

Abstract
There is an increasing awareness that diabetes has an impact on the central nervous system, with reports of impaired learning, memory and mental flexibility being more common in diabetic subjects than in the general population. Insulin-deficient diabetic mice also display learning deficits associated with defective insulin-signaling in the brain and increased activity of GSK3. In the present study, AR-A014418, a GSK3β inhibitor and TX14(A), a neurotrophic factor with GSK3 inhibitory properties, were tested against the development of learning deficits in mice with insulin-deficient diabetes. Treatments were started at onset of diabetes and continued for 10 weeks.
Treatment with AR-A014418 or TX14(A) prevented the development of learning deficits, assessed by the Barnes maze, while only AR-A014418 prevented memory deficits, as assessed by the object recognition test. Diabetes-induced increased levels of amyloid beta protein and phosphorylated tau were not significantly affected by the treatments. However the diabetes-induced decrease in synaptophysin, a presynaptic protein marker of hippocampal plasticity, was partially prevented by both treatments. These results suggest a role for GSK3 and/or reduced neurotrophic support in the development of cognitive deficits in diabetic mice that are associated with synaptic damage.
Keywords: Diabetes, Brain, GSK3, cognitive functions, synaptic plasticity
Peripheral neuropathy is the most common of the complications associated with long-term diabetes and develops in more than half of all diabetic patients. Several studies have also demonstrated a co-incidence between one or more diabetic chronic complications and impaired function of the central nervous system (Ryan and Williams 1993), suggesting that the brain is susceptible to the same processes that underlie other complications of diabetes. Studies have found a relative risk of about 1.9 to 2.3 of developing Alzheimer’s disease (AD) for diabetic patients (Ott et al. 1996; Ronnemaa et al. 2009). Most recently, diabetes was shown to increase not only the risk of dementia but also the risk of progression from mild cognitive impairment to AD (Velayudhan et al. 2010). Encephalopathy, defined as electrophysiological and structural disturbances in the brain associated with cognitive deficits, occurs in both type 1 and type 2 diabetic subjects (Biessels et al. 2008; Cukierman et al. 2005; Desrocher and Rovet 2004; Reaven et al. 1990; Ryan et al. 1993). Uncontrolled diabetes (no insulin treatment) was associated with the development of AD, while patients with controlled diabetes showed no increased dementia, suggesting a role of impaired insulin signaling in the development of neurodegeneration and AD (Xu et al. 2009b). Peripheral insulin deficiency is sufficient to cause learning deficits and AD-like features in the brain of type 1 diabetic animals (Biessels et al. 1996; Jolivalt et al. 2008). The role of insulin signaling in the brain is further supported by animal studies showing exacerbation of learning deficits, of tau deposition into tangles and of amyloid-β accumulation in mouse models of AD with concomitant insulin-deficient diabetes (Burdo et al. 2009; Jolivalt et al. 2010; Ke et al. 2009) or concomitant diet-induced insulin-resistance (Ho et al. 2004).
Disturbance of insulin signaling by diabetes disrupts assorted signaling pathways, including those mediated by phospholipase Cγ, mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K). Downstream of the PI3K/AKT pathway, glycogen synthase kinase-3 (GSK3) is an enzyme that regulates many aspects of cellular structure, function and survival and whose activity is down-regulated by phopshorylation at serine 9 and 21 (Sutherland et al. 1993). GSK3, and more particularly GSK3β, phosphorylates tau, decreasing its ability to bind and stabilize microtubules and consequently disturbing the neuronal cytoskeleton, impeding axonal transport and facilitating neurofibrillary tangle formation (review in Grundke-Iqbal and Iqbal 1989), while GSK3α, more specifically, promotes amyloid β (Aβ) formation, which is neurotoxic, inhibits axonal transport (Kasa et al. 2000) and accumulates into plaques. A number of GSK3 inhibitors are available (review Martinez 2008) and the ATP-competitive inhibitor AR-A014418 has shown potential for the treatment of AD (Bhat et al. 2003). AR-A014418 reduces tau phosphorylation in JNPL3 mice, a model of frontotemporal dementia with Parkinsonism and AD (Noble et al. 2005). Prosaptide TX14(A) is a 14-mer peptide derived from prosaposin that has neuroprotective properties in rodent models of diabetes (Calcutt et al. 1999; Jolivalt et al. 2006; Mizisin et al. 2001). Preliminary data from our group showed that TX14(A) also possesses GSK3 inhibitory properties. To determine the role of GSK3 and/or the loss of neurotrophic support in diabetic encephalopathy in a model of type 1 diabetes, insulin-deficient diabetic mice were treated with AR-A014418 or TX14(A) for 10 weeks from onset of diabetes. Both therapies prevented diabetes-associated learning deficits and synaptic damage.
Materials and Methods
Animals
Adult female Swiss Webster mice were purchased from Harlan Industries (Placentia, CA, USA). Animals were housed four to a cage with free access to food and water. All cages were maintained in a vivarium approved by the American Association for the Accreditation of Laboratory Animal Care. All study protocols were approved by the Institutional Animal Care and Use Committee of the University of California San Diego. Eight to twelve animals were used per group.
Induction of Diabetes
Insulin-deficient diabetes was induced at six to eight weeks of age by i.p. injection of streptozotocin (STZ, Sigma, St. Louis, MO) at 90mg/kg dissolved in 0.9% sterile saline solution after an overnight fast on two consecutive days. Hyperglycemia was confirmed 4 days post-STZ administration using a strip-operated glucose meter (One Touch Ultra, Lifescan, Milpitas, CA, USA) on a blood sample obtained via tail prick as well as on a blood sample obtained at the conclusion of the study. Glycemic control was tested by measuring HbA1c levels at termination of the study using a multi-test A1C system (A1C now, Bayer Healthcare, Sunnyvale, CA, USA).
Drugs
TX14(A) (Prosaptide) was supplied by Myelos Corporation in PBS solution at 4mg/ml. The ATP-competitive GSK3 inhibitor AR-A014418 (Astrazeneca) was purchased from Sigma. AR-A014418 was dissolved in PBS containing 4% DMSO. AR-A014418 was administered daily at 30 μmol/kg i.p. and TX14(A) at 1 mg/kg 3 times a week, determined from studies in diabetic painful neuropathy (Calcutt et al. 1999; Mizisin et al. 2001). Vehicle (4% DMSO in PBS) was administered to a group of control and a group of STZ-diabetic mice. Drugs and vehicle were administered at 10ml/kg i.p. for 10 weeks.
Barnes Maze test
Learning behavior was assessed using the Barnes maze test as previously described (Jolivalt et al. 2008). The Barnes maze was used as an equivalent to the Morris water maze test without the stress component. Indeed, concerns arise when studying protein phopshorylation as stress (water stress) can modulate phosphorylation of protein, including tau (Korneyev 1998; Okawa et al. 2003). Briefly, the Barnes Maze consists of a brightly lit circular white platform with 20 holes with equidistant spacing around the periphery. An escape box was placed beneath one of the holes marked by a visual cue. Before testing began the mouse was placed in the box for 1 minute. The mouse was then placed in the center of the table and allowed to explore until it either found the box or 5 minutes elapsed. The time to locate the escape box was recorded. At the end of the session, the mouse was left or placed in the escape box for an additional minute. The test was performed at 9 weeks of diabetes every day for 5 days.
Object Recognition test
The object recognition test was used to assess short-term memory and is based on the premise that rodents will explore a novel object more than a familiar one, if they remember the familiar object. The test was performed at week 10 using an 18×9 inch transparent box in a dimly lit room isolated from extraneous noise. Two identical objects are placed equidistant from each other and from the sides of the box. Mice are given 10 minutes to achieve 20 seconds of exploration time of the two objects, after which they are placed back to their home cage. Exploration is defined as the mouse facing and being within approximately 1 inch of the object. An hour later, one of the objects is replaced with a novel object and the mouse is given 5 minutes to explore. The time the mouse spend exploring the familiar and novel objects is recorded. Mice with normal cognitive function typically spend more time with the novel object.
Rota-Rod
To assess the effect of diabetes and treatments on motor functions that could affect learning and memory tests, mice were placed on a Rota-Rod (Stoelting Co., Wood Dale, IL, USA). The device accelerated from 4 to 40 RPM over 5 minutes. The amount of time spent on the rod before loss of balance was recorded over three trials. All trials were performed on the same day.
Tissue Preparation
Hippocampi and cortex were dissected within 1 min of decapitation and were homogenized in buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.5% Triton X, 1 mM EDTA, protease inhibitor cocktail). The homogenates were then centrifuged at 13,000g for 30 minutes. Clear cytosolic extracts were stored at −80°C. For tau Western-blot analysis, a portion of the cytosolic extract was boiled for 5 minutes in detergent free solution, after which the samples were centrifuged for 30 minutes to remove insoluble material. Protein concentrations were determined using the bicinchroninic acid method (BCA protein assay kit, Pierce, Rockford, IL, USA).
Western Blotting
Cytosolic extract aliquots were boiled in Laemmli LDS sample buffer (Invitrogen, Carlsbad, CA, USA). Four (tau western blot) to 15μg of extract proteins were separated on 4–12% Bis-Tris SDS-PAGE gels (Novex, Invitrogen). Separated proteins were then blotted on nitrocellulose. Blots were incubated with antibodies against phospho-GSK3β (phospho-Ser9; 1/1000, Cell Signaling technology, USA), GSK3 (1/10 000, Millipore, Billerica, MA, USA), Actin (1/2000, Sigma, Saint Louis, MO, USA), phospho-tau threonine 231 (1/3000, Biosource, Camarillo, CA, USA) and Tau-5 (1/3000, Biosource, Camarillo, CA, USA), Amyloid β (1/1000, clone 6E10, Covance/Signet Laboratories, Berkeley, CA, USA), and Synaptophysin (1/10 000, Chemicon International, Temecula, CA, USA) followed by incubation with the anti-mouse or -rabbit secondary antibody tagged with horseradish peroxidase (HRP, 1/20 000, Santa Cruz Biotechnology, Santa Cruz, CA, USA). Blots were developed using Super Signal West Pico Chemiluminescent Substrate (Thermo Scientific, Rockford, IL, USA). Densitometric scanning using Quantity One (BioRad, San Diego, CA, USA) was used for quantification. Band intensities for each protein were normalized by calculating the ratio of intensity of the band of interest to that of actin and total (non-phosphorylated) protein where applicable and percent intensity reported in graphs represents the percent of the mean intensity of the control group.
Immunofluorescence staining
Hemibrains were collected within 1 min of decapitation and post-fixed for 24 h in 4% paraformaldehyde in 0.1 M sodium phosphate buffer. Samples were embedded in paraffin and sections were cut at a thickness of 6 μm and collected onto glass slides. Aldehyde fluorescence was blocked using a solution of 0.5% sodium borohydride in 1% dibasic sodium phosphate buffer. The sections were then treated with sodium citrate antigen retrieval buffer (pH 6.0) followed by 5% normal goat serum (Vector Laboratories #S-1000, Burlingame, CA, USA) for 1 hour. Sections were incubated with synaptophysin antibody (1:2000; Millipore #MAB5258, Billerica, MA, USA) overnight at 4° C. They were then washed and incubated with Alexa Fluor 488-conjugated goat anti-mouse secondary antibody (1:500; Invitrogen #A11001, Carlsbad, CA, USA). Nuclear staining was performed using a DAPI solution at 1/20 000 (4′,6-Diamidino-2-Phenylindole, Dihydrochloride) (Invitrogen #D1306). The samples were viewed using an Olympus BX51 fluorescence microscope equipped with a QImaging Retiga 2000R camera.
Statistical analysis
Data from the Barnes maze test are expressed as group median for non-parametric data due to the cut-off limit of time set at 300s and were analyzed using repeat measure ANOVA followed by Dunnett’s post hoc test. All other data are expressed as group mean ± SEM and differences between groups were analyzed using one-way ANOVA followed by Tukey’s post-hoc test.
Results
Diabetes
Every mouse injected with STZ (90mg/kg i.p.) after an overnight fast on two consecutive days exhibited hyperglycemia (blood sugar >270 mg/dl) 4 days after STZ injection. Ten weeks later, blood glucose levels for diabetic mice were significantly higher than for non-diabetic mice and treatment with AR-A014418 or TX14(A) did not affect blood glucose levels (Table 1). Similarly, glycated hemoglobin A1c (HbA1c) was significantly elevated in STZ-diabetic mice and was not affected by either treatment (Table 1). STZ-injected mice expectedly lost some weight in the first week following the induction of diabetes but maintained a healthy weight over the 10-week period (Fig. 1). The treatment with AR-A014418 or TX14(A) did not affect the mouse body weight (Fig. 1). Occasional administration of two units of insulin (s.c.) was used to maintain healthy weight in some animals that had lost more than 20% of their starting body weight. This resulted in 1 or 2 injections for 2 or 3 mice per group for all diabetic groups during the course of the 10 weeks of the study and did not affect final blood glucose or HbA1c. The occasional insulin injection may have temporarily activated the insulin signaling pathway and therefore modulated GSK3 activity, however this treatment was limited over the course of the study and to few mice per group and if any effects were to be detected they were limited as we have detected a significant decreased phosphorylation of GSK3 in the diabetic group that also received occasional insulin injections.
Table 1.
Final blood glucose and HbA1c levels for the 4 groups of mice.
| Groups | Blood glucose (mg/dl) | HbA1c (%) |
|---|---|---|
| Control + Vehicle | 122 ± 2 | 4.6 ± 0.1 |
| STZ + Vehicle | 589 ± 5*** | 11.8 ± 0.5 *** |
| STZ + AR-A014418 | 587 ± 8*** | 12.9 ± 0.1 *** |
| STZ + TX14(A) | 584 ± 16*** | 12.8 ± 0.2 *** |
p<0.001 one-way ANOVA followed by Tukey post hoc test
Figure 1.
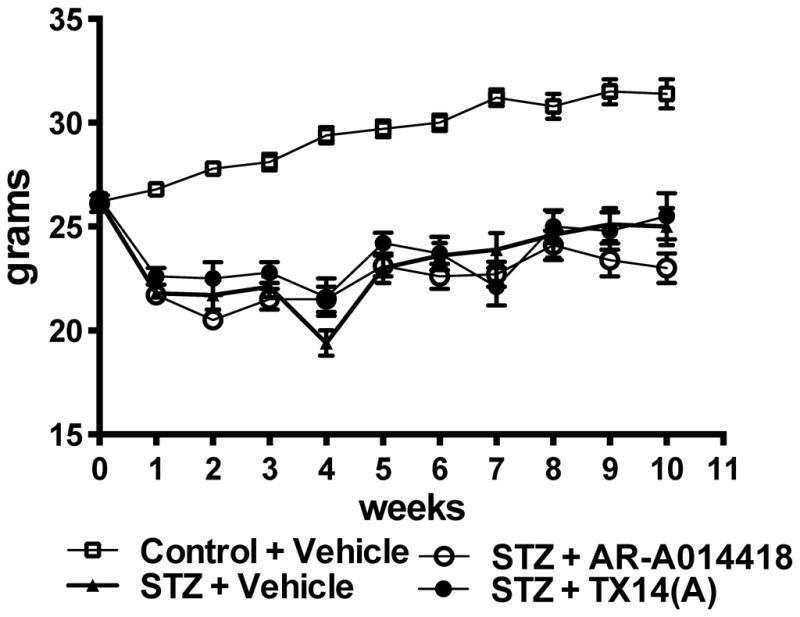
Body weight over the duration of the study for control mice receiving vehicle (open square), STZ-diabetic mice receiving vehicle (closed triangle), STZ-diabetic mice receiving AR-A014418 at 30 μmol/kg (open circle) and STZ-diabetic mice receiving TX14(A) at 1mg/kg (closed circle).
Barnes maze test
Learning abilities were testing using the Barnes maze after 9 weeks of treatment for 5 consecutive days. On the first day of testing, all 4 groups of mice found the escape box with a similar time (Fig. 2A). The time to find the escape box for control mice receiving vehicle was progressively reduced over the 5 days of testing while STZ-diabetic mice receiving vehicle find the escape box with a reduced time only by day 4 (Fig. 2A, p<0.001, repeated measures ANOVA), indicative of learning deficits for the diabetic group. After 9 weeks of treatment, diabetic mice receiving AR-A014418 (p<0.001, repeated measures ANOVA compared to STZ + Vehicle) or TX14(A) (p<0.05, repeated measures ANOVA compared to STZ + Vehicle) showed a learning curve similar to that of control mice (Fig. 2A), suggesting that both treatments significantly prevented the learning deficits induced by insulin-deficient diabetes. Using area under the curve (AUC) analysis, STZ-diabetic mice receiving vehicle showed significant (p<0.01) learning deficits in the Barnes maze compared to control mice receiving vehicle (Fig. 2B) and 9 weeks of treatment with TX14(A) partially prevented the learning deficits while treatment with AR-A014418 significantly (p<0.05) prevented the diabetic-induced deficits (Fig. 2B). The differences observed were not due to lack of exploration, as the rate of holes visited (error/min) by treated mice were similar to that of vehicle-treated diabetic mice (Fig. 2C). In addition, after 9 weeks of diabetes, no significant difference in motor ability was observed in the Rota-Rod task between any of the 4 groups (Fig. 2D).
Figure 2.

Effect of AR-A014418 or TX14(A) treatment on learning behavior in the Barnes maze test. A: Time to find the escape box in the Barnes circular maze test for control (open square) and STZ-diabetic (closed triangle) mice receiving vehicle, STZ-diabetic mice receiving AR-A014418 at 30 μmol/kg (open circle) and STZ-diabetic mice receiving TX14(A) at 1mg/kg (closed circle). Data are represented as Median for non-parametric data. B: Area under the curve (AUC) for graph A. *p<0.05, **p<0.01 by one-way ANOVA followed by Tukey post hoc test versus Control + Vehicle group. C: Number of errors performed by the mice in the Barnes maze test, *p<0.05, **p<0.01 by one-way ANOVA followed by Tukey post hoc test versus Control + Vehicle group. D: Time to fall off the rotating beam during the Rota-Rod test after 10 weeks of treatment with vehicle, AR-A014418 or TX14(A).
Object recognition test
STZ-diabetic mice showed a significantly (p<0.05) reduced time spent on the novel object in the object recognition test, demonstrating a deficit in working memory (Fig. 3). Treatment with AR-A014418 for 10 weeks significantly (p<0.05) prevented the memory deficits induced by diabetes, while treatment with TX14(A) had no effect (Fig. 3).
Figure 3.

Effect of AR-A014418 or TX14(A) treatment on memory behavior in the Object recognition test. *p<0.05, **p<0.01 by one-way ANOVA followed by Tukey post hoc test versus Control + Vehicle group.
GSK3β phosphorylation
Phosphorylation of GSK3β at the recognized inactivating site (ser 9) was significantly (p<0.05) reduced in STZ-diabetic mice brain after 10 weeks of diabetes (Fig. 4). Ten weeks of treatment with AR-A014418 or TX14(A) prevented the reduction of phosphorylated GSK3β (Fig. 4A), while there was no significant effect on total GSK3 protein expression (Fig. 4B). Similar results demonstrating increased activity of GSK3β in diabetic mice brains that was prevented by both treatments were observed for the phosphorylation of GSK3β at the activating site (tyr 216) (percent of control group intensity for WT: 100±4.2, STZ: 118.2±7.2*, STZ+ AR-A014418: 104.2±7.2, STZ+ TX14(A): 108.5±7.8, *p<0.05 vs WT).
Figure 4.
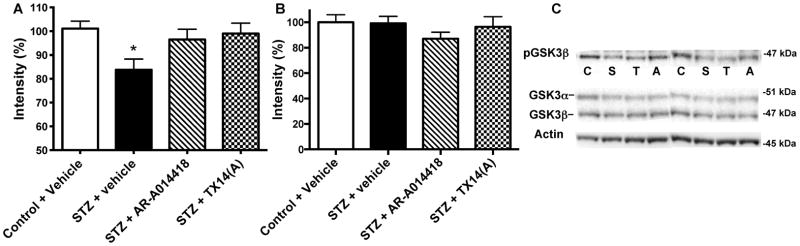
Effect of AR-A014418 or TX14(A) treatment on GSK3β phosphorylation. A: Intensity of bands from mouse brain homogenates for phosphorylated GSK3β at serine 9 (p-GSK3β) over actin. Phosphorylation of serine 9 of GSK3β isoforms results in inactivation of the enzyme. B: Intensity of bands from mouse brain homogenates for total GSK3β over actin. C: Representative Western blots performed with brain homogenates obtained from control (C) and STZ-diabetic (S) mice receiving vehicle and STZ-diabetic mice receiving AR-A014418 (A) or TX14(A) (T) for phosphorylated GSK3β (ser 9), total GSK3 and actin. Due to variable antibodies affinity, exposure times were adjusted in order to best visualize all the bands. Data are mean+SEM, n=8–10/group. *p<0.05 by one-way ANOVA versus control, followed by Tukey’s post-hoc test.
Tau phosphorylation
Normalized to total tau, tau phosphorylation at the threonine 231 site that is part of the microtubule-binding domain and phosphorylated by GSK3β, was significantly (p<0.01) increased in the brain of STZ-diabetic mice receiving vehicle (Fig. 5A) and this was not significantly prevented by treatment with AR-A014418 (Fig. 5A). Treatment with TX14(A) tended towards reducing tau phosphorylation but this effect was not significant from control nor diabetic group data (Fig. 5A).
Figure 5.
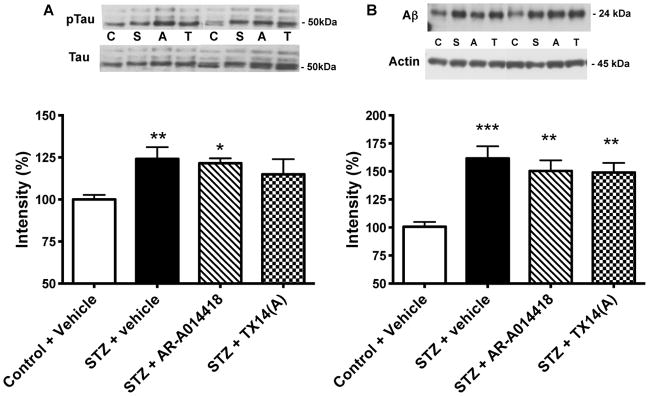
Effect of AR-A014418 or TX14(A) treatment on tau phopshorylation and amyloid β levels. A: Representative Western blot image and intensity of bands corresponding to phosphorylated tau and tau. Data are mean+SEM, n=6–8/group. The data represent the ratio ptau/tau of the intensity of the bands corresponding to phosphorylated tau at threonine 231 over the intensity of the bands corresponding to total tau. *p<0.05, **p<0.01 by one-way ANOVA versus control, followed by Tukey’s post-hoc test. B: Representative Western blot image and intensity of bands corresponding to amyloid β over actin. Data are mean+SEM, n=6–8/group. **p<0.01, ***p<0.001 by one-way ANOVA versus control, followed by Tukey’s post-hoc test
Amyloid β levels
Levels of soluble oligomers of Aβ (24kDa) were significantly (p<0.001) increased in the brain of STZ-diabetic mice receiving vehicle and 10 weeks of treatment with AR-A014418 or TX14(A) did not significantly prevent the diabetes-induced increase (Fig. 5B).
Synaptophysin levels
A reduction in intensity of synaptophysin immunoreactivity was observed in the CA1 region of the hippocampus of STZ-diabetic mice receiving vehicle compared to the control mice receiving vehicle (Fig. 6A and B). Treatment with AR-A014418 and TX14(A) for 10 weeks prevented this decrease (Fig. 6C and D).
Figure 6.
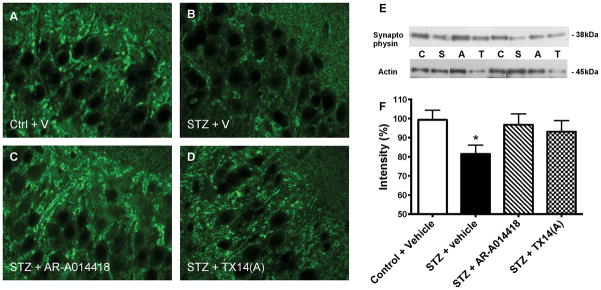
Effect of AR-A014418 or TX14(A) treatment on synaptophysin in mouse hippocampus. Synaptophysin immunoreactivity in the CA1 region of the hippocampus for control mice receiving vehicle (A), STZ-diabetic mice receiving vehicle (B), STZ-diabetic mice receiving AR-A014418 (C) and STZ-diabetic mice receiving TX14(A) (D). Bar scale=20μm. Western blot analysis of mouse brain: Intensity of bands corresponding to synaptophysin normalized to the intensity of bands corresponding to actin (E). Data are represented as mean+sem. *p<0.05 versus control + vehicle group by one-way ANOVA followed by Tukey’s post hoc test. Representative image (F) of Western blots of brain homogenates from control (C) and STZ-diabetic (S) mice receiving vehicle and STZ-diabetic mice receiving AR-A014418 (A) or TX14(A) (T).
Synaptophysin, a marker of synapse integrity, was quantified by Western blot. After 10 weeks of diabetes, synaptophysin protein levels were significantly reduced (p<0.05) in the brain of STZ-diabetic mice receiving vehicle and this reduction was partially prevented by 10 weeks of treatment with AR-A014418 or TX 14(A) (Fig. 6E and F).
Discussion
In this study, we have shown that AR-A014418 and TX14(A) prevent learning deficits associated with insulin-deficient diabetes in Swiss Webster mice without affecting glycemia levels. The prevention of learning deficits was associated with a reduction in GSK3β activity as assessed by decreased phopshorylation of Ser 9. Surprisingly, the prevention observed was not associated with significant reduction of Aβ protein levels or tau phopshorylation. However, prevention of learning deficits was associated with a prevention of synaptophysin protein decrease in the hippocampus of diabetic mice, suggesting a role of GSK3β in synaptic damage resulting in cognitive deficits.
The role of GSK3 and tau hyperphosphorylation in neurodegeneration is clearly demonstrated in transgenic mice conditionally overexpressing GSK3β (Lucas et al. 2001), and in transgenic mice overexpressing tau treated with the GSK3 inhibitor lithium (Noble et al. 2005). GSK3β overexpressing mice are characterized by a number of structural defects, neuronal stress and loss (Lucas et al. 2001), while, inversely, tau-overexpressing mice treated with lithium showed less degeneration (Noble et al. 2005). More recently, the detrimental role of active GSK3 in learning deficits in a mouse model of AD was shown using chemical inhibition of GSK3 by lithium or by crossbreeding with mice overexpressing inactive GSK3β. Similar to hAPP mice treated with lithium, expression of inactive GSK3β in the hAPP mouse model resulted in reduced Aβ and tau phopshorylation levels as well as reduced learning deficits (Rockenstein et al. 2007). Recently, lithium was shown to reverse memory impairments and restore phosphorylated GSK3β levels in rats receiving intracerebroventricular injection of STZ, but effects on Aβ and tau levels were not studied (Ponce-Lopez et al. 2011). In the current study, inhibition of GSK3β by AR-A014418 and TX14(A) did prevent learning deficits in diabetic mice but surprisingly, did not significantly affect tau phopshorylation or Aβ levels, suggesting other pathways modulating these 2 proteins in addition to GSK3. The lack of correlation between Aβ levels and learning and memory functions may be explained by a lack of toxicity of the Aβ species detected in the diabetic mouse brain. Indeed, recently, Reed et al. (2011) have shown that different sized Aβ oligomers have different effects on cognitive functions. Trimers extracted from transgenic mice, in opposition to the 56kDa oligomers, did not cause cognitive impairments when injected into rat brains (Reed et al. 2011). We have mostly detected 24kDa Aβ oligomers that, like the 12 kDa oligomer (Reed et al. 2011), may not play a critical role in cognitive impairments at that stage of the disease.
The improvement of learning ability of diabetic mice treated with AR-A014418 or TX14(A) was associated with partial prevention of decreased synaptophysin protein levels, suggesting that synaptic damage in the hippocampus is associated with spatial learning deficits and may precede tau and Aβ pathological contribution to cognitive functions decline in diabetic mice. Our findings, if somewhat controversial, are supported by the demonstration that synapse damage, as measured by synaptophysin levels, correlates better with cognitive deficits than either plaques or tangles in AD brain (DeKosky and Scheff 1990; Sze et al. 1997; Terry et al. 1991). Also, synaptic dysfunction, including long-term potentiation deficits, manifests in an age-related manner, but before plaque and tangle pathology in a transgenic mouse model of AD (Oddo et al. 2003). In diabetic rat brain, changes in synaptic plasticity were observed at anatomical and chemical levels and were partially prevented by insulin (Grillo et al. 2005; Magarinos et al. 2001). Similar to our results in diabetic mice, synaptophysin immunostaining was decreased in prefrontal cortex and CA3 from STZ rats and this was associated with learning deficits and prevented by treatment with a peptide derived from the activity-dependent neuroprotective protein (Idan-Feldman et al. 2011). In addition, consistent with the structural defects, including cerebral atrophy, detected in brain from patients affected by diabetes (Musen et al. 2006; Schmidt et al. 2004), diabetic rats and mice showed brain atrophy (Francis et al. 2008; Lupien et al. 2006), without detectable neuronal loss but with decreased synaptophysin protein levels in the cortex or hippocampus (Jolivalt et al. 2010; Jolivalt et al. 2008; Toth et al. 2006). Synaptic integrity and spine plasticity are essential to learning and memory (Xu et al. 2009a; Yang et al. 2009). The presence of both insulin receptor (Abbott et al. 1999) and GSK3 (Jiang et al. 2005; Peineau et al. 2007) at the synapse suggest a role for both proteins in structural remodeling of synapses (Lee et al. 2011). Diabetes and subsequent disruption of insulin signaling was shown to potentiate or exaggerate synaptic damage that was associated with learning deficits in transgenic mouse models of AD (Burdo et al. 2009; Jolivalt et al. 2010). Recently, a link between insulin pathway disturbances and impaired synaptic transmission as well as impaired cognitive performances has been demonstrated in mice with reduced insulin receptor expression (Nistico et al. 2012). In neuronal culture, the presence of insulin leads to an increase in the number of spines on neurons (Lee et al. 2011) and was shown to protect hippocampal neurons from Aβ-induced synaptic spine loss (De Felice et al. 2009). In insulin-deficient diabetes, as we have shown here and previously (Jolivalt et al. 2010; Jolivalt et al. 2008), insulin signaling is defective in the mouse brain and may contribute to pathological loss of spine, as observed in AD (Lee et al. 2011; Terry et al. 1991). Recently, a GSK3 inhibitor was shown not only to prevent, but also reverse, Aβ-induced inhibition of LTP in brain slice preparation, suggesting that over-activation of GSK3β is responsible for pathological synaptic plasticity (Jo et al. 2011). Taken together, these studies and our current data showing beneficial effects of both AR-A014418 and TX14(A) on synaptophysin protein levels and immunoreactivity in the hippocampus, learning deficits and activity of GSK3β without significantly affecting Aβ and phosphorylated tau levels support a pathological role of GSK3β in the synaptic function that contributes to learning deficits in diabetic mice.
In this study, despite preservation of synaptophysin levels, TX14(A) did not prevent memory deficits in diabetic mice assessed by the object recognition test contrary to AR-A014418. These results point at different mechanisms or cerebral domains participating in memory versus learning processing that we have not elucidated at this point.
Conclusions
In AD mouse models with protein overexpression (APP, tau), inhibition of GSK3 is able to reduce Aβ protein levels, tau phopshorylation, and ameliorate cognitive functions (Noble et al. 2005; Rockenstein et al. 2007). Despite sharing convergent mechanisms (Correia et al. 2012; Jolivalt et al. 2010; Jolivalt et al. 2008), our mouse model, however, does not overexpress any particular protein and therefore the deficits observed in diabetic mice may more closely resemble defects associated with human diabetes or even sporadic AD. Although Aβ and tau are major elements of AD pathology and are also increased in diabetic rodent brains, this study raises the question about the role of Aβ and tau phopshorylation in the learning and memory processes and suggests that Aβ and phosphorylated tau may be late contributors as it is possible to modulate learning abilities without significantly affecting the levels of these two proteins. Ameliorating synapse integrity may confer greater improvement than modulating Aβ and tau phosphorylation, at least in the early stages of the disease and inhibition of GSK3β may be beneficial to prevent synaptic damage and learning deficits.
Acknowledgments
This work was supported by a Career Development Award from the Juvenile Diabetes Research Foundation (JDRF) (CGJ) and by a NIH grant (AG039736) (CGJ). The authors would like to thank Dr. Nigel Calcutt for fruitful discussions on diabetes models.
Footnotes
None of the authors have a conflict of interest.
References
- Abbott MA, Wells DG, Fallon JR. The insulin receptor tyrosine kinase substrate p58/53 and the insulin receptor are components of CNS synapses. J Neurosci. 1999;19(17):7300–7308. doi: 10.1523/JNEUROSCI.19-17-07300.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat R, Xue Y, Berg S, Hellberg S, Ormo M, Nilsson Y, Radesater AC, Jerning E, Markgren PO, Borgegard T, Nylof M, Gimenez-Cassina A, Hernandez F, Lucas JJ, Diaz-Nido J, Avila J. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J Biol Chem. 2003;278(46):45937–45945. doi: 10.1074/jbc.M306268200. [DOI] [PubMed] [Google Scholar]
- Biessels GJ, Deary IJ, Ryan CM. Cognition and diabetes: a lifespan perspective. Lancet Neurol. 2008;7(2):184–190. doi: 10.1016/S1474-4422(08)70021-8. [DOI] [PubMed] [Google Scholar]
- Biessels GJ, Kamal A, Ramakers GM, Urban IJ, Spruijt BM, Erkelens DW, Gispen WH. Place learning and hippocampal synaptic plasticity in streptozotocin-induced diabetic rats. Diabetes. 1996;45(9):1259–1266. doi: 10.2337/diab.45.9.1259. [DOI] [PubMed] [Google Scholar]
- Burdo JR, Chen Q, Calcutt NA, Schubert D. The pathological interaction between diabetes and presymptomatic Alzheimer’s disease. Neurobiol Aging. 2009;30(12):1910–1917. doi: 10.1016/j.neurobiolaging.2008.02.010. [DOI] [PubMed] [Google Scholar]
- Calcutt NA, Campana WM, Eskeland NL, Mohiuddin L, Dines KC, Mizisin AP, O’Brien JS. Prosaposin gene expression and the efficacy of a prosaposin-derived peptide in preventing structural and functional disorders of peripheral nerve in diabetic rats. J Neuropathol Exp Neurol. 1999;58(6):628–636. doi: 10.1097/00005072-199906000-00007. [DOI] [PubMed] [Google Scholar]
- Correia SC, Santos RX, Carvalho C, Cardoso S, Candeias E, Santos MS, Oliveira CR, Moreira PI. Insulin signaling, glucose metabolism and mitochondria: Major players in Alzheimer’s disease and diabetes interrelation. Brain Res. 2012;1441:64–78. doi: 10.1016/j.brainres.2011.12.063. [DOI] [PubMed] [Google Scholar]
- Cukierman T, Gerstein HC, Williamson JD. Cognitive decline and dementia in diabetes--systematic overview of prospective observational studies. Diabetologia. 2005;48(12):2460–2469. doi: 10.1007/s00125-005-0023-4. [DOI] [PubMed] [Google Scholar]
- De Felice FG, Vieira MN, Bomfim TR, Decker H, Velasco PT, Lambert MP, Viola KL, Zhao WQ, Ferreira ST, Klein WL. Protection of synapses against Alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc Natl Acad Sci U S A. 2009;106(6):1971–1976. doi: 10.1073/pnas.0809158106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27(5):457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- Desrocher M, Rovet J. Neurocognitive correlates of type 1 diabetes mellitus in childhood. Child Neuropsychol. 2004;10(1):36–52. doi: 10.1076/chin.10.1.36.26241. [DOI] [PubMed] [Google Scholar]
- Francis GJ, Martinez JA, Liu WQ, Xu K, Ayer A, Fine J, Tuor UI, Glazner G, Hanson LR, Frey WH, 2nd, Toth C. Intranasal insulin prevents cognitive decline, cerebral atrophy and white matter changes in murine type I diabetic encephalopathy. Brain. 2008;131(Pt 12):3311–3334. doi: 10.1093/brain/awn288. [DOI] [PubMed] [Google Scholar]
- Grillo CA, Piroli GG, Wood GE, Reznikov LR, McEwen BS, Reagan LP. Immunocytochemical analysis of synaptic proteins provides new insights into diabetes-mediated plasticity in the rat hippocampus. Neuroscience. 2005;136(2):477–486. doi: 10.1016/j.neuroscience.2005.08.019. [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K. Neuronal cytoskeleton in the biology of Alzheimer disease. Prog Clin Biol Res. 1989;317:745–753. [PubMed] [Google Scholar]
- Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z, Peng Y, Cambareri G, Rocher A, Mobbs CV, Hof PR, Pasinetti GM. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J. 2004;18(7):902–904. doi: 10.1096/fj.03-0978fje. [DOI] [PubMed] [Google Scholar]
- Idan-Feldman A, Schirer Y, Polyzoidou E, Touloumi O, Lagoudaki R, Grigoriadis NC, Gozes I. Davunetide (NAP) as a preventative treatment for central nervous system complications in a diabetes rat model. Neurobiol Dis. 2011;44(3):327–339. doi: 10.1016/j.nbd.2011.06.020. [DOI] [PubMed] [Google Scholar]
- Jiang H, Guo W, Liang X, Rao Y. Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell. 2005;120(1):123–135. doi: 10.1016/j.cell.2004.12.033. [DOI] [PubMed] [Google Scholar]
- Jo J, Whitcomb DJ, Olsen KM, Kerrigan TL, Lo SC, Bru-Mercier G, Dickinson B, Scullion S, Sheng M, Collingridge G, Cho K. Abeta(1-42) inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3beta. Nat Neurosci. 2011;14(5):545–547. doi: 10.1038/nn.2785. [DOI] [PubMed] [Google Scholar]
- Jolivalt CG, Hurford R, Lee CA, Dumaop W, Rockenstein E, Masliah E. Type 1 diabetes exaggerates features of Alzheimer’s disease in APP transgenic mice. Exp Neurol. 2010;223(2):422–431. doi: 10.1016/j.expneurol.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolivalt CG, Lee CA, Beiswenger KK, Smith JL, Orlov M, Torrance MA, Masliah E. Defective insulin signaling pathway and increased glycogen synthase kinase-3 activity in the brain of diabetic mice: parallels with Alzheimer’s disease and correction by insulin. J Neurosci Res. 2008;86(15):3265–3274. doi: 10.1002/jnr.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolivalt CG, Ramos KM, Herbetsson K, Esch FS, Calcutt NA. Therapeutic efficacy of prosaposin-derived peptide on different models of allodynia. Pain. 2006;121(1–2):14–21. doi: 10.1016/j.pain.2005.11.013. [DOI] [PubMed] [Google Scholar]
- Kasa P, Papp H, Kovacs I, Forgon M, Penke B, Yamaguchi H. Human amyloid-beta1-42 applied in vivo inhibits the fast axonal transport of proteins in the sciatic nerve of rat. Neurosci Lett. 2000;278(1–2):117–119. doi: 10.1016/s0304-3940(99)00863-0. [DOI] [PubMed] [Google Scholar]
- Ke YD, Delerue F, Gladbach A, Gotz J, Ittner LM. Experimental diabetes mellitus exacerbates tau pathology in a transgenic mouse model of Alzheimer’s disease. PLoS One. 2009;4(11):e7917. doi: 10.1371/journal.pone.0007917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korneyev AY. Stress-induced tau phosphorylation in mouse strains with different brain Erk 1 + 2 immunoreactivity. Neurochem Res. 1998;23(12):1539–1543. doi: 10.1023/a:1020980004539. [DOI] [PubMed] [Google Scholar]
- Lee CC, Huang CC, Hsu KS. Insulin promotes dendritic spine and synapse formation by the PI3K/Akt/mTOR and Rac1 signaling pathways. Neuropharmacology. 2011;61(4):867–879. doi: 10.1016/j.neuropharm.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 2001;20(1–2):27–39. doi: 10.1093/emboj/20.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupien SB, Bluhm EJ, Ishii DN. Effect of IGF-I on DNA, RNA, and protein loss associated with brain atrophy and impaired learning in diabetic rats. Neurobiol Dis. 2006;21(3):487–495. doi: 10.1016/j.nbd.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Magarinos AM, Jain K, Blount ED, Reagan L, Smith BH, McEwen BS. Peritoneal implantation of macroencapsulated porcine pancreatic islets in diabetic rats ameliorates severe hyperglycemia and prevents retraction and simplification of hippocampal dendrites. Brain Res. 2001;902(2):282–287. doi: 10.1016/s0006-8993(01)02400-3. [DOI] [PubMed] [Google Scholar]
- Martinez A. Preclinical efficacy on GSK-3 inhibitors: towards a future generation of powerful drugs. Med Res Rev. 2008;28(5):773–796. doi: 10.1002/med.20119. [DOI] [PubMed] [Google Scholar]
- Mizisin AP, Steinhardt RC, O’Brien JS, Calcutt NA. TX14(A), a prosaposin-derived peptide, reverses established nerve disorders in streptozotocin-diabetic rats and prevents them in galactose-fed rats. J Neuropathol Exp Neurol. 2001;60(10):953–960. doi: 10.1093/jnen/60.10.953. [DOI] [PubMed] [Google Scholar]
- Musen G, Lyoo IK, Sparks CR, Weinger K, Hwang J, Ryan CM, Jimerson DC, Hennen J, Renshaw PF, Jacobson AM. Effects of type 1 diabetes on gray matter density as measured by voxel-based morphometry. Diabetes. 2006;55(2):326–333. doi: 10.2337/diabetes.55.02.06.db05-0520. [DOI] [PubMed] [Google Scholar]
- Nistico R, Cavallucci V, Piccinin S, Macri S, Pignatelli M, Mehdawy B, Blandini F, Laviola G, Lauro D, Mercuri NB, D’Amelio M. Insulin Receptor beta-Subunit Haploinsufficiency Impairs Hippocampal Late-Phase LTP and Recognition Memory. Neuromolecular medicine. 2012 doi: 10.1007/s12017-012-8184-z. in press. [DOI] [PubMed] [Google Scholar]
- Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, Gaynor K, Wang L, LaFrancois J, Feinstein B, Burns M, Krishnamurthy P, Wen Y, Bhat R, Lewis J, Dickson D, Duff K. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci U S A. 2005;102(19):6990–6995. doi: 10.1073/pnas.0500466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39(3):409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Okawa Y, Ishiguro K, Fujita SC. Stress-induced hyperphosphorylation of tau in the mouse brain. FEBS Lett. 2003;535(1–3):183–189. doi: 10.1016/s0014-5793(02)03883-8. [DOI] [PubMed] [Google Scholar]
- Ott A, Stolk RP, Hofman A, van Harskamp F, Grobbee DE, Breteler MM. Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia. 1996;39(11):1392–1397. doi: 10.1007/s001250050588. [DOI] [PubMed] [Google Scholar]
- Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, Lo E, Wu D, Saule E, Bouschet T, Matthews P, Isaac JT, Bortolotto ZA, Wang YT, Collingridge GL. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron. 2007;53(5):703–717. doi: 10.1016/j.neuron.2007.01.029. [DOI] [PubMed] [Google Scholar]
- Ponce-Lopez T, Liy-Salmeron G, Hong E, Meneses A. Lithium, phenserine, memantine and pioglitazone reverse memory deficit and restore phospho-GSK3beta decreased in hippocampus in intracerebroventricular streptozotocin induced memory deficit model. Brain Res. 2011;1426:73–85. doi: 10.1016/j.brainres.2011.09.056. [DOI] [PubMed] [Google Scholar]
- Reaven GM, Thompson LW, Nahum D, Haskins E. Relationship between hyperglycemia and cognitive function in older NIDDM patients. Diabetes Care. 1990;13(1):16–21. doi: 10.2337/diacare.13.1.16. [DOI] [PubMed] [Google Scholar]
- Reed MN, Hofmeister JJ, Jungbauer L, Welzel AT, Yu C, Sherman MA, Lesne S, LaDu MJ, Walsh DM, Ashe KH, Cleary JP. Cognitive effects of cell-derived and synthetically derived Abeta oligomers. Neurobiol Aging. 2011;32(10):1784–1794. doi: 10.1016/j.neurobiolaging.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E, Torrance M, Adame A, Mante M, Bar-on P, Rose JB, Crews L, Masliah E. Neuroprotective effects of regulators of the glycogen synthase kinase-3beta signaling pathway in a transgenic model of Alzheimer’s disease are associated with reduced amyloid precursor protein phosphorylation. J Neurosci. 2007;27(8):1981–1991. doi: 10.1523/JNEUROSCI.4321-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronnemaa E, Zethelius B, Sundelof J, Sundstrom J, Degerman-Gunnarsson M, Lannfelt L, Berne C, Kilander L. Glucose metabolism and the risk of Alzheimer’s disease and dementia: a population-based 12 year follow-up study in 71-year-old men. Diabetologia. 2009;52(8):1504–1510. doi: 10.1007/s00125-009-1393-9. [DOI] [PubMed] [Google Scholar]
- Ryan CM, Williams TM. Effects of insulin-dependent diabetes on learning and memory efficiency in adults. J Clin Exp Neuropsychol. 1993;15(5):685–700. doi: 10.1080/01688639308402589. [DOI] [PubMed] [Google Scholar]
- Ryan CM, Williams TM, Finegold DN, Orchard TJ. Cognitive dysfunction in adults with type 1 (insulin-dependent) diabetes mellitus of long duration: effects of recurrent hypoglycaemia and other chronic complications. Diabetologia. 1993;36(4):329–334. doi: 10.1007/BF00400236. [DOI] [PubMed] [Google Scholar]
- Schmidt R, Launer LJ, Nilsson LG, Pajak A, Sans S, Berger K, Breteler MM, de Ridder M, Dufouil C, Fuhrer R, Giampaoli S, Hofman A. Magnetic resonance imaging of the brain in diabetes: the Cardiovascular Determinants of Dementia (CASCADE) Study. Diabetes. 2004;53(3):687–692. doi: 10.2337/diabetes.53.3.687. [DOI] [PubMed] [Google Scholar]
- Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J. 1993;296 ( Pt 1):15–19. doi: 10.1042/bj2960015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol. 1997;56(8):933–944. doi: 10.1097/00005072-199708000-00011. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30(4):572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Toth C, Schmidt AM, Tuor UI, Francis G, Foniok T, Brussee V, Kaur J, Yan SF, Martinez JA, Barber PA, Buchan A, Zochodne DW. Diabetes, leukoencephalopathy and rage. Neurobiol Dis. 2006;23(2):445–461. doi: 10.1016/j.nbd.2006.03.015. [DOI] [PubMed] [Google Scholar]
- Velayudhan L, Poppe M, Archer N, Proitsi P, Brown RG, Lovestone S. Risk of developing dementia in people with diabetes and mild cognitive impairment. Br J Psychiatry. 2010;196(1):36–40. doi: 10.1192/bjp.bp.109.067942. [DOI] [PubMed] [Google Scholar]
- Xu T, Yu X, Perlik AJ, Tobin WF, Zweig JA, Tennant K, Jones T, Zuo Y. Rapid formation and selective stabilization of synapses for enduring motor memories. Nature. 2009a;462(7275):915–919. doi: 10.1038/nature08389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu WL, von Strauss E, Qiu CX, Winblad B, Fratiglioni L. Uncontrolled diabetes increases the risk of Alzheimer’s disease: a population-based cohort study. Diabetologia. 2009b;52(6):1031–1039. doi: 10.1007/s00125-009-1323-x. [DOI] [PubMed] [Google Scholar]
- Yang G, Pan F, Gan WB. Stably maintained dendritic spines are associated with lifelong memories. Nature. 2009;462(7275):920–924. doi: 10.1038/nature08577. [DOI] [PMC free article] [PubMed] [Google Scholar]