

Abstract
Background
The von Willebrand factor (VWF) gene is highly polymorphic, with variants correlated with VWF antigen levels, adhesion activity, clearance, and factor VIII binding. VWF mutations are detected in patients with von Willebrand disease (VWD), whereas polymorphic variants could be associated with thrombosis. However, information on the ethnic diversity of VWF variants and their association with diseases is limited.
Objectives
To characterize novel VWF variants from different ethnicities in the general population.
Patients/Methods
We analyzed samples from 1,092 subjects of 14 ethnicities available in the 1000 Genomes database for VWF variants and their potential functional impacts.
Results
We identified 2,728 SNPs and 91 insertions and deletions that had a high level of ethnic diversity, with Africans having the highest number of variants. The highest level of diversity was found in the D′ and D2 domains. Among 94 non-synonymous variants, 31 were predicted to be deleterious, including 19 that were previously associated with VWD. Most of these “VWD variants” had allele frequencies consistent with disease incidence in European subjects; but some had a significantly higher frequency in other ethnicities. The mutation R2185Q, H817Q and M740I associated with type 1 and type 2N VWD were present in more than 13% of African subjects.
Conclusions
These results highlight the complexity of VWF variations in different ethnic groups and emphasize the importance of interrogating variations on multiple ethnic backgrounds for associations with bleeding and thrombosis.
Keywords: VWF variants, VWD, ethnic diversity, 1000 Genomes Project
Introduction
Von Willebrand factor (VWF) affixed to the subendothelium mediates the initial tethering of platelets at sites of vessel injury. VWF is initially synthesized as a peptide precursor of 2,813 amino acids with well-defined domains in the order of D1-D2-D′-D3-A1-A2-A3-D4-B1-B2-C1-C2-CK. The N-terminal D domains are critical for VWF multimerization (D1-2, and D′) and contain the binding site for coagulation factor VIII (D′ and D3 domains) [1]. Binding sites for the platelet GP Ib-IX-V complex and integrin αIIbβ3 are located in the A and C domains, respectively. The absence or a low level of VWF and/or adhesive activity are associated with bleeding found in patients with von Willebrand disease (VWD) [2, 3], whereas an elevated level of VWF and/or enhanced adhesion activity is a well-established risk factor for thrombotic diseases such as myocardial infarction and stroke [4–7]. VWF also contributes to the development of atherosclerosis, especially at sites of vascular bifurcations, where blood flow is turbulent [8]. This bidirectional activity suggests that VWF is tightly regulated in order to achieve efficient hemostasis without promoting thrombosis.
The VWF gene spans ~ 180 kilobases on Chromosome 12 [9, 10] with 52 exons. Changes in the VWF gene could alter VWF biosynthesis, secretion, clearance, and adhesion activity. The ISTH-SSC VWF Online Database (http://www.vwf.group.shef.ac.uk/vwd.html) lists ~500 mutations and polymorphic variants that were associated with VWD [11]. Single nucleotide polymorphisms (SNPs) in exons, 5′ regulatory region, and introns are also reported to influence levels of VWF antigen and FVIII activity in healthy subjects [12–15]. Some of these VWF SNPs are associated with an elevated risk for thrombosis [5, 16].
Previous studies have found a large number of polymorphisms at the VWF locus [17, 18], primarily from individuals with European ancestries. A recent study by Bellissimo, D.B. et al. [19] showed that mutations previously considered to be causative for VWD in European subjects have allele frequencies up to 20% in African Americans. These variants are possible false positive in VWD associations from the initial scans, but could also be pathogenic in Europeans while non-pathogenic in other ethnicities [19]. Studying genetic variants in multiple populations is a powerful approach to enrich the number of polymorphisms (particularly rare alleles with frequency less than 1%). Ethnicity has been increasingly recognized as having a confounding effect on VWF expression, making genotypic and phenotypic association more complex [5–7]. Genetic diversity shaped by population demographics and environmental covariates should be taken into consideration in the identification and interpretation of pathogenic mutations. With the rapid dissemination of next generation sequencing (NGS) technologies, interrogating genetic polymorphisms in a large number of samples from diverse ethnic background becomes feasible [20–22]. Using this new technology, the ongoing 1000 Genomes Project (1000G) examined genomic variations among 14 ethnicities from four continents: Africa, America, Asia, and Europe [20–22]. More than 38 million genetic variants were detected in the genomes of 1,092 subjects 14 ethnicities around the world in the Phase 1 Project [23], a significant enrichment than the previous public database. Our study presents the characterization of allelic diversity at the VWF gene. This study can detect SNPs at 0.1% frequency with the power of 90% in the exome and nearly 70% across the genome [23], and allows us uncover rare ethnic-specific variants and provide new insights into the ethnic diversity of the VWF gene.
Materials and methods
1000G samples and variant datasets
We obtained VWF variants from April 2012 Integrated Variant Set release of the 1000 Genomes Project (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20110521/ALL.wgs.phase1_release_v3.20101123.snps_indels_sv.sites.vcf.gz) from 1,092 subjects of 14 ethnicities originated from four continents (Table S1, http://1000genomes.org). The 14 ethnicities are: Yoruba in Ibadan, Nigeria(YRI); African Americans in Southwest US (ASW); Luhya in Webuye, Kenya (LWK); Mexican Ancestry in Los Angeles (MXL); Colombian in Medellin Colombia (CLM); Puerto Rican in Puerto Rico (PUR); Han Chinese in Beijing (CHB); Han Chinese in South China (CHS); Japanese in Tokyo (JPT); Utah residents with Northern and Western European ancestry (CEU); Finish from Finland (FIN); English from Great Britain (GBR); Iberian in Spain (IBS), and Toscani in Italy (TSI). The exonic regions of the genome were captured and sequenced at a high coverage rate (average > 50X). The whole genome was shotgun sequenced at a low coverage rate (~2–6X). The false discovery rate (FDR) was estimated to be 1.6% for exonic SNPs, 1.8% for non-coding SNPs and <5% for indels [23].
Removal of false positive variants in the region of the VWF pseudogene
A region spanning from the position 17,161,397 to 17,185,967 on Chromosome 22 (Build 37) contains the VWF pseudogene, VWFP, and a portion of the TP TEP1 gene. This region has 97% sequence homology with exon 23–34 of the VWF gene. To remove potential influence of this homologous sequence on variations in the VWF gene, we first aligned the homologous sequences from the two regions in chromosome 12 and chromosome 22 using CLC Sequence Viewer 6.0.2. We then aligned variants in the VWF gene to the corresponding positions on the VWF pseudogene. We removed any variant that was a reference allele of the VWF pseudogene in the corresponding positions to the VWF gene, or whose origin couldn’t be identified because of identical flanking sequences for the VWF gene and VWF pseudogene. A total of 104 variants that met these criteria were considered to be potentially derived from the VWF pseudogene and removed from further analysis. The VWF variants calling file that removed possible false positive variants could be accessed at http://www.hgsc.bcm.tmc.edu/ftp-archive/VWFVariantsStudy/.
Annotation for genetic variants and data analysis
We applied ANNOVAR [24] to annotate VWF SNPs and insertions/deletions (indels). The reference genome we used was NCBI human genome build 37. The VWF gene spans from position 6,058,040 to 6,233,836 on Chromosome 12. The 5′ and 3′ un-translated regions are 255bp and 141bp, respectively. Variants that were not present in dbSNP129 before the entry of SNPs from the Genomes pilot project and phase 1 project were reported as novel.
Genetic characterization of VWF gene in different populations
The Principal Component Analysis (PCA) that summarizes high-dimensional genetic variation data to infer population structures [25] was applied to characterize the distribution of VWF variations among different ethnic groups. The frequencies of all variants in different ethnicities were calculated. The input was the frequency of variants in different ethnicities. The top two PCs were extracted for analysis.
DnaSP software was used to determine the nucleotide diversity (pi) for different VWF domains in different populations [26, 27]. where xi and xj are the respective frequencies of the ith and jth sequences, and piij is the number of nucleotide differences per nucleotide site between the ith and jth sequences [27].
Conservation scores of nucleotides in the VWF gene were from http://hgdownload.cse.ucsc.edu/goldenPath/hg19/phyloP46way/placentalMammals/. An average conservation score was calculated in order to evaluate the degree of conservation for different domains. A Chi-squared test was used to compare the distribution of allele frequencies.
Haplotype construction
The phased 1000G data VCF files (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20110521/ALL.wgs.phase1_release_v3.20101123.snps_indels_sv.sites.vcf.gz) were converted into an input format that was proper for Haploview [28]. Haploview was applied to visualize linkage disequilibrium (LD) patterns of the VWF gene in subjects of different ethnicities. LD reflects correlation among neighboring alleles to be transmitted together in a set of linked alleles [20]. LD patterns were visualized using the following settings that (1) ignored the pairwise comparison of markers that were more than 500 kb apart, (2) examined alleles found 5% or more in the population, (3) removed markers with Hardy-Weinberg disequilibrium (Fisher’s exact test, P<0.001). LD blocks were defined using a confidence interval method.
Results
Site frequency spectrum of VWF variants
We identified 2,728 SNPs and 91 indels in the VWF gene after removal of variants that are potentially from the VWF pseudogene. All of the 91 indels were in intronic regions. Among the VWF SNPs, 2,573 were intronic (94.3%), 146 exonic (5.4%), 2 in the 5′ untranslated regions (UTR) (0.07%), and 7 at splice sites (0.26%, 6 are within coding regions) (Table 1). The majority (75.1%) was defined as novel (95.9% and 64.1% of variants in the MAF<0.01 and MAF=0.01–0.1, respectively, Figure 1). These variants have not been interrogated by previous VWF related functional studies, demonstrating the value of using the 1000G for enriching the polymorphism knowledge of particular genes of interest.
Table 1.
VWF Variants in 1000G Subjects
| Variants Type | Number of Variants | Shared (%with MAF<0.01) | ||||
|---|---|---|---|---|---|---|
| AFR* (n=246) (%AFR specific) | AMR (n=181) (%AMR specific) | ASN (n=286) (%ASN specific) | EUR (n=379) (%EUR specific) | Overall (n=1092) (%with MAF<0.01) | ||
| Silent | 32 (43.7) | 23 (13.0) | 20 (45.0) | 21 (23.8) | 55 (73.7) | 8 (0) |
| Missense | 47 (63.8) | 36 (41.7) | 19 (73.7) | 23 (47.8) | 91 (85.3) | 4 (0) |
| Intronic | 1816 (32.4) | 1395 (9.6) | 913 (31.1) | 1127 (19.3) | 2573 (64.1) | 541 (2.8) |
| Splice | 4 (50.0) | 4 (25.0) | 2 (50.0) | 2 (0) | 7† (85.7) | 1 (0) |
| UTR-5 | 1 (0) | 1 (0) | 0 | 1 (100.0) | 2 (100) | 0 (0) |
| Indels | 86 (2.3) | 86 (0) | 57(1.8) | 70 (0) | 91 (19.8) | 52 (7.7) |
| Total | 1986 (32.1) | 1545 (9.9) | 1011 (30.6) | 1244 (18.9) | 2819 (63.6) | 606 (3.1) |
AFR: Africa, AMR: America, ASN: Asia, and EUR: Europe.
Six of the seven variants at splice sites were exonic ones.
Figure 1. Distribution of novel and known SNPs based on minor allele frequency.
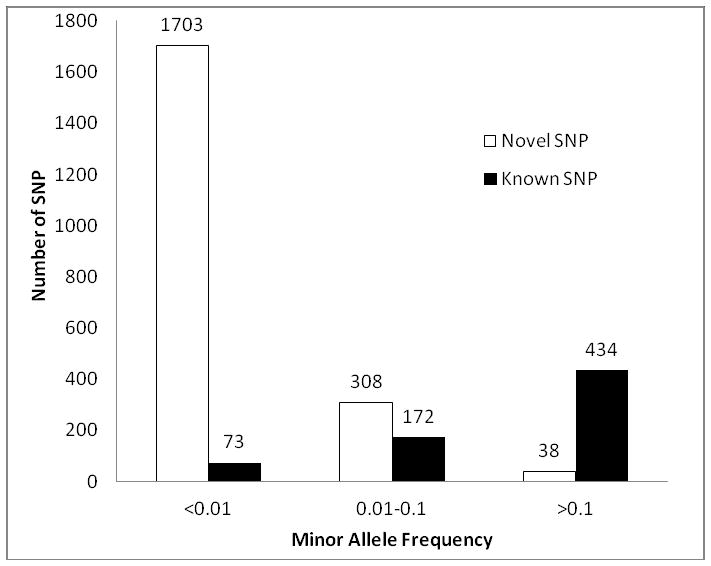
We defined novel VWF SNPs as those that were not present in the dbSNP129 data set before the entry of 1000G data.
When data were stratified into four continental groups, we identified 1,986, 1,545, 1,011 and 1,244 variants in subjects from Africa, America, Asia, and Europe, respectively (Table 1). As expected, Africans harbor more genetic polymorphisms compared to non-Africans across all annotation categories. The proportion of variants shared among populations varied by annotation categories, ranging from 4.4% (4/91) for missense SNPs to 21.0% (541/2573) for intronic SNPs (Table 1). Most of the shared variants had an allele frequency that was higher than 1% (Table 1). Compared to SNPs, 57.1% of indels were shared among populations. This high sharing rate could be attributed to a more stringent method that filtered variants with a minor allele frequency of < 0.5%.
Overall, 45.7% (1,288/2,819) of the variants were specific to one of the four populations, where Africa and Asia had the highest proportion of missense SNPs that were specific to the two continents: 63.8% and 73.7% respectively.
Functional impacts of VWF variants by computation
Ninety-one of the 152 exonic SNPs were non-synonymous (59.9%), 55 were synonymous (36.2%) and 6 were at a splicing site (3.9%) (Table 1, and see Table S2 for a detailed list). We further analyzed their potential for functional impacts using two widely used in silico approaches: the SIFT [29] and Polyphen-2 [30]. The former [29] identifies critical amino acids by their conservation in a specific protein family, whereas the latter [30] compares the difference in a protein structure between a wild-type and variants. For the 91 non-synonymous and three coding splicing site non-synonymous SNPs, 36 were predicted to be deleterious by SIFT, 44 by PolyPhen-2, and 31 by both models (Table 2, see Table S2 for the full list of predictions on the 152 variants). When closely examined, the 31 variants that were predicted deleterious by both programs were distributed in all domains except the A1 and CK domains. After correction for domain sizes, no particular clustering pattern was found (data not shown).
Table 2.
Deleterious Mutations Predicted by SIFT and PolyPhen-2 Models†
| Position | Exon | Domain | Alleles | Amino Acids Changes | Minor Allele Frequency (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| AFR (n=246) | AMR (n=181) | ASN (n=286) | EUR (n=379) | Overall (n=1092) | |||||
| 6219687 | 5 | D1 | G/T | L129M* | 1.42 | 0 | 0 | 0 | 0.32 |
| 6219557 | 5 | D1 | C/T | D172G | 0 | 0 | 0.17 | 0 | 0.05 |
| 6184558 | 7 | D1 | A/G | R273W* | 0 | 0.28 | 0 | 0 | 0.05 |
| 6182865 | 8 | D1 | C/G | S306C | 0.2 | 0 | 0 | 0 | 0.05 |
| 6182850 | 8 | D1 | A/G | T311I | 0 | 0.28 | 0 | 0 | 0.05 |
| 6182808 | 8 | D1 | A/C | C325F | 0.2 | 0 | 0 | 0 | 0.05 |
| 6181569 | 9 | D1 | A/G | T346I | 2.03 | 0 | 0 | 0 | 0.46 |
| 6174374 | 11 | D2 | C/T | G408R | 0.2 | 0 | 0 | 0 | 0.05 |
| 6174350 | 11 | D2 | A/G | R416W | 0 | 0.28 | 0 | 0 | 0.05 |
| 6172212 | 13 | D2 | A/G | R481C | 0 | 0 | 0.17 | 0 | 0.05 |
| 6153591 | 18 | D′ | A/G | P770S | 0 | 0 | 0.35 | 0 | 0.09 |
| 6153587 | 18 | D′ | A/G | M771T | 0.2 | 0 | 0 | 0 | 0.05 |
| 6143965 | 20 | D′ | C/G | C858W | 0 | 0 | 0.17 | 0 | 0.05 |
| 6140615 | 21 | D3 | C/T | G939R | 0.2 | 0 | 0 | 0 | 0.05 |
| 6132811 | 25 | D3 | A/G | T1122M | 0 | 0 | 0.52 | 0 | 0.14 |
| 6127837 | 28 | A2 | A/G | R1583W* | 0 | 0.28 | 0 | 0 | 0.05 |
| 6127833 | 28 | A2 | C/T | Y1584C* | 0 | 0.55 | 0 | 0.53 | 0.27 |
| 6122775 | 32 | A3 | C/T | Y1831C | 0 | 0 | 0 | 0.13 | 0.05 |
| 6103699 | 36 | D4 | C/G | I2046M | 0 | 0.28 | 0 | 0 | 0.05 |
| 6103650 | 36 | D4 | A/G | P2063S* | 0 | 0.83 | 0 | 0.79 | 0.41 |
| 6103587 | 36 | D4 | C/G | L2084V | 0 | 0 | 0 | 0.13 | 0.05 |
| 6103202 | 37 | D4 | A/G | L2142F | 0 | 0 | 0.35 | 0 | 0.09 |
| 6094771 | 39 | D4 | A/G | R2287W* | 2.24 | 0 | 0 | 0 | 0.50 |
| 6094250 | 40 | B1 | A/G | R2313C | 0 | 0.28 | 0 | 0 | 0.05 |
| 6091089 | 42 | B2 | A/G | R2384W | 0 | 0.55 | 0 | 0 | 0.09 |
| 6085388 | 43 | C1 | A/C | Q2442H | 0 | 0 | 0.52 | 0 | 0.14 |
| 6085353 | 43 | C1 | G/T | T2454N | 0 | 0 | 0.17 | 0 | 0.05 |
| 6085324 | 43 | C1 | A/G | R2464C* | 0 | 0 | 0 | 0.13 | 0.05 |
| 6078434 | 45 | C2 | A/G | P2558S | 0.2 | 0 | 0 | 0 | 0.05 |
| 6061604 | 49 | CK-C2 | A/C | V2690F | 0 | 0.28 | 0 | 0 | 0.05 |
| 6061559 | 49 | CK-C2 | C/T | G2705R | 0.81 | 7.18 | 0 | 5.54 | 3.3 |
For PolyPhen-2, effects of probable and possible damages are classified as deleterious.
Previously reported variants
Association of VWF variants with VWD
The functional impact of these VWF variants was also examined by their association with VWD (http://www.vwf.group.shef.ac.uk/). We identified 19 variants previously associated with or determined to be causative for VWD (Table 3, Table S3–S4) [31–34]. Seven of them (36.8%) —L129M, M576I, M740I, H817Q, R924Q, R2185Q and R2287W— had an allele frequency of > 1% in one population, but not in others (Table 3). M7401I, H817Q and R2185Q were found in 19.5%, 13.8%, and 20.7% of AFR subjects, respectively, whereas their allele frequencies in non-AFR subjects were consistent with previous reports [31, 33, 35]. M740I was found by screening families with type 2M VWD with an Italian ancestry and in 3 additional cases in the European study [32, 33]. It was consistently co-segregated with R1205H in the European study of VWD patietns, but none of the individuals in the 1000G had R1205H variant. H817Q was found to affect the binding of mature VWF to coagulation factor VIII in a European patient with type 1 VWD, who was compound heterozygous also for R782W [35]. R2185Q was found in one index European patient and no function study was performed [31].
Table 3.
VWF Variants Associated with VWD*
| VWD type | Nucleotide Change | Amino Acid Change | Exon | Domain | Minor Allele Frequency (%) | Overall Frequency (%) | |||
|---|---|---|---|---|---|---|---|---|---|
| AFR (n=246) | AMR (n=181) | ASN (n=286) | EUR (n=379) | ||||||
| 1 | CTG-aTG | L129M† | 5 | D1 | 1.42 | 0 | 0 | 0 | 0.32 |
| 1/3 | CGG-tGG | R273W | 7 | D1 | 0 | 0.28 | 0 | 0 | 0.05 |
| 1 | ATG-ATt | M576I | 14 | D2 | 0 | 1.93 | 0 | 0 | 0.32 |
| 1/2M | ATG-ATa | M740I† | 17 | D2 | 19.51** | 0.83 | 0 | 0 | 4.53 |
| 2N | CCG-CtG | P812L | 18 | D′ | 0 | 0 | 0 | 0.26 | 0.09 |
| 1 | CAT-CAa | H817Q† | 19 | D′ | 13.82** | 0.28 | 0 | 0 | 3.16 |
| 1/2N | CGG-CaG | R854Q† | 20 | D′ | 0 | 0.55 | 0 | 0.13 | 0.14 |
| 1/2N | CGG-CaG | R924Q† | 21 | D3 | 0 | 0.55 | 0 | 2.64 | 1.01 |
| 1/2A | CGG-tGG | R1583W | 28 | A2 | 0 | 0.28 | 0 | 0 | 0.05 |
| 1 | TAC-TgC | Y1584C† | 28 | A2 | 0 | 0.55 | 0 | 0.53 | 0.27 |
| 2A | GGA-aGA | G1672R | 28 | A2 | 0 | 0 | 0.52 | 0.13 | 0.18 |
| 1 | GTC-aTC | V1760I | 30 | A3 | 0 | 0 | 0 | 0.26 | 0.09 |
| Unclassified | TCA-aCA | S1731T | 30 | A3 | 0 | 0 | 0 | 0.13 | 0.05 |
| 1/3 | CCA-tCA | P2063S† | 36 | D4 | 0 | 0.83 | 0 | 0.79 | 0.41 |
| 1 | CGG-CaG | R2185Q† | 37 | D4 | 20.73** | 2.21 | 0 | 0 | 5.04 |
| 1 | CGG-tGG | R2287W† | 39 | D4 | 2.24 | 0 | 0 | 0 | 0.5 |
| 1 | CGC-CaC | R2313H† | 40 | B1 | 0.2 | 0.28 | 0 | 0.13 | 0.14 |
| 1 | CGC-tGC | R2464C | 43 | C2 | 0 | 0 | 0 | 0.13 | 0.05 |
| 1 | ACG-AtG | T2647M† | 48 | C2 | 0 | 0 | 0 | 0.92 | 0.32 |
Based on ISTH-SSC VWF Online Database
Variants with high allele frequencies in African, but low in other populations.
Variants that were also reported by Bellissimo, D.B. et al.[19]
Among the 19 VWD variants, 7 were also predicted by computations as deleterious (annotated with asterisk in Table 2). When deleterious variants were further annotated with 1000G allele frequencies, 4 (L129M, T346I, R2287W, and G2705R) had MAF greater than 1% in one of the four continents. This is higher than what we would expect given the prevalence of the VWD (estimates ranging from 0.01% – 0.8%) [36, 37]. L129M, T346I, and R2287W were found only in AFR subjects (Table 2). L129M and R2287W were initially identified as Type 1 VWD mutations by screening patients with European ancestry [31, 32]. A likely explanation is that their VWD propensity is specific to non-AFR population, but this propensity is modified by environmental and/or genetic factors in AFR subjects. G2705R segregated with MAFs of 5.5% and 7.2% for EUR and AMR subjects, respectively (Table 2). Because AMR consists of a number of admixed ethnicities (MXL, COL, and PUR), a proportion of alleles is of European origin. It is therefore likely that G2705R in AMR is originated from European founders. It may also be false positive prediction by both SIFT and PolyPhen-2 because of a relatively high population frequency.
Patterns of ethnic diversity among VWF domains
We next quantified the nucleotide diversity among exonic VWF domains by calculating pi [27]. The D′ domain had the highest nucleotide diversity, followed by the D2 and D4 domains (Figure 2). Alleles with MAF>10% in the three domains may have accounted for this higher nucleotide diversity (Table S3). Many of the variants (60%) found at high frequency in these three domains were synonymous (Table S3). The conservation scores, calculated by comparing human sequences against 46 species, found that D2 and D′ had a similar level of sequence conservation as compared to other VWF domains (1.0 and 1.1 versus 0.9, 0.9 and 1.0 for the B1, B2 and CK domains), suggesting that the D2 and D′ domains are not targeted for positive selection.
Figure 2. Nucleotide diversity among VWF domains.
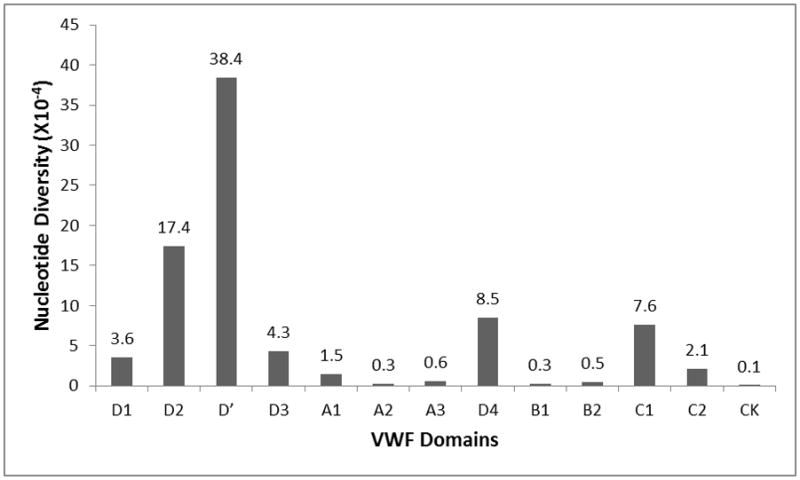
Nucleotide diversity (pi) was calculated across the entire study population using DnaSP software.
The nucleotide diversity of VWF domains also varied among four continents. As expected, AFR subjects were more polymorphic than others (Table S4). Some of the domains had too few variants (B1, B2, CK, Table S2) to have statistical power for comparison of nucleotide diversity. This limitation could in part account for the low nucleotide diversity in these domains.
The diversity of intronic SNPs was more evenly distributed throughout the VWF gene, with those in introns 29–32 (flanking exons that encode the A3 domain) being the least variable (Figure S1). The ascertainment complexity in the 1000G could account for this low diversity because intronic SNPs were detected only by the low pass sequencing (average read depth of coverage = ~2–6X/subject), whereas exonic SNPs were detected by both low pass and exome sequencing.
Genetic Characteristics in Populations
We performed PCA to study the genetic relationship among 13 ethnicities (14 subjects from Iberian in Spain (IBS) were excluded because of an insufficient sample size). The 1st and 2nd PCs accounted for 96.8% of total variance. When the 1st and 2nd principal components were examined, subjects could be divided into three groups: ASN, AMR/EUR, and AFR. The ASW was between AFR and AMR/EUR, but close to AFR (Figure S1), indicating its closer relationship with Africans. This finding is consistent with the genome wide expectation and historical record that ~80% of African American ancestries were closely related to western Africans [38].
Haplotypes of VWF SNPs among subjects from the four continents showed distinct LD patterns. LD blocks from Africans had a much shorter range than those from other continents (Figure S4). Africans had 83 haplotype blocks in the VWF gene with the longest being 4 kb of nucleotides, whereas Asian, American and European subjects had 29, 42, 40 LD blocks, with the longest blocks of 21kb, 13kb and 19kb, respectively. The four European ethnicities had a similar LD structure that was different from the three African ethnicities (Figure S4 & S5).
Discussion
We have examined the genetic variation and ethnic diversity of the VWF gene using the 1000G dataset (Table 1 and Figure 1). As expected, there were more variations in Africans than other ethnic groups (Table 1). Bellissimo, D.B, et al [19] have recently reported 14 variants that were previously associated with VWD, but have a minimal influence on plasma VWF antigen. We identified 11 of these 14 variants in the 1000G dataset. For the rest, R1342C was not detected in 1000G, and V1229G and N1231T were removed because of possible contamination from the VWF pseudogene. By working on a much larger sample size with more diversified ethnic background, we extended the Bellissimo’s study by identifying a larger number of non-synonymous SNPs (69 out of 94 were not in dbSNP129), including 19 listed in the ISTH VWF database (Table 3 and TableS3).
The allelic diversity of VWF variants was further interrogated by PCA (Figure S2) and analysis for linkage disequilibrium (Figure S3–S5). The four European ethnicities had very similar LD patterns that differed from the African ethnicities. The three Asian ethnicities (CHS, CHB, and JPT) can be grouped together on the PCA panel, indicating their similar genetic structures. Together, these data demonstrate that the VWF gene is ethnically diversified at a level that has not been reported before. This global analysis of diversity in the VWF gene provides necessary background information for understanding the presence of “VWD variants” in disease and non-disease populations. We further showed that the nucleotide diversity of exonic VWF variants was highest in the D′ and D2 domains (Figure 2). The functional impacts of these variants have not been examined experimentally, but one could speculate that this higher genetic diversity in the D′ and D2 domains could result in variations in VWF multimer patterns among subjects from different ethnicities.
A diversified presence of the “VWD mutations” in four continents (Table 3) raises three critically important and related questions. First, do these variants alter cellular and biochemical properties of VWF consistent with a VWD phenotype? The answer is yes, at least for some, as demonstrated in vitro for recombinant S1731T, Y1584C, and R273W variants that result in intracellular retention and the lack of high molecular weight multimers [39–41]. However, question remains as whether these genetic variations cause a mild phenotype that requires additional mutations that co-segregate (i.e. in strong LD) or environmental triggers to present a bleeding phenotype. For the former, 57.9% of the VWD variants found in the 1000G cohort are in the D domains that are likely to generate a mild phenotype as compared to those found in the A1 domain, where the binding site for the platelet GP Ib-IX-V complex is located. For the latter, M740I were found to co-segregate with R1205H in three European patients with type 1/2M VWD [32, 33]. This mutation has an allele frequency of 19.5% in Africans, but none has R1205H co-segregation.
Second, can these mutations cause VWD in an ethnic-specific manner? The question is raised because very few VWF variants, especially non-synonymous SNPs, are shared among subjects from different continents (Table 1, 3). M740I, H817Q and R2185Q, which were originally reported in European patients with VWD, have minor allele frequencies of >10% in Africans. There may be several reasons for this ethnic diversity in disease association: 1) These mutations result in low VWF antigen that is partially compensated by a high baseline VWF antigen level found in Africans and African Americans [42]; 2) they are VWD-causing only when they are confined in ethnic specific haplotypes; and 3) they arose recently to escape negative selection pressures in a more diverse gene pool such as Africans [20, 22]. These possibilities can be further investigated in larger cohorts of healthy subjects and VWD patients with available VWF antigen and adhesive activity.
Third, in addition to 19 VWF variants that were previously associated with VWD, 31 non-synonymous variants were also considered to be deleterious by two computational programs that evaluate the potential for affecting functions by a specific variant based on structural differences between wild type and the variant, and their conservations in a protein family. The biological impact of these deleterious variants remained to be verified experimentally, but one can speculate that they induce mild-to-moderate alterations in the VWF structure or folding that are insufficient to cause a catastrophic phenotype. This is especially possible for variations in the D domains, where changes in multimer patterns and interaction with FVIII may not severely or directly affect VWF interaction with the platelet GP Ib-IX-V complex as mutants in the A domains found in most of type 2 VWD. However, these mild-to-moderate variants can be additive to influence VWF structure and function when they reside in specific haplotypes together with other variants.
Comparing to SNPs that have been extensively studied in the past, indels are new variants for which functional significance remains to be investigated. We identified 91 indels all in introns so that no frame-shift for the coding sequence is expected. Biological impacts of these 91 indels are unknown, but could potentially result in changes in the rate of VWF gene splicing and microRNA binding. Some may also serve as markers for other genetic variations.
In summary, in this comprehensive study of VWF variants in different ethnicities, we have identified 2,728 VWF SNPs and 91 indels in 1000G subjects with 75.1% being novel. The D′ and D2 domains had the highest level of nucleotide diversity. Furthermore, 19 non-synonymous SNPs that have previously been associated with VWD in Europeans are detected in 1000G subjects and some have different allele frequencies among four populations. Results from this study demonstrate that the VWF gene is ethnically diverse but this ethnic complexity and its contribution to diseases requires further study in large cohorts. Molecular diagnostic panels for VWD may also benefit by considering ethnic diversity in linking a specific VWF variant to a bleeding phenotype.
Supplementary Material
Acknowledgments
The authors thank the participants of the 1000 Genomes Project for their work and contributions.
This work is supported by the grants HG003273, HG005211-01, MH089175, HL71895 and HL085769 from the National Institute of Health. Q.Y. Wang is a recipient of the Chinese Scholarship Council.
Footnotes
Addendum
Q.Y. Wang: designed the study, analyzed the data, and wrote the manuscript;
J. Song: analyzed the data;
R.A. Gibbs and E. Boerwinkle: designed the study;
F.L. Yu and J.F. Dong: developed the hypothesis, designed the study, analyzed the data and wrote the manuscript.
Disclosure of Conflict of Interest:
The authors declare no relevant financial interests.
References
- 1.Hassan MI, Saxena A, Ahmad F. Structure and function of von Willebrand factor. Blood Coagul Fibrinolysis. 2012;23:11–22. doi: 10.1097/MBC.0b013e32834cb35d. [DOI] [PubMed] [Google Scholar]
- 2.Sadler JE, Budde U, Eikenboom JC, Favaloro EJ, Hill FG, Holmberg L, Ingerslev J, Lee CA, Lillicrap D, Mannucci PM, Mazurier C, Meyer D, Nichols WL, Nishino M, Peake IR, Rodeghiero F, Schneppenheim R, Ruggeri ZM, Srivastava A, Montgomery RR, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006;4:2103–14. doi: 10.1111/j.1538-7836.2006.02146.x. [DOI] [PubMed] [Google Scholar]
- 3.James P, Lillicrap D. The role of molecular genetics in diagnosing von Willebrand disease. Semin Thromb Hemost. 2008;34:502–8. doi: 10.1055/s-0028-1103361. [DOI] [PubMed] [Google Scholar]
- 4.van Schie MC, van Loon JE, de Maat MP, Leebeek FW. Genetic determinants of von Willebrand factor levels and activity in relation to the risk of cardiovascular disease: a review. J Thromb Haemost. 2011;9:899–908. doi: 10.1111/j.1538-7836.2011.04243.x. [DOI] [PubMed] [Google Scholar]
- 5.Gottesman RF, Cummiskey C, Chambless L, Wu KK, Aleksic N, Folsom AR, Sharrett AR. Hemostatic factors and subclinical brain infarction in a community-based sample: the ARIC study. Cerebrovasc Dis. 2009;28:589–94. doi: 10.1159/000247603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Folsom AR, Rosamond WD, Shahar E, Cooper LS, Aleksic N, Nieto FJ, Rasmussen ML, Wu KK. Prospective study of markers of hemostatic function with risk of ischemic stroke. The Atherosclerosis Risk in Communities (ARIC) Study Investigators. Circulation. 1999;100:736–42. doi: 10.1161/01.cir.100.7.736. [DOI] [PubMed] [Google Scholar]
- 7.Folsom AR, Wu KK, Rosamond WD, Sharrett AR, Chambless LE. Prospective study of hemostatic factors and incidence of coronary heart disease: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 1997;96:1102–8. doi: 10.1161/01.cir.96.4.1102. [DOI] [PubMed] [Google Scholar]
- 8.Methia N, Andre P, Denis CV, Economopoulos M, Wagner DD. Localized reduction of atherosclerosis in von Willebrand factor-deficient mice. Blood. 2001;98:1424–8. doi: 10.1182/blood.v98.5.1424. [DOI] [PubMed] [Google Scholar]
- 9.Ginsburg D, Handin RI, Bonthron DT, Donlon TA, Bruns GA, Latt SA, Orkin SH. Human von Willebrand factor (vWF): isolation of complementary DNA (cDNA) clones and chromosomal localization. Science. 1985;228:1401–6. doi: 10.1126/science.3874428. [DOI] [PubMed] [Google Scholar]
- 10.Collins CJ, Underdahl JP, Levene RB, Ravera CP, Morin MJ, Dombalagian MJ, Ricca G, Livingston DM, Lynch DC. Molecular cloning of the human gene for von Willebrand factor and identification of the transcription initiation site. Proc Natl Acad Sci U S A. 1987;84:4393–7. doi: 10.1073/pnas.84.13.4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hampshire DJ, Goodeve AC. The international society on thrombosis and haematosis von Willebrand disease database: an update. Semin Thromb Hemost. 2011;37:470–9. doi: 10.1055/s-0031-1281031. [DOI] [PubMed] [Google Scholar]
- 12.Campos M, Sun W, Yu F, Barbalic M, Tang W, Chambless LE, Wu KK, Ballantyne C, Folsom AR, Boerwinkle E, Dong JF. Genetic determinants of plasma von Willebrand factor antigen levels: a target gene SNP and haplotype analysis of ARIC cohort. Blood. 2011;117:5224–30. doi: 10.1182/blood-2010-08-300152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith NL, Chen MH, Dehghan A, Strachan DP, Basu S, Soranzo N, Hayward C, Rudan I, Sabater-Lleal M, Bis JC, de Maat MP, Rumley A, Kong X, Yang Q, Williams FM, Vitart V, Campbell H, Malarstig A, Wiggins KL, Van Duijn CM, et al. Novel associations of multiple genetic loci with plasma levels of factor VII, factor VIII, and von Willebrand factor: The CHARGE (Cohorts for Heart and Aging Research in Genome Epidemiology) Consortium. Circulation. 2010;121:1382–92. doi: 10.1161/CIRCULATIONAHA.109.869156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Campos M, Buchanan A, Yu F, Barbalic M, Xiao Y, Chambless LE, Wu KK, Folsom AR, Boerwinkle E, Dong JF. Influence of single nucleotide polymorphisms in factor VIII and von Willebrand factor genes on plasma factor VIII activity: the ARIC Study. Blood. 2012;119:1929–34. doi: 10.1182/blood-2011-10-383661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harvey PJ, Keightley AM, Lam YM, Cameron C, Lillicrap D. A single nucleotide polymorphism at nucleotide -1793 in the von Willebrand factor (VWF) regulatory region is associated with plasma VWF:Ag levels. Br J Haematol. 2000;109:349–53. doi: 10.1046/j.1365-2141.2000.02000.x. [DOI] [PubMed] [Google Scholar]
- 16.Smith NL, Rice KM, Bovill EG, Cushman M, Bis JC, McKnight B, Lumley T, Glazer NL, van Hylckama Vlieg A, Tang W, Dehghan A, Strachan DP, O’Donnell CJ, Rotter JI, Heckbert SR, Psaty BM, Rosendaal FR. Genetic variation associated with plasma von Willebrand factor levels and the risk of incident venous thrombosis. Blood. 2011;117:6007–11. doi: 10.1182/blood-2010-10-315473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mancuso DJ, Tuley EA, Westfield LA, Worrall NK, Shelton-Inloes BB, Sorace JM, Alevy YG, Sadler JE. Structure of the gene for human von Willebrand factor. J Biol Chem. 1989;264:19514–27. [PubMed] [Google Scholar]
- 18.Sadler JE, Ginsburg D. A database of polymorphisms in the von Willebrand factor gene and pseudogene. For the Consortium on von Willebrand Factor Mutations and Polymorphisms and the Subcommittee on von Willebrand Factor of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost. 1993;69:185–91. [PubMed] [Google Scholar]
- 19.Bellissimo DB, Christopherson PA, Flood VH, Gill JC, Friedman KD, Haberichter SL, Shapiro AD, Abshire TC, Leissinger C, Hoots WK, Lusher JM, Ragni MV, Montgomery RR. VWF mutations and new sequence variations identified in healthy controls are more frequent in the African-American population. Blood. 2012;119:2135–40. doi: 10.1182/blood-2011-10-384610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, Schaffner SF, Yu F, Bonnen PE, de Bakker PI, Deloukas P, Gabriel SB, Gwilliam R, Hunt S, Inouye M, Jia X, Palotie A, Parkin M, Whittaker P, Chang K, Hawes A, Lewis LR, et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–8. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.The 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Novembre J, Stephens M. Interpreting principal component analyses of spatial population genetic variation. Nat Genet. 2008;40:646–9. doi: 10.1038/ng.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 27.Nei M, Li WH. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci U S A. 1979;76:5269–73. doi: 10.1073/pnas.76.10.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 29.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–4. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.James PD, Notley C, Hegadorn C, Leggo J, Tuttle A, Tinlin S, Brown C, Andrews C, Labelle A, Chirinian Y, O’Brien L, Othman M, Rivard G, Rapson D, Hough C, Lillicrap D. The mutational spectrum of type 1 von Willebrand disease: Results from a Canadian cohort study. Blood. 2007;109:145–54. doi: 10.1182/blood-2006-05-021105.. [DOI] [PubMed] [Google Scholar]
- 32.Goodeve A, Eikenboom J, Castaman G, Rodeghiero F, Federici AB, Batlle J, Meyer D, Mazurier C, Goudemand J, Schneppenheim R, Budde U, Ingerslev J, Habart D, Vorlova Z, Holmberg L, Lethagen S, Pasi J, Hill F, Hashemi Soteh M, Baronciani L, et al. Phenotype and genotype of a cohort of families historically diagnosed with type 1 von Willebrand disease in the European study, Molecular and Clinical Markers for the Diagnosis and Management of Type 1 von Willebrand Disease (MCMDM-1VWD) Blood. 2007;109:112–21. doi: 10.1182/blood-2006-05-020784. [DOI] [PubMed] [Google Scholar]
- 33.Castaman G, Missiaglia E, Federici AB, Schneppenheim R, Rodeghiero F. An additional unique candidate mutation (G2470A; M740I) in the original families with von Willebrand disease type 2 M Vicenza and the G3864A (R1205H) mutation. Thromb Haemost. 2000;84:350–1. [PubMed] [Google Scholar]
- 34.Kroner PA, Friedman KD, Fahs SA, Scott JP, Montgomery RR. Abnormal binding of factor VIII is linked with the substitution of glutamine for arginine 91 in von Willebrand factor in a variant form of von Willebrand disease. J Biol Chem. 1991;266:19146–9. [PubMed] [Google Scholar]
- 35.Kroner PA, Foster PA, Fahs SA, Montgomery RR. The defective interaction between von Willebrand factor and factor VIII in a patient with type 1 von Willebrand disease is caused by substitution of Arg19 and His54 in mature von Willebrand factor. Blood. 1996;87:1013–21. [PubMed] [Google Scholar]
- 36.Sadler JE, Mannucci PM, Berntorp E, Bochkov N, Boulyjenkov V, Ginsburg D, Meyer D, Peake I, Rodeghiero F, Srivastava A. Impact, diagnosis and treatment of von Willebrand disease. Thromb Haemost. 2000;84:160–74. [PubMed] [Google Scholar]
- 37.Werner EJ, Broxson EH, Tucker EL, Giroux DS, Shults J, Abshire TC. Prevalence of von Willebrand disease in children: a multiethnic study. J Pediatr. 1993;123:893–8. doi: 10.1016/s0022-3476(05)80384-1. [DOI] [PubMed] [Google Scholar]
- 38.Tian C, Hinds DA, Shigeta R, Kittles R, Ballinger DG, Seldin MF. A genomewide single-nucleotide-polymorphism panel with high ancestry information for African American admixture mapping. Am J Hum Genet. 2006;79:640–9. doi: 10.1086/507954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ribba AS, Loisel I, Lavergne JM, Juhan-Vague I, Obert B, Cherel G, Meyer D, Girma JP. Ser968Thr mutation within the A3 domain of von Willebrand factor (VWF) in two related patients leads to a defective binding of VWF to collagen. Thromb Haemost. 2001;86:848–54. [PubMed] [Google Scholar]
- 40.O’Brien LA, James PD, Othman M, Berber E, Cameron C, Notley CR, Hegadorn CA, Sutherland JJ, Hough C, Rivard GE, O’Shaunessey D, Lillicrap D. Founder von Willebrand factor haplotype associated with type 1 von Willebrand disease. Blood. 2003;102:549–57. doi: 10.1182/blood-2002-12-3693. [DOI] [PubMed] [Google Scholar]
- 41.Allen S, Abuzenadah AM, Hinks J, Blagg JL, Gursel T, Ingerslev J, Goodeve AC, Peake IR, Daly ME. A novel von Willebrand disease-causing mutation (Arg273Trp) in the von Willebrand factor propeptide that results in defective multimerization and secretion. Blood. 2000;96:560–8. [PubMed] [Google Scholar]
- 42.Miller CH, Dilley A, Richardson L, Hooper WC, Evatt BL. Population differences in von Willebrand factor levels affect the diagnosis of von Willebrand disease in African-American women. Am J Hematol. 2001;67:125–9. doi: 10.1002/ajh.1090. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.