

Abstract
Objective:
To identify the genetic variant that causes autosomal dominantly inherited motor neuron disease in a 4-generation Israeli-Arab family using genetic linkage and whole exome sequencing.
Methods:
Genetic linkage analysis was performed in this family using Illumina single nucleotide polymorphism chips. Whole exome sequencing was then undertaken on DNA samples from 2 affected family members using an Illumina 2000 HiSeq platform in pursuit of potentially pathogenic genetic variants that comigrate with the disease in this pedigree. Variants meeting these criteria were then screened in all affected individuals.
Results:
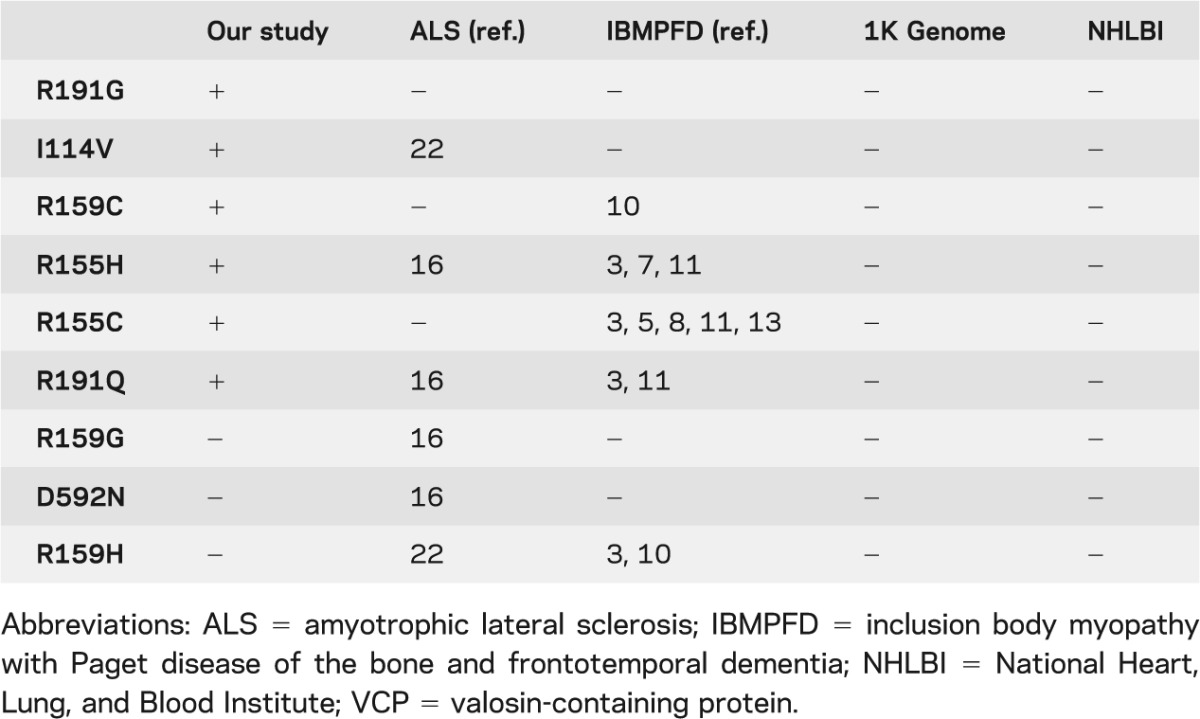
A novel mutation (p.R191G) in the valosin-containing protein (VCP) gene was identified in the index family. Direct sequencing of the VCP gene in a panel of DNA from 274 unrelated individuals with familial amyotrophic lateral sclerosis (FALS) revealed 5 additional mutations. Among them, 2 were previously identified in pedigrees with a constellation of inclusion body myopathy with Paget disease of the bone and frontotemporal dementia (IBMPFD) and in FALS, and 2 other mutations (p.R159C and p.R155C) in IBMPFD alone. We did not detect VCP gene mutations in DNA from 178 cases of sporadic amyotrophic lateral sclerosis.
Conclusions:
We report a novel VCP mutation identified in an amyotrophic lateral sclerosis family (p.R191G) with atypical clinical features. In our experience, VCP mutations arise in approximately 1.5% of FALS cases. Our study supports the view that motor neuron disease is part of the clinical spectrum of VCP-associated disease.
In 2001, a locus on chromosome 9p13.3-12 was genetically linked to 3 dominantly inherited disease traits: inclusion body myopathy (IBM) with Paget disease of the bone and frontotemporal dementia (IBMPFD).1 Subsequently, mutations in the valosin-containing protein (VCP) gene were identified as the genetic cause of IBMPFD; 38 IBMPFD families and 19 missense VCP mutations have been reported to date (table e-1 on the Neurology® Web site at www.neurology.org).2–15 Recently, a study using whole exome sequencing identified a VCP mutation (p.R191Q) in a family in which some individuals presented clinically with amyotrophic lateral sclerosis (ALS). The frequency of VCP mutations in familial ALS (FALS) patients is estimated at approximately 2%.16
Herein, we report that genetic linkage analysis coupled with whole exome sequencing from 2 patients in an Israeli-Arab family with dominantly inherited ALS identified a novel VCP gene mutation comigrating with affected individuals; subsequently, we identified VCP gene mutations in 5 additional families with ALS.
METHODS
Subjects
Standard protocol approvals, registrations, and patient consents were obtained. All studies were performed under an ethically approved institutional protocol. In addition to the index family, 178 FALS cases, 178 sporadic ALS cases, and 184 controls were screened for VCP mutations. Additionally, 96 patients with FALS were screened for exon 5 of VCP. We define ALS as familial when (a) it is encountered in more than one individual within a pedigree, and (b) in at least one member of the family the diagnosis was confirmed by a neurologist. SOD1, TARDBP, and FUS mutations were excluded. For VCP screening, control samples were obtained from Coriell Cell Repositories (Coriell Institute, Camden, NJ).
Linkage analysis
Genetic linkage analysis was performed in our index family (FALS682) diagnosed with ALS by El Escorial criteria17 (figure 1). Genotypes for 6 affected individuals and 7 unaffected relatives, which allowed reconstruction of a seventh ALS case, were generated for >900,000 known genetic variants using the Genome-Wide Human SNP 6.0 Array (www.affymetrix.com). Data were reformatted using Alohamora; multipoint logarithm (decadic) of the odds (LOD) scores were calculated using FASTLINK (http://www.ncbi.nlm.nih.gov/CBBresearch/Schaffer/fastlink.html).18
Figure 1. Pedigree of valosin-containing protein (VCP) family FALS682.
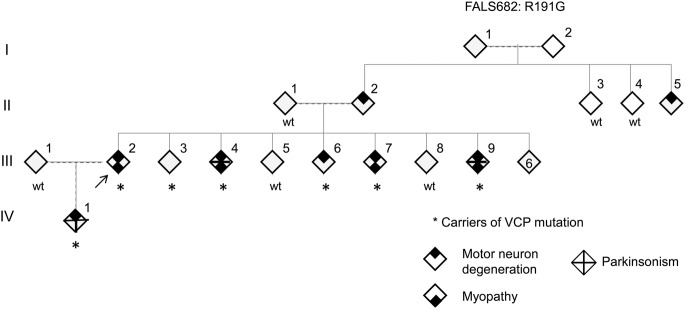
The arrow indicates the proband. Individuals III:2 and III:4 underwent full exome sequencing. Phenotype status is denoted by symbols as indicated. Individuals III:4 and III:9 had motor neuron disease, myopathy, and parkinsonism. Genders have been anonymized. FALS = familial amyotrophic lateral sclerosis.
Chromosomal segments with LOD scores >1.0 were reviewed for expressed elements; genetic variants from whole exome sequencing were excluded for segments where the LOD scores were negative.
Whole exome sequencing
We performed whole exome sequencing on 2 individuals (III:2 and III:4) from FALS682 (figure 1) using the 37-MB Agilent SureSelect Human All Exon Kit (Agilent Technologies, Inc., Santa Clara, CA) and an Illumina Genome Analyzer II (Illumina, Inc., San Diego, CA) with paired-end reads. Our coverage of the targeted region was 74× and 78×, with 95% and 91% of the targeted regions having at least 5× coverage. DNA was aligned to the reference genome (NCBI Build 36 Ensembl release 50) using the BWA software.19 SAMtools was used for variant identification using the pileup command with the –c option and default settings.20 The variants were then filtered using SAMtools’ variation filter with the default settings but removing the filter for a maximum allowed coverage per variant by setting it to 10 million. All single nucleotide variations were screened for quality by requiring a single nucleotide polymorphism quality score of at least 20, a consensus score of at least 20, and at least 3 reads supporting the variant; for indels, an indel quality score of 50, an indel consensus score of 20, at least 3 reads supporting the variant, and a variant/reference read ratio between 0.2 and 5 were required. As a control for this study, we used both the Single Nucleotide Polymorphism database (dbSNP) and a database of 823 full exome and genome sequences obtained in the Duke University Center for Human Genome variation to exclude common variants. Using SVA (SequenceVariantAnalyzer) software (http://www.svaproject.org/), we then grouped rare variants into categories of differing functional significance.21
Mutational analysis
DNA was isolated from venous blood. Whole genome amplification was performed on the DNA samples using the Illustra GenomiPhi V2 DNA Amplification Kit (catalog no. 25-6600-31; GE HealthCare, Pittsburgh, PA). VCP exons 1 through 17 were amplified by PCR with primers that were designed using Primer 3.0 software (table e-2).
RESULTS
Linkage analysis in FALS682
Analysis of genotypes from 13 members of FALS682 (6 affected, 7 unaffected) documented 27 loci with multipoint LOD scores >1.0 (table e-3). LOD scores were also generated using affected individuals only (figure e-1).
Mutational analysis
We next sought mutations identified by full exome sequencing of 2 affected individuals that were (a) present under the linkage peaks, and (b) subsequently verified to comigrate with the disease when the mutation in question was tested in the full pedigree. Four variants fulfilled these criteria: manose-binding lectin (protein c) 2, soluble (COLECII); syntrophin beta 1 (SNTB1); thumb-domain containing 2 (THUMPD2); and VCP (table e-4). VCP was the only of these 4 genes whose variants were previously documented to be associated with the multisystem complex of disorders in our family (ALS, myopathy, parkinsonism). Accordingly, our evaluations subsequently focused on the VCP gene in which there was a heterozygous mutation at bp 35,055,253 converting a cytosine to a guanosine, changing the codon (CGA>GGA) and the corresponding amino acid (arginine 191 > glycine).
We next completed mutational screening of all exons of VCP in 178 unrelated FALS and 178 sporadic ALS DNA samples. This yielded 4 additional mutations only among FALS cases: R155H in FALS001, R155C in FALS614, R159C in FALS007, and I114V in FALS354 (table 1, figure e-2). Because we subsequently learned that affected individuals from family FALS614 have previously been reported to have the R155C VCP gene mutation, this family is not described in detail here.2 Because most of the mutations were found in exon 5 of VCP, we subsequently screened an additional set of 96 FALS DNA samples specifically for mutations in exon 5, identifying R191Q in FALS385 (figure e-2). None of these mutations were observed in 184 control subjects, dbSNP (hg18), 1000 Genomes (http://www.1000genomes.org/home), or in approximately 3,500 European and 2,000 Afro-American DNA samples, as tabulated in the summarized National Heart, Lung, and Blood Institute database (http://evs.gs.washington.edu/EVS/).
Table 1.
Main clinical features of VCP families in this report
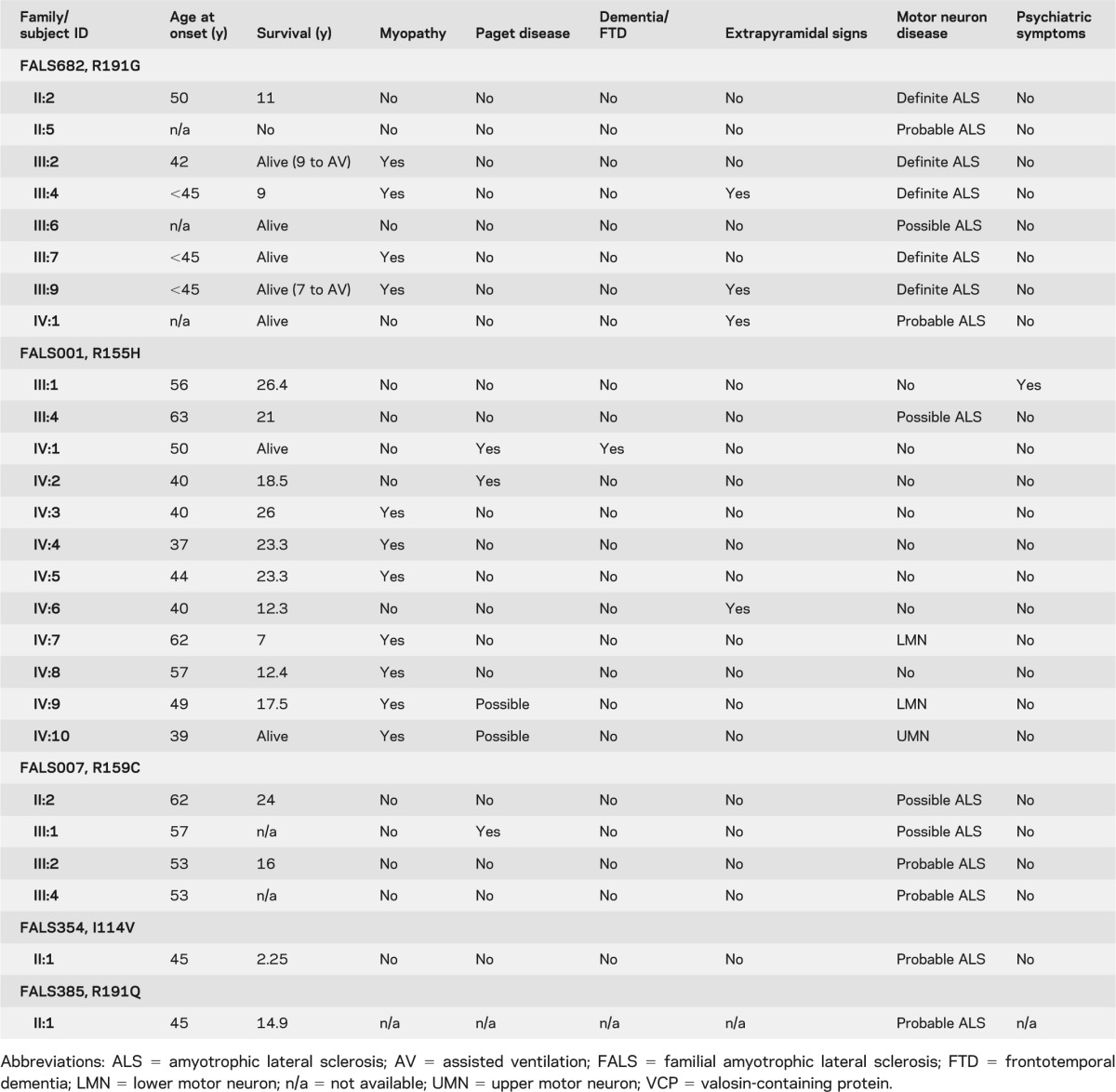
Clinical description of VCP families
The pedigrees and clinical profiles for these families with VCP gene mutations are shown in figure 1, figure e-2, and table 1, and summarized briefly here.
FALS682, R191G
The proband (III:2) is a 56-year-old Israeli-Arab patient followed for 11 years since presenting with muscle cramps beginning at age 40 and progressive nasality of speech that started at age 42 (figure 1). The proband's parents (II:1 and II:2) are remotely related. The patient was referred to a neurologist because of proximal weakness, predominantly in the lower extremities, and difficulty climbing stairs, running, and walking. A nasal voice but no difficulty swallowing was also observed. Other cranial nerves were normal. Marked hyperlordosis and a waddling gait suggested an underlying myopathy; there was weakness and atrophy of some shoulder girdle muscles and lower cervical and upper thoracic paraspinal muscles. Fasciculations were seen in these muscles and in the pectorals and vastus lateralis bilaterally. Reflexes were brisk but not pathologic. There were no sensory, cerebellar, autonomic, or cognitive problems. Creatine phosphokinase levels ranged between 500 and 800 IU (upper normal limit: 150 IU). Alkaline phosphatase was normal. Motor and sensory nerve conduction studies were normal. On the first EMG, the motor units in proximal muscles (deltoid, supraspinatus, and quadriceps bilaterally) had both myopathic and neurogenic features (both small and enlarged polyphasic) with reduced recruitment and a few fibrillations, positive sharp waves, and fasciculations of complex morphology, the latter suggesting reinnervation. During the subsequent years, the weakness and atrophy progressed to involve distal muscles. Pyramidal signs including bilateral upgoing toes became evident, and the patient became pseudobulbar with severe dysphagia and unintelligible speech. Repeated EMGs demonstrated widespread lower motor neuron involvement with complex, unstable fasciculations in many muscles including thoracic paraspinals. More than 8 years after disease onset, urinary and fecal incontinence complicated the clinical picture and, after 1 more year, respiratory insufficiency required tracheotomy and assisted ventilation. The patient retains normal cognition. Ancillary investigations excluded Kennedy syndrome, hexosaminidase-A deficiency, Pompe disease, SOD1 mutations, and Paget disease of the bone.
The proband's parent (II:2) had a nasal voice similar to that of the patient and, since the age of 50 years, severe dysphagia, progressive tetraparesis with muscle atrophy, and fasciculations in the lower extremities. II:2 died at the age of 61 years, completely anarthric, wheelchair ridden, and nourished by feeding gastrostomy. In addition, II:5 had slowly progressive ALS features. No DNA from these 2 individuals is available. The patient is the eldest among 14 siblings; III:4, III:7, and III:9 presented during the last years with a clinical picture of definite ALS similar to that of this patient. For many years, they experienced insidiously progressing dysphonia with nasal speech that was perceived as a "family trait" and not as a health problem. Mild dysphagia for liquids was sporadically noticed. In each, the onset of limb weakness occurred before age 45 years, with a proximal distribution involving mainly the pelvic girdle, a waddling gait, muscle cramps, and proximal muscle atrophy affecting the paraspinal muscles or producing scapular winging. All complained of lower back pain and a “rigid spine.” In each, the creatine phosphokinase was normal or moderately elevated and the alkaline phosphatase was within normal limits. In 2 patients, early EMGs revealed the same mixed myopathic-neuropathic motor units as III:2. Over time, both upper and lower motor neuron features became widespread, fulfilling the clinical and electrophysiologic El Escorial criteria for definite ALS. All affected individuals have normal behavior, memory, and judgment; 2 (III:4 and III:9) had parkinsonian features. Disease progression was rather slow: 7 years to assisted ventilation (III:9) and 9 years to death (III:4); patient III:7, after 6 years of follow-up, is sporadically using a wheelchair but is independent for most daily life activities.
III:6 and IV:1 fulfill clinical and EMG criteria for possible and probable ALS; among other features, both have gait problems and hyperlordosis and IV:1 has nasal speech, mild swallowing problems, and mild parkinsonian features. After a follow-up of 3 and 4 years, they are independent, active, and cognitively normal.
FALS001, R155H
The mutation R155H was identified in the proband of family FALS001 (IV:10) (figure e-2a). At 50 years of age, this individual developed asymmetric proximal weakness, mainly in the lower limbs, and complained of pain in the back and legs. EMG studies showed myopathic changes as well as fibrillations and positive waves. The proband's parent (III:4), who had been diagnosed with typical limb-onset ALS at the age of 63 years, died before the proband developed the first neuromuscular symptoms; therefore, the DNA was not collected. Other members of this family presented with features of a multisystem disorder. In their fifth decade, subjects IV:3, IV:4, IV:5, IV:7, IV:8, and IV:9 developed muscle weakness and atrophy in a limb girdle distribution, with scapular winging, weakness of neck flexor muscles, and fasciculations. In most affected individuals, EMG again revealed a myopathic pattern as well as abnormal insertional activity, fibrillations, and positive sharp waves. Muscle biopsy on subject IV:5 disclosed marked distortion of the normal architecture, variation in fiber size, and abundant atrophic angulated fibers indicating denervation. A few fibers contained rimmed vacuoles suggesting IBM. In addition, Paget disease of the bone (IV:1 and IV:2), frontotemporal dementia (IV:1), psychiatric symptoms (III:1), parkinsonism (IV:6), and heart block that required pacemaker placement (IV:9) were other distinctive clinical features in this family. The R155H mutation comigrated with the disease traits as shown in figure e-2a.
FALS007, R159C
The VCP gene mutation R159C was identified in the proband of family FALS007 (III:1 in figure e-2b) who was affected with a slowly progressive, limb-onset motor neuron disease classified as possible ALS by El Escorial criteria. Neurologic examination revealed spasticity and hyperreflexia in the lower extremities, in addition to gait difficulties and marked bilateral thenar atrophy. Paget disease of the bone was diagnosed in this patient before neuromuscular symptoms started. Subjects II:1, II:2, III:2, and III:4 were also diagnosed with ALS. Individual III:4 was confirmed to carry the same VCP mutation as the proband. No DNA was available from the other affected relatives. Of note, III:3, IV:1, IV:2, and IV:3, whose ages at DNA donation were 68, 37, 31, and 25 years, respectively, were identified as subjects at risk because they carried the mutation (figure e-2b) without clinical symptoms. At least for IV:1, IV:2, and IV:3, being asymptomatic may reflect their relatively young age when DNA was donated.
FALS385, R191Q
p.R191Q was identified in the proband of family FALS385 (II:1), a 45-year-old patient who developed typical ALS. Her medical history was unremarkable except for 2 miscarriages. DNA from individual I:1, also affected, could not be collected (figure e-2c).
FALS354, I114V
This mutation was identified in subject II:1. At age 45 years, II:1 (figure e-2d) developed rapidly progressive weakness in the upper limbs with severe interosseous muscle atrophy, fasciculations, and slightly hyperactive reflexes in both arms. Fasciculations appeared in the tongue, with weakness in the neck muscles and lower limbs. Death occurred 2 years after onset with a diagnosis of probable ALS (EL Escorial criteria). Importantly (see below), his unaffected parent was also confirmed to carry the mutation; no DNA was obtained from the affected parent who died of ALS before the proband developed the first symptoms (figure e-2d).
DISCUSSION
The combination of genetic linkage analysis and whole exome sequencing in family FALS682 disclosed a novel missense variant (R191G) in exon 5 of VCP that cosegregated with the disease. This mutation has not been previously described in either ALS or IBMPFD syndrome. Mutational screening of VCP in a large set of FALS cases revealed 5 additional mutations, 4 in exon 5 and previously described in IBMPFD (R159C, R155H, R155C, and R191Q) and 1 in exon 4 (I114V) (table 2). Two of these mutations (R159C and R155C) have not previously been described in ALS, in contrast with R155H, R191Q, and I114V.16,22 Each of these mutations was shown to comigrate with the disease in our families, with 2 exceptions: R191Q (FALS385) and I114V (FALS354). It is quite likely that R191Q can cause ALS, given that it was recently reported to comigrate with ALS within an Italian family with ALS.16 In contrast, I114V was identified in the proband in FALS354, whose affected parent died of ALS and whose unaffected parent carried the I114V mutation but was asymptomatic. It is possible that this is a nonpathogenic polymorphism as previously suggested22 although, as noted above, it is not found in extensive databases covering several thousand control alleles. Lastly, R159C mutation was identified in 2 affected individuals (III:1 and III:4) in FALS007 but also in subject III:3, for whom no evidence of disease has been reported at age 68 years, which shows an incomplete penetrance for this mutation in this family.
Table 2.
VCP mutations identified in ALS to date
The pedigrees reported herein were all initially identified as families with ALS. However, because most of the VCP mutations identified in these families had previously been reported in the context of IBMPFD, we further investigated the families for other features of IBMPFD. Thus, in FALS001, the first individual to come to clinical attention (III:4) carried the diagnosis exclusively of ALS. Subsequently, the proband in this family (IV:10) and others (IV:3, IV:4, IV:5, IV:7, IV:8, and IV:9) presented with a clinical picture of IBM. Moreover, the proband had intense pain consistent with the bone pain of Paget disease, as did 2 other affected members of his family (IV:1 and IV:2). In the same family, frontotemporal dementia was diagnosed in subject IV:1, who was also a carrier of the R155H mutation. V:1 and V:2 were asymptomatic carriers. The R159C mutation comigrated within FALS007 among affected members diagnosed with ALS without evidence of dementia or muscle disease. However, subject III:1 in that family had been diagnosed with Paget disease before the onset of neuromuscular symptoms. Clinical features other than ALS were not observed in FALS385 and FALS354, perhaps because these pedigrees were small and, as mentioned, because of the uncertain pathogenic role or reduced penetrance of I114V.
A set of striking clinical findings (psychiatric symptoms, cardiomyopathy, cataracts, liver disease, akinetic rigid syndrome, and myoclonus) have been described in some IBMPFD pedigrees.4,5,7,11,15,23,24 We observed parkinsonism in III:4, III:9, and IV:1 in FALS682 and IV:6 in FALS001. III:1 and IV:9 in FALS001 presented with psychiatric symptoms and heart block, respectively; although these individuals are obligatory carriers, we cannot be certain that these 2 conditions are a consequence of R155H mutation.
The VCP gene encodes an abundant, 97-kDa, multifunctional adenosine triphosphatase involved in multiple cellular processes. Importantly, it has been implicated in protein degradation via both the ubiquitin-proteasome and autophagy pathways, and in membrane fusion, transcriptional activation, apoptosis, and molecular chaperoning.25–28 The 6 VCP mutations detected in this series are predominantly in the CDC48 substrate binding domain or the associated L1 linker between the CDC48 and D1 domains (figure 2). These residues are highly conserved across species; these mutations were predicted to be pathogenic by Pmut (http://mmb2.pcb.ub.es:8080/PMut/), with the exception of I114V, which was predicted to be neutral.
Figure 2. Valosin-containing protein (VCP) mutations and crystal structure of VCP protein.

(A) VCP mutations identified in inclusion body myopathy with Paget disease of the bone and frontotemporal dementia (IBMPFD) syndrome and amyotrophic lateral sclerosis (ALS): VCP mutations identified in individuals with the full IBMPFD syndrome are displayed in black. Mutations identified in this study in individuals with ALS (and other components of IBMPFD) are in red; those reported previously are in blue,16 whereas mutations detected both in the previous report and in the present study are in green. As illustrated, most of the mutations fall within the CDC48 polyubiquitin binding domain and the associated linker region L1 suggesting that these mutations interfere with the interaction of the VCP protein with polyubiquitinated substrates, and consequently with protein degradation by the proteasome. (B) Crystal structure of VCP protein showing the clustered locations of the mutated residues detected in familial ALS pedigrees in this study. The arginine at position 191 (implicated in the mutations p.R191G and in p.R191Q) is on a flexible loop corresponding to the linker between CDC48 and D1 domains (L1). The arginines at positions 155 and 159 (p.R155H and p.R159C) and the isoleucine at position 114 (p.I114V) are in β sheets within CDC48 domain. ADP = adenosine diphosphate; ATP = adenosine triphosphate; ATPase = adenosine triphosphatase.
From a pathologic point of view, ubiquitin and TAR DNA-binding protein 43 positive inclusions have been identified in both ALS and frontotemporal dementia, as well as in IBM; in IBM, their distribution is prominently in myonuclei.24,29 Ubiquitinated inclusions have been described in the osteoclasts of patients affected with Paget disease of the bone.30 These findings suggest that ubiquitin-dependent pathways have an important role in the pathogenesis of all these entities.
The major observation from this study is that VCP mutations cause some cases of FALS (approximately 2% of our pedigrees). Additionally, we report a novel VCP gene mutation that clusters in the same domain of the VCP protein as most of the other mutations. In contrast with data from FALS, we failed to identify any VCP mutation in sporadic ALS, suggesting that the contribution of VCP to the sporadic form of ALS is marginal.31 These findings emphasize the importance of screening for other elements of the IBMPFD syndrome in the context of FALS. Furthermore, the diversity of phenotypes associated with genetic variants in the VCP gene is consistent with the view that ALS is a heterogeneous disorder. It is striking that mutations in at least 3 different genes can cause ALS with frontotemporal dementia (VCP, TARDBP, and C9orf72).32–34 Presumably, a nuanced understanding of the molecular distinctions and points of convergence in these diverse forms of ALS will ultimately bear directly upon strategies for treating ALS.
Supplementary Material
ACKNOWLEDGMENT
Sample contributions were obtained through the auspices of many individuals (see contributors appendix on the Neurology® Web site at www.neurology.org). The authors acknowledge the assistance of Dr. Daryl Bosco in generating figure 2 illustrating the structure of the VCP protein and Dr. Jerry Mendell for providing clinical information for FALS614. The authors also thank all of the patients and their families for participating in this investigation.
Glossary
- ALS
amyotrophic lateral sclerosis
- dbSNP
Single Nucleotide Polymorphism database
- FALS
familial amyotrophic lateral sclerosis
- IBM
inclusion body myopathy
- IBMPFD
inclusion body myopathy with Paget disease of the bone and frontotemporal dementia
- LOD
logarithm (decadic) of the odds
- VCP
valosin-containing protein
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
P. González-Pérez: sequencing analysis, interpretation of data, drafting of the manuscript. E.T. Cirulli: exome sequencing analysis, revision of the manuscript. V.E. Drory: collection of clinical data, genetic database, revision of the manuscript. R. Dabby: collection of clinical data, revision of the manuscript. P. Nisipeanu: collection of clinical data, revision of the manuscript. R.L. Carasso: collection of clinical data, revision of the manuscript. M. Sadeh: collection of clinical data, revision of the manuscript. A. Fox: genetic databases, revision of the manuscript. B.W. Festoff: collection of clinical data and review of the manuscript. P.C. Sapp: linkage study, revision of the manuscript. D. Mckenna-Yasek: collection of clinical data, revision of the manuscript. D.B. Goldstein: exome sequencing analysis, revision of the manuscript. R. Brown: design and conceptualization of the study, drafting and revision of the manuscript. S.C. Blumen: design of the study, collection of clinical data, drafting and revision of the manuscript.
Study funding
Supported by National Institute for Neurological Disease and Stroke (grants 1RC2NS070342-02 to R.H.B. and D.G.; 5RO1-NS050557-05 to R.H.B.). R.H.B. also receives support from the Angel Fund, the ALS Association, Project ALS, P2ALS, Pierre L. de Bourgknecht ALS Research Foundation, the Al-Athel ALS Foundation, and the ALS Therapy Alliance. P.G.-P. receives support from the Alfonso Martin Escudero Foundation (Madrid). P.S. was supported through the auspices of Dr. H. Robert Horvitz, an Investigator at the Howard Hughes Medical Institute in the Department of Biology at the Massachusetts Institute of Technology.
DISCLOSURE
P. González-Pérez, E. Cirulli, V. Drory, R. Dabby, P. Nisipeanu, R. Carasso, M. Sadeh, A. Fox, B. Festoff, P. Sapp, D. Mckenna-Yasek, and D. Goldstein report no disclosures. R. Brown is a consultant to Biogen Idec and Iperian; he is a cofounder of AviTx, Inc. S. Blumen reports no disclosures. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Kovach MJ, Waggoner B, Leal SM, et al. Clinical delineation and localization to chromosome 9p13.3-p12 of a unique dominant disorder in four families: hereditary inclusion body myopathy, Paget disease of the bone, and frontotemporal dementia. Mol Genet Metab 2001;74:458–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Watts GD, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 2004;36:377–381 [DOI] [PubMed] [Google Scholar]
- 3.Haubenberger D, Bittner RE, Rauch-Schorny S, et al. Inclusion body myopathy and Paget disease is linked to a novel mutation in the VCP gene. Neurology 2005;65:1304–1305 [DOI] [PubMed] [Google Scholar]
- 4.Guyant-Marèchal L, Laquerrière A, Duyckaerts C, et al. Valosin-containing protein gene mutations: clinical and neuropathological features. Neurology 2006;67:644–651 [DOI] [PubMed] [Google Scholar]
- 5.Watts GD, Thomasova D, Ramdean SK, et al. Novel VCP mutations in inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia. Clin Genet 2007;72:420–426 [DOI] [PubMed] [Google Scholar]
- 6.Viassolo V, Previtali SC, Schiatti E, et al. Inclusion body myopathy, Paget’s disease of the bone and frontotemporal dementia: recurrence of the VCP R155H mutation in an Italian family and implications for genetic counselling. Clin Genet 2008;74:54–60 [DOI] [PubMed] [Google Scholar]
- 7.Gidaro T, Modoni A, Sabatelli M, Tasca G, Broccolini A, Mirabella M. An Italian family with inclusion-body myopathy and frontotemporal dementia due to mutation in VCP gene. Muscle Nerve 2008;37:111–114 [DOI] [PubMed] [Google Scholar]
- 8.Djamshidian A, Schaefer J, Haubenberger D, et al. A novel mutation in the VCP gene (G157R) in a German family with inclusion-body myopathy with Paget disease of bone and frontotemporal dementia. Muscle Nerve 2009;39:389–391 [DOI] [PubMed] [Google Scholar]
- 9.Bersano A, Del Bo R, Lamperti C, et al. Inclusion body myopathy and frontotemporal dementia caused by a novel VCP mutation. Neurobiol Aging 2009;30:752–758 [DOI] [PubMed] [Google Scholar]
- 10.Stojkovic T, Hammouda el H, Richard P, et al. Clinical outcome in 19 French and Spanish patients with valosin-containing protein myopathy associated with Paget’s disease of bone and frontotemporal dementia. Neuromuscul Disord 2009;19:316–323 [DOI] [PubMed] [Google Scholar]
- 11.Kumar KR, Needham M, Mina K, et al. Two Australian families with inclusion body myopathy, Paget’s disease of bone and frontotemporal dementia: novel clinical and genetic findings. Neuromuscul Disord 2010;20:330–334 [DOI] [PubMed] [Google Scholar]
- 12.Kim EJ, Park YE, Kim DS, et al. Inclusion body myopathy with Paget disease of bone and frontotemporal dementia linked to VCP p.Arg155Cys in a Korean family. Arch Neurol 2011;68:787–796 [DOI] [PubMed] [Google Scholar]
- 13.Palmio J, Sandell S, Snominen T, et al. Distinct distal myopathy phenotype caused by VCP gene mutation in a Finnish family. Neuromuscul Disord 2011;21:551–555 [DOI] [PubMed] [Google Scholar]
- 14.Fanganiello RD, Kimonis VE, Corte CC, Nitrini R, Passos-Bueno MR. A Brazilian family with hereditary inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia. Braz J Med Biol Res 2011;44:374–380 [DOI] [PubMed] [Google Scholar]
- 15.Rohrer JD, Warren JD, Reiman D, et al. A novel exon 2 I27V VCP variant is associated with dissimilar clinical syndromes. J Neurol 2011;258:1494–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson JO, Mandrioli J, Benatar M, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010;68:857–864 Erratum in: Neuron 2011;69:397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brooks BR, Miller RG, Swash M, et al. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:293–299 [DOI] [PubMed] [Google Scholar]
- 18.Ruschendorf F, Nurnberg P. ALOHOMORA: a tool for linkage analysis using 10K SNP array data. Bioinformatics 2005;21:2123–2125 [DOI] [PubMed] [Google Scholar]
- 19.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics 2009;25:2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ge D, Ruzzo EK, Shianna KV, et al. SVA: software for annotating and visualizing sequenced human genomes. Bioinformatics 2011;27:1998–2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koppers M, van Blitterwijk MM, Vlam L, et al. VCP mutations in familial and sporadic amyotrophic lateral sclerosis. Neurobiol Aging 2012;33:837.e7–837.e13 [DOI] [PubMed] [Google Scholar]
- 23.Hübbers C, Clemen C, Kesper K, et al. Pathological consequences of VCP mutations on human striated muscle. Brain 2007;130:381–393 [DOI] [PubMed] [Google Scholar]
- 24.Weihl CC, Pestronk A, Kimonis VE. Valosin-containing protein disease: inclusion body myopathy with Paget’s disease of the bone and fronto-temporal dementia. Neuromuscul Disord 2009;19:308–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vij N. AAA ATPase p97/VCP: cellular functions, disease and therapeutic potential. J Cell Mol Med 2008;12:2511–2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolf DH, Stolz A. The Cdc 48 machine in endoplasmic reticulum associated protein degradation. Biochim Biophys Acta 2012;1823:117–124 [DOI] [PubMed] [Google Scholar]
- 27.Ju JS, Fuentealba RA, Miller SE, et al. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol 2009;187:875–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song C, Wang Q, Li CC. Characterization of the aggregation-prevention activity of p97/valosin-containing protein. Biochemistry 2007;46:14889–14898 [DOI] [PubMed] [Google Scholar]
- 29.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar dementia and amyotrophic lateral sclerosis. Science 2006;314:130–133 [DOI] [PubMed] [Google Scholar]
- 30.Nalbandian A, Donkervoort S, Dec E, et al. The multiple faces of valosin-containing protein-associated diseases: inclusion body myopathy with Paget’s disease of bone, frontotemporal dementia, and amyotrophic lateral sclerosis. J Mol Neurosci 2011;45:522–531 [DOI] [PubMed] [Google Scholar]
- 31.Abramzon Y, Johnson JO, Scholz SW, et al. Valosin-containing protein (VCP) mutations in sporadic amyotrophic lateral sclerosis. Neurobiol Aging 2012;33:2231.e1–2231.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008;319:1668–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeJesus-Hernández M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of c9orf72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72:245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Renton AE, Maujonie E, Waite A, et al. A hexanucleotide repeat expansion in c9orf72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72:257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.