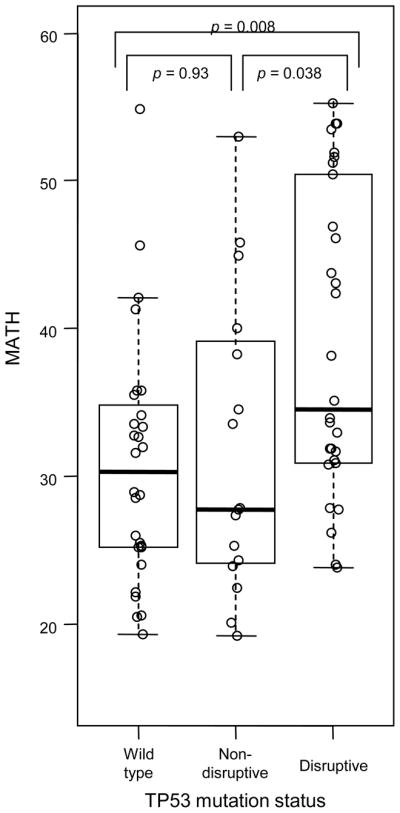
Boxplots (thick horizontal lines, medians; whiskers to farthest points within 1 interquartile range of box) and individual MATH values (circles) for HNSCC grouped by TP53 mutation status. p-values are for Wilcoxon rank-sum tests.
Second, we examined the relation of intratumor heterogeneity to HPV status. HPV infection contributes to a large and growing number of HNSCC cases,15 with HPV-positive HNSCC usually discovered at a younger patient age, having a lower overall mutation rate,10 exhibiting fewer large-scale genomic copy-number changes,19 and typically having better outcomes than HPV-negative tumors.14, 15 With the p53 and p16-pRb tumor suppressor pathways inactivated by HPV gene products,20 somatic mutations silencing these pathways are not needed before development of invasive tumors. The less extensive history of mutation and selection in HPV-positive than in HPV-negative tumors at time of presentation leads to the hypothesis that HPV-positive tumors would have less intratumor heterogeneity.
Consistent with this hypothesis, MATH was lower in HPV-positive than in HPV-negative cases (p = 0.011; Fig. 3, left versus center). To rule out a simple explanation based on the lack of TP53 mutations in these HPV-positive tumors,10 we also restricted analysis to tumors having wild-type TP53. There was still a significant difference in MATH between HPV-positive and HPV-negative cases having wild-type TP53 (p = 0.047; Fig. 3, center versus right).
Fig. 3. Relation of MATH to HPV status in HNSCC.
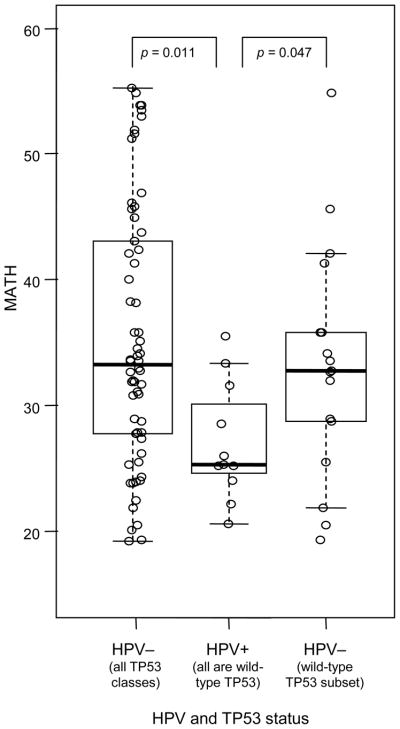
Boxplots and individual MATH values as in Fig. 2 for HNSCC grouped by HPV status. All HPV-negative cases on the left; HPV-negative cases having wild-type TP53 on the right.
Third, we examined the relation of intratumor heterogeneity to cigarette use. Exposure to cigarette smoke not only is a mutagenic risk factor for HNSCC but also is associated with worse outcome following therapy, with each pack-year of cigarette use increasing the hazard ratio for relapse or death by 1%.16 Increased exposure to mutagens as measured by pack-years, generating clonal subpopulations of cells thought to underlie progression and field cancerization in HPV-negative HNSCC,21 would be predicted to lead to greater intratumor heterogeneity.
Consistent with this hypothesis, MATH was significantly associated with cigarette pack-years, among smokers having HPV-negative tumors, when TP53 mutation status was taken into account (Table 1). Each 10 extra pack-years was associated with an increase of 1.1 MATH units in a tumor.
Table 1. Relation of MATH to disruptive TP53 mutation and pack-years in cigarette smokers with HPV-negative HNSCC.
Bivariate linear model of MATH against disruptive TP53 mutation status (versus any other TP53 status) and cigarette pack-years (as continuous variable) for 51 HPV-negative tumors from cigarette smokers. F-test, p = 0.004.
| MATH relation to: | Coefficient | p-value |
|---|---|---|
| Disruptive TP53 | 7.64 | 0.004 |
| Pack-years | 0.11/yr | 0.033 |
Note that for classifications of HNSCC by each of these clinical variables, the class having the highest heterogeneity as assessed by MATH values was also the class associated with the worst outcome. Intratumor heterogeneity as measured by MATH values thus is associated with clinically significant aspects of tumor biology, consistent with worse outcomes for more-heterogeneous tumors, although a relation of MATH to patient outcomes within each of these classifications remains to be established.
DISCUSSION
The relation of MATH to clinically important variables and the simplicity of calculating MATH from NGS results support evaluation of MATH as a biomarker of intratumor heterogeneity in studies of treatment outcomes, both in HNSCC and in other cancers. In contrast to the extensive tumor dissection or single-cell analysis used in studies that identify cell populations, MATH has a unique combination of advantages for routine application. It does not require fresh tissue; it is a quantitative measure available whenever tumor and matched normal DNA can be sequenced by next-generation methods. Unlike methods that analyze pre-selected markers, MATH is based on whatever mutations NGS identifies, at a genomic resolution much finer than provided by karyotyping, CGH, or SNP arrays. MATH implicitly incorporates CNA and includes an inherent correction for the normal tissue inevitably present in a tumor.
Future work will need to establish the relation of MATH scores to individual patient outcomes, not just to outcome classes, in survival models that take into account the diversity of tumor types and treatment regimens in HNSCC. (Clinical treatments and outcomes were not published for the data set examined here.10) For even if high intratumor heterogeneity is generally associated with worse outcomes, the specific relation of high heterogeneity to outcome may differ among combinations of tumor types with surgical, radiological, or chemotherapeutic treatments. The NGS data being collected for the Tumor Cancer Genome Atlas project22 might be used for retrospective analysis of MATH in HNSCC, provided that clinical annotations are sufficiently detailed. Prospectively, MATH could be included as a candidate biomarker in clinical studies and trials. MATH could similarly be evaluated in cancers other than HNSCC. Translational research that combines analysis of MATH with studies on the mechanisms that lead to intratumor heterogeneity may point the way to clinical strategies that specifically target heterogeneous tumors.
Our results with this HNSCC data set provide important guidance for such work on evaluating and extending MATH as a prognostic biomarker. First, the relation of the precision of MATH values to the number of mutated loci (Fig. 1D) means that broader genome coverage than provided by exome sequencing, such as targeted capture of non-exonic sequences or whole-genome sequencing, might improve heterogeneity analysis of HPV-positive OPSCC, which typically have low numbers of mutations,10 or of cancers that have lower mutation rates than HNSCC. Second, given the difficulties in distinguishing true mutations with low mutant-allele fractions from sequence-read errors, the criteria for identifying tumor-specific mutated loci must be consistent among tumors whose MATH values are being compared. Third, although explicitly including information on locus-specific CNA did not provide advantages over MATH calculated directly from mutant-allele fractions in these HNSCC (Supplementary Table S1), this issue must be reassessed in future applications.
With its emphasis on overall genetic diversity regardless of which genes are mutated, MATH should provide an important complement to analysis of mutations or expression of specific genes in tumors. The heterogeneity measured by MATH could also be much more consistent among different portions of a tumor than are the sets of mutated loci themselves. For example, although 2 regions of a single renal-cell carcinoma sequenced in depth by Gerlinger et al.7 differed substantially in terms of specific mutated genes, the MATH values of these regions were highly similar (based on loci with mutant-allele fractions > 0.01 in their Supplementary Table 3, MATH values were: region R4, 25.6; region R9, 25.9). Thus assessment of MATH might be less sensitive to the tumor-sampling problems that can fundamentally limit identification of specific gene profiles,7 and should allow assessment of heterogeneity per se separately from mutation or expression profiles as risk factors. We suspect that tumors with high MATH, as an overall measure of genetic diversity, will be those most likely to harbor clinically significant subpopulations of cells even if those populations are not directly represented in the analyzed sample.
Direct comparison of MATH with individual outcomes following uniform treatment will ultimately determine whether this measure of intratumor genetic heterogeneity provides a clinically useful biomarker in HNSCC and other cancer types. For example, tumors with higher MATH may have more diverse populations of cells prone to metastasis and treatment failure than their less-heterogeneous counterparts. Similarly, genetically diverse tumors with higher MATH may be those most likely to exhibit a poor response or relapse earlier following targeted therapy. Analysis of MATH will be straightforward in these studies, as NGS of tumors becomes more widespread in research and clinical practice.
Supplementary Material
Acknowledgments
Funding Sources: The Norman Knight Fund, the Flight Attendant Medical Research Institute, the National Institute of Dental and Craniofacial Research (R01 DE022087), and the National Cancer Institute (R21 CA119591).
Role of the funding sources: financial support only.
Footnotes
Suggestions for reviewers:
Robert L. Ferris, MD, PhD, University of Pittsburgh, ferrisrl@upmc.edu
Jeffrey N Myers, MD, PhD, MD Anderson Cancer Center, jmyers@mdanderson.org
Ezra Cohen, MD, The University of Chicago, ecohen@medicine.bsd.uchicago.edu
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Salk JJ, Fox EJ, Loeb LA. Mutational heterogeneity in human cancers: origin and consequences. Annu Rev Pathol. 2010;5:51–75. doi: 10.1146/annurev-pathol-121808-102113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12(5):323–334. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 3.Maley CC, Galipeau PC, Finley JC, Wongsurawat VJ, Li X, Sanchez CA, et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet. 2006;38(4):468–473. doi: 10.1038/ng1768. [DOI] [PubMed] [Google Scholar]
- 4.Jovanovic L, Delahunt B, McIver B, Eberhardt NL, Grebe SK. Most multifocal papillary thyroid carcinomas acquire genetic and morphotype diversity through subclonal evolution following the intra-glandular spread of the initial neoplastic clone. J Pathol. 2008;215(2):145–154. doi: 10.1002/path.2342. [DOI] [PubMed] [Google Scholar]
- 5.Park SY, Gonen M, Kim HJ, Michor F, Polyak K. Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype. J Clin Invest. 2010;120(2):636–644. doi: 10.1172/JCI40724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467(7319):1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472(7341):90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486(7403):395–399. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333(6046):1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.R Core Team. [Accessed on Sept. 7, 2012.];R: A Language and Environment for Statistical Computing. http://www.r-project.org.
- 12.Poeta ML, Manola J, Goldwasser MA, Forastiere A, Benoit N, Califano JA, et al. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N Engl J Med. 2007;357(25):2552–2561. doi: 10.1056/NEJMoa073770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skinner HD, Sandulache VC, Ow TJ, Meyn RE, Yordy JS, Beadle BM, et al. TP53 disruptive mutations lead to head and neck cancer treatment failure through inhibition of radiation-induced senescence. Clin Cancer Res. 2012;18(1):290–300. doi: 10.1158/1078-0432.CCR-11-2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fakhry C, Westra WH, Li S, Cmelak A, Ridge JA, Pinto H, et al. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J Natl Cancer Inst. 2008;100(4):261–269. doi: 10.1093/jnci/djn011. [DOI] [PubMed] [Google Scholar]
- 15.Chaturvedi AK, Engels EA, Pfeiffer RM, Hernandez BY, Xiao W, Kim E, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol. 2011;29(32):4294–4301. doi: 10.1200/JCO.2011.36.4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. 2010;363(1):24–35. doi: 10.1056/NEJMoa0912217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358(6381):15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 18.Xu Y. Induction of genetic instability by gain-of-function p53 cancer mutants. Oncogene. 2008;27(25):3501–3507. doi: 10.1038/sj.onc.1211023. [DOI] [PubMed] [Google Scholar]
- 19.Smeets SJ, Braakhuis BJ, Abbas S, Snijders PJ, Ylstra B, van de Wiel MA, et al. Genome-wide DNA copy number alterations in head and neck squamous cell carcinomas with or without oncogene-expressing human papillomavirus. Oncogene. 2006;25(17):2558–2564. doi: 10.1038/sj.onc.1209275. [DOI] [PubMed] [Google Scholar]
- 20.Marur S, D’Souza G, Westra WH, Forastiere AA. HPV-associated head and neck cancer: a virus-related cancer epidemic. Lancet Oncol. 2010;11(8):781–789. doi: 10.1016/S1470-2045(10)70017-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Califano J, van der Riet P, Westra W, Nawroz H, Clayman G, Piantadosi S, et al. Genetic progression model for head and neck cancer: implications for field cancerization. Cancer Res. 1996;56(11):2488–2492. [PubMed] [Google Scholar]
- 22.National Cancer Institute. The Cancer Genome Atlas. [Accessed on Sept. 7, 2012.];Head and Neck Squamous Cell Carcinoma. http://cancergenome.nih.gov/cancersselected/headandneck.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.