

Abstract
Background
One of the major hurdles in studying diabetes pathophysiology is the lack of adequate methodology that allows for direct and real-time determination of glucose transport and metabolism in cells and tissues. In this article, we present a new methodology that adopts frequency-domain fluorescence lifetime imaging microscopy (FD-FLIM) to visualize and quantify the dynamics of intracellular glucose within living cells using a biosensor protein based on fluorescence resonance energy transfer (FRET).
Method
The biosensor protein was developed by fusing a FRET pair, an AcGFP1 donor and a mCherry acceptor to N- and C- termini of a mutant glucose-binding protein (GBP), respectively. The probe was expressed and biosynthesized inside the cells, offering continuous monitoring of glucose dynamics in real time through fluorescence lifetime imaging microscopy (FLIM) measurement.
Results
We transfected the deoxyribonucleic acid of the AcGFP1-GBP-mCherry sensor into murine myoblast cells, C2C12, and continuously monitored the changes in intracellular glucose concentrations in response to the variation in extracellular glucose, from which we determined glucose uptake and clearance rates. The distribution of intracellular glucose concentration was also characterized. We detected a high glucose concentration in a region close to the cell membrane and a low glucose concentration in a region close to the nucleus. The monoexponential decay of AcGFP1 was distinguished using FD-FLIM.
Conclusions
This work enables continuous glucose monitoring (CGM) within living cells using FD-FLIM and a biosensor protein. The sensor protein developed offers a new means for quantitatively analyzing glucose homeostasis at the cellular level. Data accumulated from these studies will help increase our understanding of the pathology of diabetes.
Keywords: biosensor; continuous lifetime imaging; diabetes; FLIM; frequency domain, FRET, GBP, glucose sensor
Introduction
In the wake of an exponential global rise in diabetes, extensive studies have been performed regarding diabetes pathology. However, the mechanisms responsible for blood glucose regulation, which is key in diabetes treatment, have not been fully elucidated as yet. In particular, the complete molecular mechanisms underlying cellular glucose uptake and clearance in diabetes remain obscure. Characterization of these molecular events has been impaired due, in part, to the scarcity of reliable methodologies that can directly monitor glucose dynamics within living cells. Currently, monitoring intracellular glucose dynamics largely relies on nuclear magnetic resonance spectroscopy and inferring glucose uptake from complex glucose clamp and tracer infusion techniques, which indirectly measure intracellular glucose levels.1–4 Another technique, positron emission tomography, is routinely used to estimate glucose metabolism in localized tissue regions, but it does not provide estimates of intracellular glucose dynamics.5,6Many approaches have been tested to circumvent these problems. One in particular, fluorescence resonance energy transfer (FRET), has been applied for developing a number of fluorescent sensor proteins that can be used to monitor blood glucose.7 These sensor proteins, or glucose indicator proteins (GIPs), can be expressed inside the cells to visualize glucose dynamics through FRET-imaging microscopy measurements.8–10
In a FRET glucose sensor protein, enhanced cyan fluorescent protein (ECFP) and enhanced yellow fluorescent protein (EYFP) are linked by a glucose-binding protein (GBP).11,12 When glucose binds to the GBP, it triggers rearrangement of the flap region located on one side of the hinge β-sheet, leading to an alteration in the distance between the ECFP and EYFP and thus generating a FRET signal that can be used for glucose measurement.7,11,12 In essence, these proteins can be used to determine intracellular glucose levels through FRET microscopy measurement,8,13,14 a technique that has been adapted for studying glucose uptake and clearance in a variety of cells under various conditions, offering new insights into glucose metabolism in cells.
However, while FRET intensity-based imaging microscopy measurement is useful for end-point analysis, it is not suitable for the continuous monitoring of glucose dynamics within living cells because of the photobleaching of ECFP and EYFP during repeated exposures for microscopic imaging measurements. For instance, because ECFP is prone to excessive photobleaching, the FRET signal reduces significantly during measurement.15 Overall, photobleachingand crosstalk between the ECFP and the EYFP—caused by their largely overlapping excitation and emission spectra—warrant elaborate estimations of correction factors.16,17
Fluorescence lifetime imaging microscopy (FLIM) measure-ment offers a complementary method to intensity-based FRET analysis. In FLIM, changes in FRET efficiency can be determined by measuring changes in the lifetime of a donor. This lifetime is independent of probe concentration but depends on the chemical and physical properties of the local environment, including the energy transfer from a donor to its acceptor in FRET.18,19 Because only the donor’s lifetime is monitored, the crosstalk between a donor and its acceptor, such as that between ECFP and EYFP, does not affect the lifetime measurement.20 This FLIM approach has been validated by the spectral imaging of similar GFP molecules and interactions between protein and deoxyribonucleic acid (DNA) in living cells.21,22
There are two ways to perform FLIM: time-domain FLIM (TD-FLIM) and frequency-domain FLIM (FD-FLIM).21–23 Time-domain-FLIM is extremely slow to implement and cannot achieve the short acquisition time required for visualizing the dynamics of intracellular molecules. On the other hand, in FD-FLIM, fluorescence lifetime is determined by measuring the phase shift and amplitude reduction of the fluorescence using a detector equipped with a gain modulator. Compared with TD-FLIM, FD-FLIM is simpler and much more cost-effective because it uses a sinusoidally modulated light-emitting diode (LED) light source for nanosecond (ns) measurements. In addition, FD-FLIM has been used in quantitative pH imaging conducted on a FRET sensor protein developed by Esposito and coauthors.24 Despite such advances in FD-FLIM, using a fluorescent sensor protein for imaging the dynamics of intracellular glucose, has yet to be realized, to the best of our knowledge. In this work, we adopted FD-FLIM to investigate the feasibility of applying this technique for real-time determination of intracellular glucose dynamics within living cells using a FRET glucose probe. The work presented herein offers a new CGM molecular sensor platform.
Methods
GIP Construction
The construction of a GIP has been described previously;10 however, in brief, the sensor protein was constructed by attaching Aequorea coerulescens green fluorescent protein (AcGFP1) and a red fluorescent protein (mCherry) to the N- and C- termini of a mutant GBP (GBPcys).7,11,12 The resultant plasmid is referred to as pTA-AcGFP1-GBPcys-mCherry. To express GIPs in mammalian cells, the AcGFP1-GBPcys-mCherry fusion gene was subcloned to pcDNA3.1 plasmid (Life Technologies Corporation, Carlsbad, CA).
Ratiometric FRET Imaging Microscopy Measurement
Murine myoblast cells, C2C12 (ATCC® CRL-1772™), were routinely maintained at 37 oC and 5% CO2 in Dulbecco’s Modified Eagles’s Medium (Mediatech, Inc., Manassas, VA). For imaging, cells were seeded onto 40 mm circular glass cover slips. To express GIPs, plasmid pcDNA3.1AcGFP1-GBPcys-mCherry was transiently transfected into C2C12 cells when they were 50–60% confluent, using Polyfect Transfection Reagent (QIAGEN Inc., Valencia, CA). Imaging was performed at 24–48 h post-transfection using an inverted fluorescence microscope (Olympus IX71,® Japan), described in detail elsewhere.10
Frequency-Domain FLIM
The FD-FLIM System (LIFA, Lambert Instruments, The Netherlands) consists of a signal generator, a 3-W LED as the excitation source, and an intensified charge-coupled device (CCD) (LI²CAM, Lambert Instruments) equipped with a multichannel plate (MCP)-based image intensifier. Both the LED and signal generator are computer controlled using LI-FLIM 1.2.7 software (Lambert Instruments). The intensified CCD camera was mounted onto an inverted phase contrast fluorescence microscope (Olympus IX51,® Japan). A schematic diagram of the FD-FLIM system is illustrated in Figure 1. The lifetime of the AcGFP1 was detected by means of the LED light source (485 nm), a filter cube entailing a 470/40-nm band-pass excitation filter, a 495LP dichroic mirror, and a 525/50-nm band-pass emission filter (Chroma Technology Corporation, Vermont). The ECFP was detected with use of the LED (445 nm), a filter cube encompassing a 436/20-nm band-pass excitation filter, a 455LP dichroic mirror, and a 480/40-nm band-pass emission filter (Chroma Technology Corporation). To perform FD-FLIM, the excitation light from the LED was modulated in intensity at a frequency of 1–100 MHz, leading to the intensity modulation of its induced fluorescence. Due to the decay of the light emission, the fluorescence exhibited a phase shift (delay in time) and a decrease in modulation depth with respect to the excitation light.25 This phase shift and decrease in modulation depth depended on the decay constants of a fluorophore. To extract the phase shift and the decrease in modulation depth from the emission signal relative to the excitation signal, the sensitivity of the image intensifier was modulated at the same frequency. Signals were detected at each phase step during a short integration period of the CCD camera. When emission signals were in phase with the sensitivity of the intensifier, a high detector signal level was measured. Likewise, when emission signals were out of phase with the sensitivity of the intensifier, a low detector signal level was measured. A sine function was fitted to the intensity-versus-phase-delay data, pixel by pixel. Lifetime values were extracted from the phase delay in fluorescence and the modulation of emission compared with excitation, and these values were used to determine changes in the lifetime of the sensor protein in response to variation in intracellular glucose.
Figure 1.
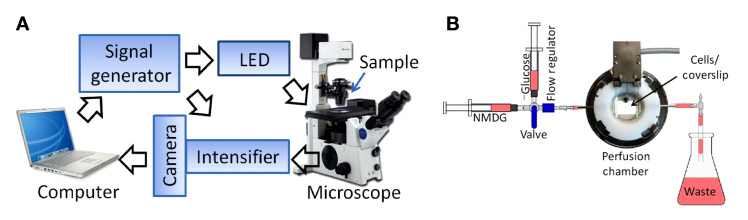
Schematic diagram of the FD-FLIM imaging system to determine intracellular glucose dynamics using a fluorescent glucose sensor protein. (A) The FD-FLIM system was integrated into an inverted phase contrast fluorescence microscope (Olympus IX51). The LED (excitation light source) was mounted in the standard arc lamp house that is connected to the excitation light port of the microscope, whereas an image intensifier in combination with a digital CCD camera was attached to the camera port of the microscope. A signal generator delivered modulation signals to the LED and image intensifier. The intensifier, camera, LED, and signal generator are all computer controlled. (B) Illustration of a medium perfusion chamber designed for live-cell time-lapse FD-FLIM
Live-Cell Time-Lapse FLIM
To perform live-cell time-lapse FLIM, GIP-expressing C2C12 cells were grown on cover slips in a gas-tight, temperature-controlled perfusion chamber (Model FCS2®, Bioptechs Inc., Butler, PA). A medium containing various concentrations of glucose was perfused through the chamber at a flow rate of 1.0–1.5 ml/min using a gravity-fed six-way perfusion device (Warner Instruments, Hamden, CT) controlled by a Cheminert® multiposition valve with a microelectric actuator (VICI® Valco Instru-ments, Houston, TX) and a flow regulator (Warner Instruments, Hamden, CT) (Figure 1B). The perfusion solution (pH = 7.2) was composed of 140 mM sodium chloride, 5 mM potassium chloride, 1.1 mM magnesium chloride, 2.5 mM calcium chloride, and 10 mM 4-(2-hydroxyethyl)-1-piperazine-ethanesulfonic acid (also known as HEPES) supplemented with either 10 mM glucose or 10 mM N-methyl-d-glucamine (NMDG), a glucose analog. The height as well as the volume of the chamber was fixed with a 0.5-mm-thick gasket.
Fluorescence lifetime was measured by modulating the LED excitation light and the detector-intensifier gain at a frequency of 40 MHz. Each experiment was completely recorded with an image stack that includes both the reference phase stack and the sample phase stack. An image stack of the reference or sample phase stack is a series of 12 images recorded at 12 different phases at the same modulation frequency. Generally, 12 images were captured at phase intervals of 30° with a random order and a background image (without excitation). Twelve images were selected to ensure a good compromise between the speed and accuracy of the measurement. The fluorescence lifetime for a single pixel was calculated by recording the phase shift and the decrease in modulation depth of the fluorescence light emitted by the sample. By recording a reference, the software identified the phase of the system (i.e., entire setup) and the modulation for a lifetime of t = 0 ns. Every deviation in phase and modulation of emitted fluorescence from the sample corresponded to a certain lifetime. An ideal reference is a material or solution with a uniformly distributed concentration of fluorescent molecules and a single lifetime component that can be used with the same filter cube as that of the sample. In this study, a fluorescein solution with a known lifetime was used to record a reference phase stack. A 10 µm amount of fluorescein (Sigma-Aldrich, St. Louis, MO) dissolved in 100 mM of Tris (hydroxymethyl)aminomethane (Tris) buffer (pH 10) with a known single lifetime of 4.00 ns served as the reference fluorophore for calibration. This value was used to deduce the system modulation and phase for a lifetime of 0.0 ns. A Tris buffer was used to further dilute the fluorescein to the appropriate concentration. To obtain the most accurate lifetime measurements, it is best to maintain the intensifier-gain and exposure-time settings at the same level or within a very close range between a reference recording and a sample recording. As a result, the sample fluorescence image recordings are optimized and completed before reference lifetime images are recorded. By changing the concentration of the fluorescein solution, we easily matched the exposure time and MCP voltage between the sample and the reference.
A 16-bit, 2 × 2 binned mode was used for all measure-ments because 16 bits offer the best intensity resolution supported by the LI2CAM. This camera has actually a 12-bit resolution that is stretched to 16 bits. The 2 × 2 binning could decrease the spatial resolution and increase the sensitivity of the measurement. The 2 × 2 binning of a 1.4-megapixel chip is usually not the limiting factor for spatial resolution. The image intensifier usually determines its maximum resolution when used for FLIM imaging. We set the gain for the camera at 1. Camera exposure time and MCP voltage were adjusted for different samples according to the manufacturers’ instructions. The exposure time ranged from 100 ms to 400 ms, depending on the level of expression of the sensor protein at MCP voltage 400–600 V. Background correction was performed automatically by subtracting an image obtained with blocked excitation, but, otherwise, the settings used were identical with the sample settings.
Results
Lifetimes of the ECFP and AcGFP1 were determined in the C2C12 cells (Figure 2). The lifetime of the ECFP determined from phase shifting was 2.3 ns, whereas its modulation lifetime was 3.0 ns, with the valid pixel-value of the region of interest (ROI) = 677. Differences inthe calculated lifetimes indicated that the ECFP exhibited a multiexponential decay in C2C12 cells.26 By contrast, the AcGFP1 donor exhibited monoexponential decay. The normalized count (count in a class divided by total number of observations; in this case relative counts were normalized to sum one) curve of the phase shift over-lapped with its normalized count curve of modulation change, indicating a single exponential lifetime of 2.70 ns, with the valid pixel-value of the ROI = 1217 (Figure 2C).
Figure 2.

Lifetime maps of AcGFP1 and ECFP expressed in C2C12 cells. Cells were transiently transfected with pcDNA3.1-AcGFP1-GBP and pECFP- expressing plasmids, respectively. The FD-FLIM was performed at 24 h post- transfection. (A) and (D) are fluorescence micrographs of AcGFP1 and ECFP; (B) and (E) are pseudocolored lifetime maps of AcGFP1 and ECFP; (C) and (F) are normalized-count curves of both the phase and modulation lifetime of AcGFP1 and ECFP, respectively. Lifetime Lookup Table: 2.0–3.0 ns
FLIM imaging of AcGFP1-GBPcys-mCherry or AcGFP1-GBPcys-expressing cells was performed at 48 h post-transfection in the absence of extracellular glucose (Figure 3). The lifetime of AcGFP1 detected from AcGFP1-GBPcys-expressing cells was 2.70 ns whereas the lifetime decreased to 2.10 ns in AcGFP1-GBPcys-mCherry-expressing cells. A FRET efficiency of 22% was estimated for this biosensor based in the equation:
Figure 3.
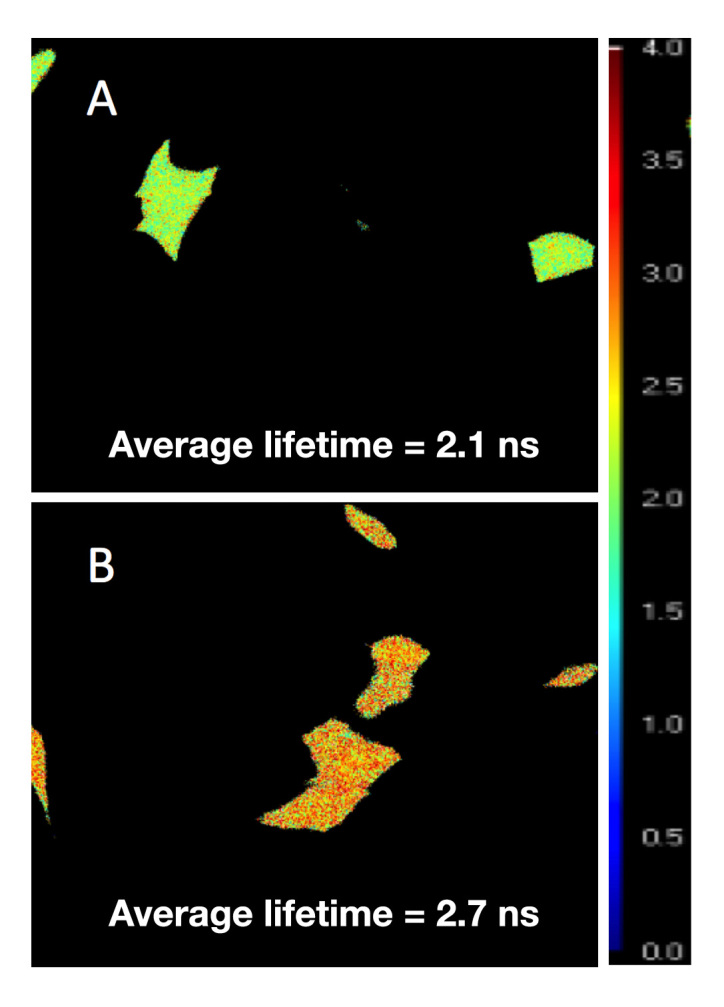
Fluorescence lifetime imaging of C2C12 cells expressing AcGFP1-GBPcys-mCherry and AcGFP1-GBPcys, respectively. C2C12 cells were transfected transiently with either pcDNA3.1-AcGFP1-GBPcys-mCherry (A) or pcDNA3.1-AcGFP1-GBPcys (B). FD-FLIM was performed at 48 h post- transfection. Lifetime Lookup Table: 0.0–4.0 ns
| (1) |
where τa is the lifetime of AcGFP1 determined from AcGFP1-GBPcys-mCherry- expressing cells, and τm is the lifetime of AcGFP1 measured from AcGFP1-GBPcys-expressing cells. Based on the value of E, separation distance was estimated between AcGFP1 from mCherry in the AcGFP1-GBPcys-mCherry sensor protein without glucose binding. Because R0 (Förster distance) depends on the spectral properties of both donor and acceptor,27 it can be given that:
| (2) |
where J is the spectral overlap integral, qD is the donor fluorescence quantum yield, n is the refractive index of the medium (assumed to be 1.3 for the calculation),28 κ2 is the orientation factor (assumed to be 0.667 corresponding to the random donor and acceptor orientation).29 J (nm4M-1cm-1) can be determined as follows:
| (3) |
where εA(λ) is the (wavelength-dependent) extinction coefficient of the acceptor (M-1 cm-1), fD(λ) is the (wavelength-dependent) donor fluorescence intensity (arbitrary units), and λ is the wavelength (nm). The qD of 0.82 for AcGFP1 and the maximal extinction coefficient of the acceptor εA of 72,000 M-1 cm-1 30 were adopted to scale the measured mCherry absorbance spectrum to obtain εA(λ). FRETView software31–33 was adopted to deduce the R0 of the AcGFP-mCherry pair to be 55.9 Å. On the other hand,
| (4) |
where R is the distance between the donor and acceptor. By substituting both E and R0 values into Equation (4), we estimated the R value; i.e., in the absence of glucose binding, the AcGFP1 was separated from mCherry at a distance of approximately 68.9 Ǻ. When glucose was bound to the protein, the donor lifetime increased to 2.26 ns, corresponding to a 72.8-Ǻ separation between fluorophores.
Next, we examined the effect of photobleaching on continuous FD-FLIM. We carried out a time-lapse lifetime imaging of C2C12 cells expressing AcGFP1-GBPcys protein. Both fluorescent intensity and lifetime of AcGFP1 were measured every 12 s for 1000 s. The bath solution containing either 10 mM glucose or 10 mM NMDG was perfused into the cell culture chamber, as shown in Figure 4. The fluorescence intensity of AcGFP1 dropped 12% continuously during imaging (Figure 4). By contrast, the lifetime of AcGFP1 remained stable during imaging, suggesting the superiority of lifetime imaging over intensity imaging for quantifying the dynamics of intracellular molecules.
Figure 4.
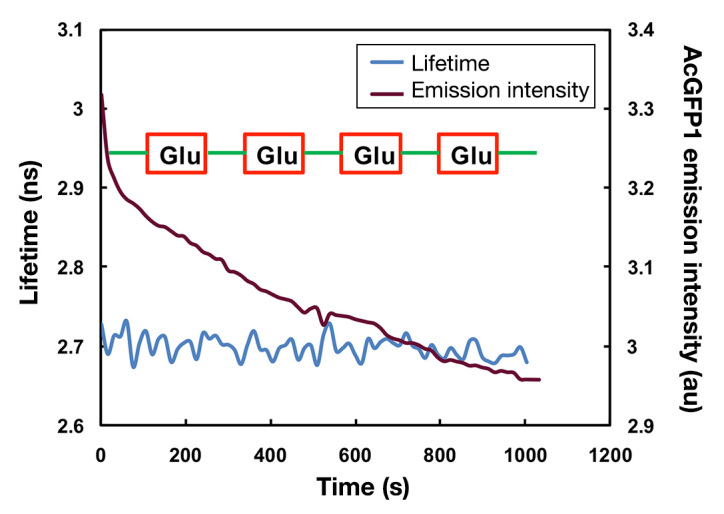
The effect of photobleaching on continuous microscopy imaging of AcGFP1 in C2C12 cells. C2C12 cells were transfected transiently with pcDNA3.1-AcGFP1-GBPcys, and both intensity and lifetime imaging were performed in a time-lapse mode with the continuous perfusion of either 10 mM glucose (red) or 10 mM NMDG bath solution (green)
After confirming stability of FLIM detection, we quantified the dynamics of cellular glucose uptake and clearance in response to variation in extracellular glucose using FLIM. The AcGFP1-GBPcys-mCherry sensor protein was transiently transfected into C2C12 cells. A bath solution containing either 10 mM glucose or 10 mM NMDG was flown into the cell culture chamber, and changes in intracellular glucose concentrations were continuously monitored by measuring changes in the lifetime of AcGFP1 (Figure 5A). The cellular glucose uptake and clearance rates in response to variation in extracellular glucose concentration were determined by fitting lifetime measurements with first-order exponential regression (Figure 5B and 5C). The lifetime of AcGFP1 was observed to rise with the increase in intracellular glucose concentration, indicating glucose uptake. By contrast, the lifetime dropped after removal of the extracellular glucose when perfusing NMDG. The response of intra-cellular glucose concentration to changes in extracellular glucose concentration was observed within 30 s. Glucose uptake (rate constant at 45 s) also appeared to be faster than its clearance (rate constant at 88 s).
Figure 5.

FD-FLIM of glucose uptake and clearance dynamics within living cells. C2C12 cells were transfected transiently with pcDNA3.1-AcGFP1-GBPcys-mCherry. FD-FLIM was performed at 24 h post-transfection in a time-lapse mode with continuous perfusion of either 10 mM glucose (red arrows) or 10 mM NMDG (green arrows). (A) Changes in lifetime of AcGFP1-GBPcys-mCherry sensor protein in response to glucose concentrations in the bath solution. The glucose uptake (B) and clearance (C) rates were determined by fitting lifetime measurements with first-order exponential nonlinear regression. Experiments were carried out in multiple batches (n = 4), and various cells from each batch were monitored for glucose response
Finally, we determined whether there was any glucose concentration gradient within the living cells. Compared to intensity-based FRET, the FD-FLIM detected the spatial distribution of molecules with much higher resolution.18,34 Both intensity and lifetime FRET imaging microscopy measurement were performed on cells expressing the AcGFP1-GBPcys-mCherry sensor protein in the presence of 10 mM glucose. Two ROIs, i.e., a region close to the cell membrane and a region close to the nucleus, were selected in a single cell (Figure 6A and 6C). By plotting fluorescence intensity of AcGFP1 vs each pixel in these two regions, we observed no difference in the pattern of intensity distribution between these two regions (Figure 6B). The lifetime of AcGFP1 determined in ROI1 (395 pixels), i.e., a region close to the cell membrane was 2.34 ns, whereas the lifetime of AcGFP1 in ROI2 (338 pixels), i.e., a region close to the nucleus, was 2.12 ns (Figure 6D), indicating a low glucose concentration in the area close to the nucleus. It was manifest that glucose uptake occurred in the interface between the cell and the cell-culture medium; thus, an elevated concentration of glucose as opposed to the ROI close to the nucleus. However, the FRET intensity imaging microscopy measurement failed to reveal this difference (Figure 6B).
Figure 6.

FD-FLIM of glucose concentration gradients in cytoplasm of C2C12 cells. C2C12 cells were transfected with pcDNA3.1-AcGFP1-GBPcys-mCherry. FD-FLIM was performed at 24 h post-transfection in the presence of 10 mM glucose. (A) Monochromatic intensity FRET image and (C) pseudocolored lifetime map. Fluorescence intensity (B) and lifetime (D) of AcGFP1 were determined for two ROIs in a single C2C12 cell. Pseudocolor Lifetime Look-up Table: 2.20–2.34 ns
Discussion
Thus far, intracellular glucose monitoring has relied on radioisotope-labeled glucose analogs and fluorescence-intensity imaging. Measurement that depend on isotopic analogs do not reveal the dynamics of glucose uptake and clearance in living cells,2 whereas fluorescence intensity- imaging methods are affected by issues such as photobleaching.18 Although a number of GFP-based glucose sensor proteins have been developed,7,9,11 the application of these sensor proteins for continuous glucose monitoring within living cells has been limited. The most widely employed fluorophore, ECFP, is known to exhibit light-induced variations in its lifetime that are not proportional to decreases in steady-state fluorescence intensities and that vary from cell to cell.35 Furthermore, a concentration-dependent quenching of ECFP in the absence and presence of EYFP significantly diminishes the accuracy of the detection using ECFP/EYFP.36,37
These drawbacks can be overcome by choosing fluorescence proteins that are more tolerant to photobleaching. Here, we investigated the feasibility of pairing AcGFP1 withmCherry for FD-FLIM and then successfully demonstrated, for the first time, that FD-FLIM of the AcGFP1-GBPcys-mCherry sensor protein monitored changes in intracellularglucose concentrations in response to variations in extracellular glucose levels. In essence, the fluorescent sensor protein developed herein allowed for the determination of glucose uptake and clearance rate through FD-FLIM. To minimize the effect of photo-bleaching on the lifetime of the AcGFP1-GBPcys-mCherry glucose sensor, we adopted a random 12-phase image acquisition approach.38 With this approach we observed that the lifetime of AcGFP1 remained stable during time-lapse FRET imaging. This result is consistent with the work of others35 and is significant because the visualization of intracellular molecular events has long been limited to intensity-based FRET imaging microscopy and TD-FLIM. FD-FLIM is, in fact, a better approach than TD-FLIM because although TD-FLIM [particularly time-correlated single-photon counting (TCSPC)] can provide a very high resolution in a wide-field setup, additional signal processing is needed to obtain the position of a photon. If a special detector is used with TCSPC, it would also require a photocathode, a MCP, and a delay-line system or a four-element anode system at the output. TD-FLIM offers high counting efficiency and good temporal resolution. However, the system makes it difficult to reach a high count rate and also requires complex detectors that are not widely available.39–42
Another finding in this study is that, compared to the multiexponential decay of ECFP,43 AcGFP1 exhibits only monoexponential decay, which makes it suitable for continuous intracellular glucose monitoring through FD-FLIM (Figure 3). The dissociation constant of the sensor protein for glucose was estimated to be 0.13 mM,10 making it suitable for use in monitoring intracellular glucose concentrations because the mean intracellular glucose concentration is approximately 0.10–0.20 mM in both healthy humans and those with diabetes.3
We have also found that FRET-FLIM is less affected by light scattering and the local concentration of sensor proteins,38,44–47 making it ideal for three-dimensional (3D) imaging. Such 3D imaging can be achieved using a spinning-disk confocal microscope where samples are scanned simultaneously by thousands of pinholes, resulting in a virtually instantaneous image with more than a 10-fold reduction in photobleaching.48,49 The integration of a spinning-disk unit into a frequency-domain FLIM instrument could reduce artifacts considerably while maintaining the advantages of a wide field, especially for FLIM on objects with a 3D lifetime structure.50,51 Realization of this measurement would potentially allow for the continuous monitoring of intracellular cellular glucose concentrations in animal models, offering a new tool for studying the pathology of diabetes.
Conclusions
We demonstrated the feasibility of using FD-FLIM to monitor the dynamics of intracellular glucose concentration changes in response to extracellular glucose concentrations. Our experimental results revealed that glucose uptake by the C2C12 cells was relatively faster than its clearance. Our results are in agreement with the observations made by other groups using time-lapse intensity measurements in live cells.8,13 This sensor could potentially be used for both in vitro and in vivo continuous glucose monitoring. Intracellular glucose monitoring under various pathophysiological conditions in different cells and tissues could accelerate the research on drug discovery and diabetes treatment. The FD-FLIM developed herein can be combined with the spinning-disk confocal unit for 3D FD-FLIM imaging of intra-cellular glucose concentrations in animal models.
Glossary
- (AcGFP1)
Aequorea coerulescens green fluorescent protein, (a.u.) arbitrary unit
- (CCD)
charge-coupled device
- (CGM)
continuous glucose monitoring continuous glucose monitoring continuous glucose monitoring
- (DNA)
deoxyribonucleic acid
- (ECFP)
enhanced cyan fluorescent protein
- (EYFP)
enhanced yellow fluorescent protein
- (FD-FLIM)
frequency-domain fluorescence lifetime imaging microscopy
- (FLIM)
fluorescence lifetime imaging microscopy
- (FRET)
fluorescence resonance energy transfer
- (GBP)
glucose-binding protein
- (GIP)
glucoseindicator protein
- (LED)
light-emitting diode
- (MCP)
multichannel plate
- (NMDG)
N-methyl-d-glucamine nanosecond
- (ROI)
region of interest
- (TD-FLIM)
time-domain fluorescence lifetime imaging microscopy, (Tris) tris(hydroxymethyl)aminomethane
Funding
This research was partially supported by National Institutes of Health Grant EB006378-01 and Arkansas Biosciences Institute Grant 0402-27504-21-726.
References
- 1.Foley JE, Cushman SW, Salans LB. Intracellular glucose concentration in small and large rat adipose cells. Am J Physiol Endocrinol Metab. 1980;238(2):E180–5. doi: 10.1152/ajpendo.1980.238.2.E180. [DOI] [PubMed] [Google Scholar]
- 2.Cline GW, Jucker BM, Trajanoski Z, Rennings AJM, Shulman GI. A novel 13C NMR method to assess intracellular glucose concentration in muscle, in vivo. Am J Physiol Endocrinol Metab. 1998;274(2):E381–9. doi: 10.1152/ajpendo.1998.274.2.E381. [DOI] [PubMed] [Google Scholar]
- 3.Cline GW, Petersen KF, Krssak M, Shen J, Hundal RS, Trajanoski Z, Inzucchi S, Dresner A, Rothman DL, Shulman GI. Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes. N Engl J Med. 1999;341(4):240–246. doi: 10.1056/NEJM199907223410404. [DOI] [PubMed] [Google Scholar]
- 4.Ravikumar B, Stewart A, Kita H, Kato K, Duden R, Rubinsztein DC. Raised intracellular glucose concentrations reduce aggregation and cell death caused by mutant huntingtin exon 1 by decreasing mTOR phosphorylation and inducing autophagy. Hum Mol Genet. 2003;12(9):985–994. doi: 10.1093/hmg/ddg109. [DOI] [PubMed] [Google Scholar]
- 5.Takagi S, Takahashi W, Shinohara Y, Yasuda S, Ide M, Shohtsu A, Seio T. Quantitative PET cerebral glucose metabolism estimates using a single non-arterialized venous-blood sample. Ann Nucl Med. 2004;18(4):297–302. doi: 10.1007/BF02984467. [DOI] [PubMed] [Google Scholar]
- 6.Smith GS, Ma Y, Dhawan V, Gunduz H, Carbon M, Kirshner M, Larson J, Chaly T, Belakhleff A, Kramer E, Greenwald B, Kane JM, Laghrissi-Thode F, Pollock BG, Eidelber D. Serotonin modulation of cerebral glucose metabolism measured with positron emission tomography (PET) in human subjects. Synapse. 2002;45(2):105–112. doi: 10.1002/syn.10088. [DOI] [PubMed] [Google Scholar]
- 7.Ye K, Schultz JS. Genetic engineering of an allosterically based glucose indicator protein for continuous glucose monitoring by fluorescence resonance energy transfer. Anal Chem. 2003;75(14):3451–3459. doi: 10.1021/ac034022q. [DOI] [PubMed] [Google Scholar]
- 8.Fehr M, Lalonde S, Lager I, Wolff MW, Frommer WB. In vivo imaging of the dynamics of glucose uptake in the cytosol of COS-7 cells by fluorescent nanosensors. J Biol Chem. 2003;278(21):19127–19133. doi: 10.1074/jbc.M301333200. [DOI] [PubMed] [Google Scholar]
- 9.Fehr M, Okumoto S, Deuschle K, Lager I, Looger LL, Persson J, Kozhukh L, Lalonde S, Frommer WB. Development and use of fluorescent nanosensors for metabolite imaging in living cells. Biochem Soc Trans. 2005;33(Pt 1):287–290. doi: 10.1042/BST0330287. [DOI] [PubMed] [Google Scholar]
- 10.Jin S, Veetil JV, Garrett JR, Ye K. Construction of a panel of glucose indicator proteins for continuous glucose monitoring. Biosens Bioelectron. 2011;26(8):3427–3431. doi: 10.1016/j.bios.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garrett JR, Wu X, Jin S, Ye K. pH-insensitive glucose indicators. Biotechnol Prog. 2008;24(5):1085–1089. doi: 10.1002/btpr.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Veetil JV, Jin S, Ye K. A glucose sensor protein for continuous glucose monitoring. Biosens Bioelectron. 2010;26:1650–1655. doi: 10.1016/j.bios.2010.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.John SA, Ottolia M, Weiss JN, Ribalet B. Dynamic modulation of intracellular glucose imaged in single cells using a FRET-based glucose nanosensor. Pflugers Arch. 2008;456(2):307–322. doi: 10.1007/s00424-007-0395-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fehr M, Takanaga H, Ehrhardt DW, Frommer WB. Evidence for high-capacity bidirectional glucose transport across the endoplasmic reticulum membrane by genetically encoded fluorescence resonance energy transfer nanosensors. Mol Cell Biol. 2005;25(24):11102–11112. doi: 10.1128/MCB.25.24.11102-11112.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tramier M, Zahid M, Mevel JC, Masse MJ, Coppey-Moisan M. Sensitivity of CFP/YFP and GFP/mCherry pairs to donor photo-bleaching on FRET determination by fluorescence lifetime imaging microscopy in living cells. Microscopy Res Tech. 2006;69(11):933–939. doi: 10.1002/jemt.20370. [DOI] [PubMed] [Google Scholar]
- 16.Thaler C, Vogel SS. Quantitative linear unmixing of CFP and YFP from spectral images acquired with two-photon excitation. Cytometry A. 2006;69A(8):904–911. doi: 10.1002/cyto.a.20267. [DOI] [PubMed] [Google Scholar]
- 17.Zal T, Gascoigne NR. Photobleaching-corrected FRET efficiency imaging of live cells. Biophy J. 2004;86(6):3923–3939. doi: 10.1529/biophysj.103.022087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Periasamy A, editor. Methods in cellular imaging. New York: Oxford University Press; 2001. [Google Scholar]
- 19.Herman B, Wodnicki P, Kwon S, Periasamy A, Gordon G, Mahajan N, Wang X. Recent developments in monitoring calcium and protein interactions in cells using fluorescence lifetime microscopy. Journal of Fluorescence. 1997;7(1):85–91. [Google Scholar]
- 20.Elangovan M, Day RN, Periasamy A. Nanosecond fluorescence resonance energy transfer-fluorescence lifetime imaging microscopy to localize the protein interactions in a single living cell. J Microsc. 2002;205(1):3–14. doi: 10.1046/j.0022-2720.2001.00984.x. [DOI] [PubMed] [Google Scholar]
- 21.Harpur AG, Wouters FS, Bastiaens PI. Imaging FRET between spectrally similar GFP molecules in single cells. Nat Biotechnol. 2001;19(2):167–169. doi: 10.1038/84443. [DOI] [PubMed] [Google Scholar]
- 22.Cremazy FG, Manders EM, Bastiaens PI, Kramer G, Hager GL, van Munster EB, Verschure PJ, Gadella TJ, Jr., van Driel R. Imaging in situ protein-DNA interactions in the cell nucleus using FRET-FLIM. Exp Cell Res. 2005;309(2):390–396. doi: 10.1016/j.yexcr.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 23.Chang CW, Sud D, Mycek MA. Fluorescence lifetime imaging microscopy. Methods Cell Biol. 2007;81:495–524. doi: 10.1016/S0091-679X(06)81024-1. [DOI] [PubMed] [Google Scholar]
- 24.Esposito A, Gralle M, Dani MA, Lange D, Wouters FS. pHlameleons: a family of FRET-based protein sensors for quantitative pH imaging. Biochemistry. 2008;47(49):13115–13126. doi: 10.1021/bi8009482. [DOI] [PubMed] [Google Scholar]
- 25.Squire A, Bastiaens PI. Three dimensional image restoration in fluorescence lifetime imaging microscopy. J Microsc. 1999;193(Pt 1):36–49. doi: 10.1046/j.1365-2818.1999.00427.x. [DOI] [PubMed] [Google Scholar]
- 26.Schneider PC, Clegg RM. Rapid acquisition, analysis, and display of fluorescence lifetime-resolved images for real-time applications. Review of Scientific Instruments. 1997;68(11):4107–4119. [Google Scholar]
- 27.Lakowicz JR. Principles of fluorescence spectroscopy. New York: Springer; 2006. p. 954. [Google Scholar]
- 28.Shcherbo D, Souslova EA, Goedhart J, Chepurnykh TV, Gaintzeva A, Shemiakina II, Gadella TW, Lukyanov S, Chudakov DM. Practical and reliable FRET/FLIM pair of fluorescent proteins. BMC Biotechnol. 2009;9:24. doi: 10.1186/1472-6750-9-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goldman RD, Spector DL. Live cell imaging: a laboratory manual. New York: Cold Spring Harbor Laboratory Press; 2005. p. 631. [Google Scholar]
- 30.Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22(12):1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 31.Stevens N. FRETView Plugin. http://nathan.instras.com/projects/fretview/index.html. Accessed October 9, 2012.
- 32.Stevens N. FRETView 1.0 help manual. http://nathan.instras.com/projects/fretview/help.html. Accessed Sept. 24, 2012.
- 33.Stevens N, Dyer J, Marti AA, Solomon M, Turro NJ. FRETView: a computer program to simplify the process of obtaining fluorescence resonance energy transfer parameters. Photochem Photobiol Sci. 2007;6(8):909–911. doi: 10.1039/b702631e. [DOI] [PubMed] [Google Scholar]
- 34.Duncan RR, Bergmann A, Cousin MA, Apps DK, Shipston MJ. Multi-dimensional time-correlated single photon counting (TCSPC) fluorescence lifetime imaging microscopy (FLIM) to detect FRET in cells. J Microsc. 2004;215(1):1–12. doi: 10.1111/j.0022-2720.2004.01343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tramier M, Zahid M, Mevel JC, Masse MJ, Coppey-Moisan M. Sensitivity of CFP/YFP and GFP/mCherry pairs to donor photo-bleaching on FRET determination by fluorescence lifetime imaging microscopy in living cells. Microsc Res Tech. 2006;69(11):933–939. doi: 10.1002/jemt.20370. [DOI] [PubMed] [Google Scholar]
- 36.Grailhe R, Merola F, Ridard J, Couvignou S, Le Poupon C, Changeux JP, Laguitton-Pasquier H. Monitoring protein interactions in the living cell through the fluorescence decays of the cyan fluorescent protein. ChemPhysChem. 2006;7(7):1442–1454. doi: 10.1002/cphc.200600057. [DOI] [PubMed] [Google Scholar]
- 37.Van Munster EB, Gadella TW. Fluorescence lifetime imaging microscopy (FLIM). Adv Biochem Eng Biotechnol. 2005;95:143–175. doi: 10.1007/b102213. [DOI] [PubMed] [Google Scholar]
- 38.Van Munster EB, Gadella TW., Jr Suppression of photobleaching-induced artifacts in frequency-domain FLIM by permutation of the recording order. Cytometry A. 2004;58(2):185–194. doi: 10.1002/cyto.a.20013. [DOI] [PubMed] [Google Scholar]
- 39.Elson D, Requejo-Isidro J, Munro I, Reavell F, Siegel J, Suhling K, Tadrous P, Benninger R, Lanigan P, McGinty J, Talbot C, Treanor B, Webb S, Sandison A, Wallace A, Davis D, Lever J, Neil M, Phillips D, Stamp G, French P. Time-domain fluorescence lifetime imaging applied to biological tissue. Photochem Photobiol Sci. 2004;3(8):795–801. doi: 10.1039/b316456j. [DOI] [PubMed] [Google Scholar]
- 40.Peter M, Ameer-Beg SM. Imaging molecular interactions by multiphoton FLIM. Biol Cell. 2004;96(3):231–236. doi: 10.1016/j.biolcel.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 41.Krishnan RV, Masuda A, Centonze VE, Herman B. Quantitative imaging of protein-protein interactions by multiphoton fluorescence lifetime imaging microscopy using a streak camera. J Biomed Opt. 2003;8(3):362–367. doi: 10.1117/1.1577574. [DOI] [PubMed] [Google Scholar]
- 42.Becker W, Bergmann A. Lifetime imaging techniques for optical microscopy. http://www.boselec.com/products/documents/TCSPCforMicroscopy.pdf. Accessed Oct. 9, 2012.
- 43.Tramier M, Gautier I, Piolot T, Ravalet S, Kemnitz K, Coppey J, Durieux C, Mignotte V, Coppey-Moisan M. Picosecond-hetero-FRET microscopy to probe protein-protein interactions in live cells. Biophys J. 2002;83(6):3570–3577. doi: 10.1016/S0006-3495(02)75357-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen Y, Mills JD, Periasamy A. Protein localization in living cells and tissues using FRET and FLIM. Differentiation. 2003;71(9-10):528–541. doi: 10.1111/j.1432-0436.2003.07109007.x. [DOI] [PubMed] [Google Scholar]
- 45.Festy F, Ameer-Beg SM, Ng T, Suhling K. Imaging proteins in vivo using fluorescence lifetime microscopy. Mol Biosyst. 2007;3(6):381–391. doi: 10.1039/b617204k. [DOI] [PubMed] [Google Scholar]
- 46.Wallrabe H, Periasamy A. Imaging protein molecules using FRET and FLIM microscopy. Curr Opin Biotechnol. 2005;16(1):19–27. doi: 10.1016/j.copbio.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 47.Koo V, Hamilton PW, Williamson K. Non-invasive in vivo imaging in small animal research. Cell Oncol. 2006;28(4):127–139. doi: 10.1155/2006/245619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lambert Instruments. LIFA confocal spinning disk. http://www.lambertinstruments.com/products/flim-systems/item/lifa-confocal-spinning-disk. Accessed Oct. 9, 2012.
- 49.Wang E, Babbey CM, Dunn KW. Performance comparison between the high-speed Yokogawa spinning disc confocal system and single-point scanning confocal systems. J Microsc. 2005;218(Pt 2):148–159. doi: 10.1111/j.1365-2818.2005.01473.x. [DOI] [PubMed] [Google Scholar]
- 50.Van Munster EB, Goedhart J, Kremers GJ, Manders EM, Gadella TW., Jr. Combination of a spinning disc confocal unit with frequency-domain fluorescence lifetime imaging microscopy. Cytometry A. 2007;71(4):207–214. doi: 10.1002/cyto.a.20379. [DOI] [PubMed] [Google Scholar]
- 51.Buranachai C, Kamiyama D, Chiba A, Williams B, Clegg R. Rapid frequency-domain FLIM spinning disk confocal microscope: lifetime resolution, image improvement and wavelet analysis. J Fluoresc. 2008;18(5):929–942. doi: 10.1007/s10895-008-0332-3. [DOI] [PubMed] [Google Scholar]