

Abstract
Type 2 diabetes mellitus (T2DM) is one of the most prevalent diseases worldwide. Current treatments are often associated with off-target effects and do not significantly impact disease progression. New therapies are therefore urgently needed to overcome this social burden. Glucagon-like peptide-1 (GLP-1), an incretin hormone, has been used to control T2DM symptomatology. However, the administration of peptide or proteins drugs is still a huge challenge in the pharmaceutical field, requiring administration by parenteral routes. This article reviews the main hurdles in oral administration of GLP-1 and focuses on the strategies utilized to overcome them.
Keywords: exenatide; glucagon-like peptide-1; glucagon-like peptide-1 analogs; liraglutide, oral delivery systems; type 2 diabetes mellitus
Introduction
Type 2 diabetes mellitus (T2DM) is a complex metabolic disorder, being one of the most prevalent worldwide chronic diseases that continuously increases in numbers and impact.1 It is characterized by resistance to insulin action or inadequate insulin secretion due to pancreatic β-cell dysfunction, which causes abnormal glucose levels, resulting from the interaction between genetic andenvironmental factors.2 Additionally, T2DM is the maincause of microvascular and macrovascular complications.3
Current treatments for T2DM consist of nonpharmaco-logical actions such as diet and exercise and the use of oral pharmacological agents. However, these drugs are usually associated with side effects, including weight gain and hypoglycemia, resulting in poor patient compliance. Therefore, novel approaches for diabetes therapy are urgently needed.2,3 With the increased knowledge of the pathophysiology of T2DM, several strategies based on the action of glucagon-like peptide-1 (GLP-1), an incretin hormone responsible for inducing glucose-dependent stimulation of insulin secretion, have been developed. Glucagon-like peptide-1 and new analogs are currently used in clinical protocols as subcutaneous injections, this route of administration being a limitation for peptides or proteins drugs.4–6
Glucagon-Like Peptide-1 and Glucagon-Like Peptide-1 Analogs for Control of Diabetes
Glucagon-like peptide-1 is a 30 amino-acid peptide (3355.67 Da) derived from a proglucagon gene that is secreted by neuroendocrine L cells of the ilium and colon. The mechanisms that lead to its secretion are regulated by meal intake and are described elsewhere.7 Briefly, L cells are stimulated directly by nutrients that contact with their apical surface and indirectly by a variety of neural and endocrine factors because of the contact made by neuronal and vascular tissue with their basolateral side.8,9 Glucagon-like peptide-1 diffuses across the basal lamina into the lamina propria and enters into circulation via intestinal capillaries, draining into the hepatic portal vein and then into the systemic circulation.10,11 Its elimination occurs by renal clearance through glomerular filtration and catabolism.8,12
Glucagon-like peptide-1 acts by binding to its G protein-coupled receptor [glucagon-like peptide-1 receptor (GLP-1R)],which activates the GLP-1R signaling pathway in a glucose-dependent manner, i.e., only during hyper-glycemia.8,13 Its receptor is widely expressed in pancreatic islets, where GLP-1 stimulates insulin secretion, decreases glucagon concentration, and suppresses its release,12,14 also stimulating neogenesis and proliferation of β cells, which increases pancreas mass and inhibits cell apoptosis.13,15–17 Nevertheless, GLP-1R is also expressed in the gastrointestinal tract (GI), in which GLP-1 acts by reducing the rate of gastric emptying;18 in the brain, as a neurotransmitter in the hypothalamus, which regulates and leads to satiety,19 in the heart, having direct protective effects;20 and within liver, kidney, muscle and adipose tissue (Figure 1).8,21 In patients with T2DM, it is known that the activity of GLP-1 is diminished, but the number of GLP receptors is unaltered.4–6
Figure 1.
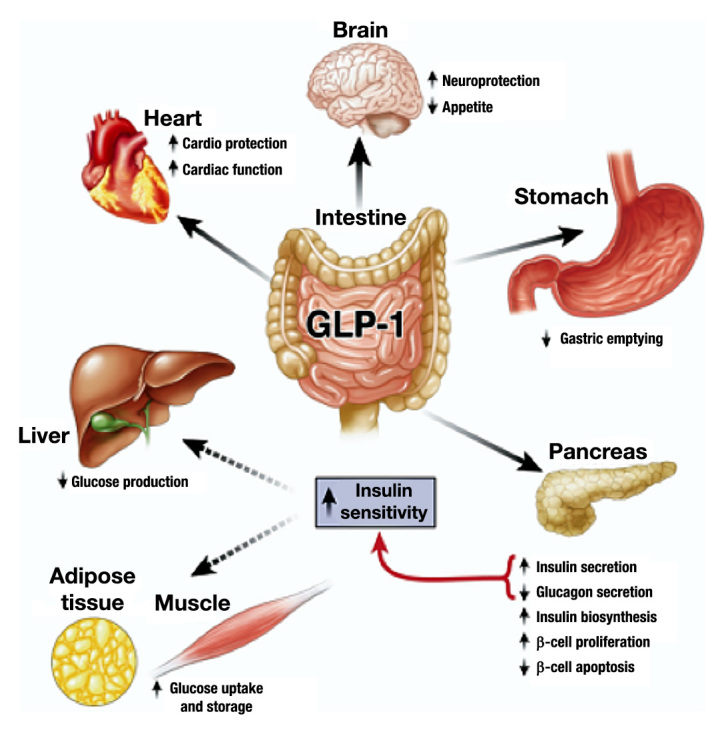
Glucagon-like peptide-1 actions by direct interaction (solid arrows) with GLP-1Rs on specific tissues and in liver, fat, and muscle most likely occur through indirect mechanisms (dashed arrows). Reprinted with permission from Gastroenterology.8
Given that GLP-1 is not related to hypoglycemia or weight gain and its effects have great value in T2DM treatment, GLP-1 has been in the pipeline for diabetes therapy.5,15,22 However, GLP-1 is limited due to the metabolic instability caused once dipeptidyl peptidase-4 (DPP-4) enzyme rapidly cleaves to its N terminal. The rapid cleavage to the secreted GLP-1 is almost immediate, resulting in a short half-life of less than 2 min. Dipeptidyl peptidase-IV is mainly located on the luminal surface of the endothelial cells, which means that a large portion of the GLP-1 that leaves the gut is already degraded to the inactive metabolite,8,9,12 and only 25% of the GLP-1 secreted reaches the portal circulation. Additionally, 40% to 50% of GLP-1 that bypasses gut inactivation is metabolized in the liver, and only the remaining 10% to 15% enters the systemic circulation. The metabolism of GLP-1 is described in more detail by Holst and coauthors.9,23
To overcome premature GLP-1 metabolism, long-acting GLP-1 analogs have been developed to resist DPP-4 degradation. Alternatively, DPP-4 inhibitors are also being considered to concomitant administration with GLP-1 analogs.24 However, the action of GLP-1 analogs are dependent on the concentration of endogenous GLP-1 and the number of receptors, which provides much higher pharmacological levels than the DPP-4 inhibitors.25,26
In 2005 and 2010, two GLP-1 analogs, exenatide (Byetta®) and liraglutide (Victoza®), were approved by the Food and Drug Administration for treatment of T2DM.27 Exenatide (4186.6 Da) is the synthetic form of exendin 4,a peptide isolated from the salivary gland of Gila monster (Heloderma suspectum), showing 53% homology to GLP-1. It has a half-life of 2.4 h and acts by binding GLP-1R at least with the same affinity as native GLP-1.6,15,26 On the other hand, liraglutide (3751.2 Da) shows 97% homology to GLP-1, being very similar to native peptide, and differs by only one amino acid substitution, being further linked by a fatty acid side chain. This fatty acid portion allows a noncovalent binding with serum albumin, which increases the half-life of the peptide for approximately 13 h, resulting from the delayed degradation by DPP-4 and its clearance. Regardless of the modifications, the ability of binding with GLP-1R remains the same.
Both exenatide and liraglutide are delivered parenterally and have been proven to improve glycemic control.6,27,28 Many other GLP-1 analogs are in late clinical develop-ment,29 thus new agents are expected to reach the market in the future.
Although alternatives have been created to work around the short half-life of GLP-1, the use of GLP-1 analogs have also limited therapeutic utility because it must be administered continuously by parenteral routes. Oral administration is necessary and could prove safe and effective; it would mimic endogenous secretion of GLP-1 and provide more convenience, ease of administration, and comfort, which would increase patients’ compliance to the treatment.10,30,31 However, because of their poor oral bioavailability, many scientists and the pharmaceutical industry are focusing their research efforts to deliver therapeutically active GLP-1.
Barriers to Oral Glucagon-Like Peptide-1 and Glucagon-Like Peptide-1 Analog Delivery
As a gatekeeper of the human body, the GI tract has several barriers that thwart drugs from being absorbed.32,33 It provides an optimum environment for the entry of nutrients and for digestion but is a hostile environment to pathogens and xenobiotics, making the absorption of oral-active drugs a real challenge.32,34,35
The variable pH along the GI tract (pH ranging from 1–3 to 6.5–8) is the first impasse to peptide absorption. This variation induces several modifications on peptides that can lead to the loss of their conformation or even to their partial destruction before reaching the intestine.30,36,37 Another main risk for GLP-1 and its analogs absorption is their rapid degradation by enzymes secreted throughout the GI lumen, like pepsine, pancreatic enzymes, and peptidases and enzymes from the intestinal flora.30,38,39
Any peptide that survives passage through the chemical and enzymatic degradation must then face the absorption barriers, such as intestinal epithelial cells and the mucus layer. The main component of this layer are mucin chains, cross linked and tangled with each other like a net, forming a semipermeable barrier with an elastic, robust, and viscous gel consistency. It protects the surface of the epithelium and influences drug absorption through its composition and thickness along the GI tract, allowing only water and small molecules to cross it,30,35,39–41 as described in more detail by Ensign and coauthors40 and Johansson and coauthors.42 It also has a negative charge at neutral pH, which causes an electrostatic repulsion with proteins, preventing their contact with intestinal cells.30,36
After passage through the mucus, GLP-1 and its analogs face a second absorption barrier, the intestinal epithelium cells. These cells consist predominantly of enterocytes, primarily responsible for absorption and transport of molecules; goblet cells, of glandular origin, that are the second most prevalent cell type in the intestine that function only to produce mucus;30,43 and M cells, specialized for antigen uptake, that reside in Peyer’s patches (lymphoid regions).30,44,45 These cells provide another possible gateway for oral delivery of peptides as for nanoparticles and microparticles because they are relatively less protected by mucus and have a high trans-cytotic capacity.40
Drug permeation may therefore occur either through a transcellular or paracellular route. In the first one, the transport of drugs occurs through intestinal cells, either passively or carried, mediated by specific transporters.46,47 In this type of permeation, drugs must have specific physiochemical properties, such as lipophilicity and molecular weight. Moreover, drug degradation from intracellular organelles like lysosomes, as well as the existence of efflux transporters, may prevent drugs fromreaching the bloodstream.30,34,35 Because GLP-1 and its analogs are hydrophilic peptide drugs, the paracellular route is the most probable route for their absorption.34,48,49 The absorption may occur between cells across the intestinal cell junctions. In contrast to transcellular, the paracellular route depends not on lipophilicity, but rather on the size of peptides, its ionic charge, and has the great advantage of absence of proteolytic activity.34,47,49 However, this kind of transport is usually minimal, not only because it represents less than 1% of the intestinal epithelium, but also due to the tight junctions (TJs) existing between the intestinal cells that limit the transport of small hydrophilic molecules. The molecular weight cutoff for this kind of permeation is usually considered 200 Da,50 which is not the case for GLP-1 or its analogs, with a molecular weight above 3 kDa.30,46,48,49,51
To increase peptide oral bioavailability, many strategies involving absorption enhancers and proteolytic inhibitors have been developed.38,52–54 Although such approaches are very promising, they do not have the full confidence of clinicians and competent authorities. Many peptides are used for the treatment of chronic diseases, and their implications in long-term therapy may be a concern for patient health.52,55 Absorption enhancers, such as chitosan or bile salts, improve the permeation of proteins drugs, increasing the paracellular transport by modulating TJ permeability.56 However, the use of this strategy is limited by the fact that, once TJs are open, transport through the GI tract is enhanced not only for the therapeutic drugs, but also for toxic molecules.53,55,57,58 Likewise, the GI tract contains numerous enzymes with specific target sites, which makes the use of enzyme inhibitors extremely difficult to achieve. Moreover, in long-term therapy, the use of enzyme inhibitors can be a problem because it can influence normal absorption of protein nutrients and can also affect the absorption of other peptides/proteins.37,38,53,55 Thus different approaches should be considered to increase oral bioavailability of GLP-1 and its analogs.
Oral Delivery Systems for Glucagon-Like Peptide-1 and Glucagon-Like Peptide-1 Analogs
The best approach to overcome all aforementioned problems is to modify the chemical structure of GLP-1 by adding novel functional groups or using new pharma-ceutical dosage forms that, instead of modifying the characteristics of the GI tract, will modify the peptide physicochemical properties or apply delivery carrier systems that protect the peptides and promote the crossing of biological barriers.52,59 This approach can also provide a higher bioavailability of the peptides when orally administered with delivery systems, as long as the biological activity is maintained without damaging the biological barriers.55
Byotinylated Glucagon-Like Peptide-1
One possible strategy to increase membrane permeability is to modify the peptide surface by adding a site-specific bioconjugation, such as fatty acids and vitamins that promote contact between the peptide and the apical membrane of enterocytes. This way, the absorption into cells is facilitated, allowing the drug to cross the intestinal epithelium.60–63
In this attempt, Youn and coauthors48 proposed an oral GLP-1 analog chemically modified with biotin. Because it is not synthesized by the human body, biotin must be obtained through diet, being actively transported to enterocytes by sodium-dependent multivitamin transport.64–66 In this study, biotin was added to the backbone of GLP-1 in lysine residues that, aside from promoting their transportation through epithelium, influence the trypsin proteolytic action in these peptides. Monobiotinylation and bibiotinylation (Lys26,34-biotin-GLP-1) were tested. Both of them had the same biotin-target sites, showing higher Caco-2 cell permeability (3.6-fold and 5.4-fold, respectively) and greater enzymatic resistance (monobiotinylated GLP-1 had a half-life 2.3-fold and 1.7-fold in rat intestine fluid and in homogenate, respectively, and bibiotinylated had a half-life 8.5-fold and 3.5-fold, respectively). These results revealed that the bibiotinylation was the best approach. Bioactivity tests showed that, despite the peptide modifications, the insulinotropic activity was well preserved (94.5%) and was not an impairment, which can be explained by the fact that its N terminal was well preserved.48 Moreover, under optimized conditions, bibiotinylated GLP-1 showed higher glucose-lowering potentials (nine-fold) than GLP-1 in diabetic mice after oral administration (Figure 2). These optimal conditions comprised a gastric neutralizer that mimics an enteric coating that allows peptides to reach the intestine undergoing the harsh stomach environment and 50% of its composition being propylene glycol, which decreases the exposure to GI tract enzymes.48
Figure 2.
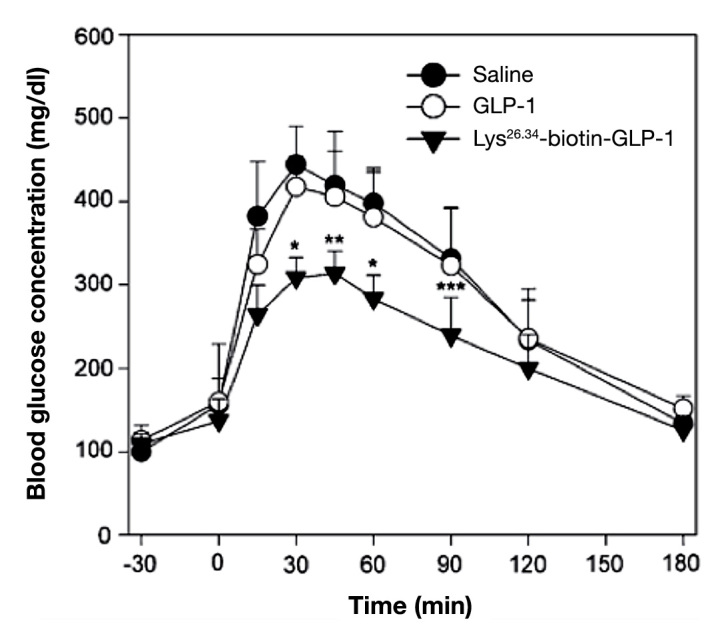
Oral hypoglycemic efficacy profiles of GLP-1 and Lys26,34-biotin-GLP-1 in diabetic mice (*p < .007, **p < .01, and ***p < .05). Reprinted with permission from European Journal of Pharmaceutics and Biopharmaceutics.48
In another work, exenatide-4 was used in similar experiences as described earlier. As what happened to the GLP-1, bibiotinylated exanatide-4 (DB-Ex-4) had the same biological activity, better stability (8.4-fold in trypsin and 9.0-fold in intestinal fluid), and higher hypoglycemic degrees (5.3-fold) than exanatide-4 in diabetic mice (Figure 3).67
Figure 3.
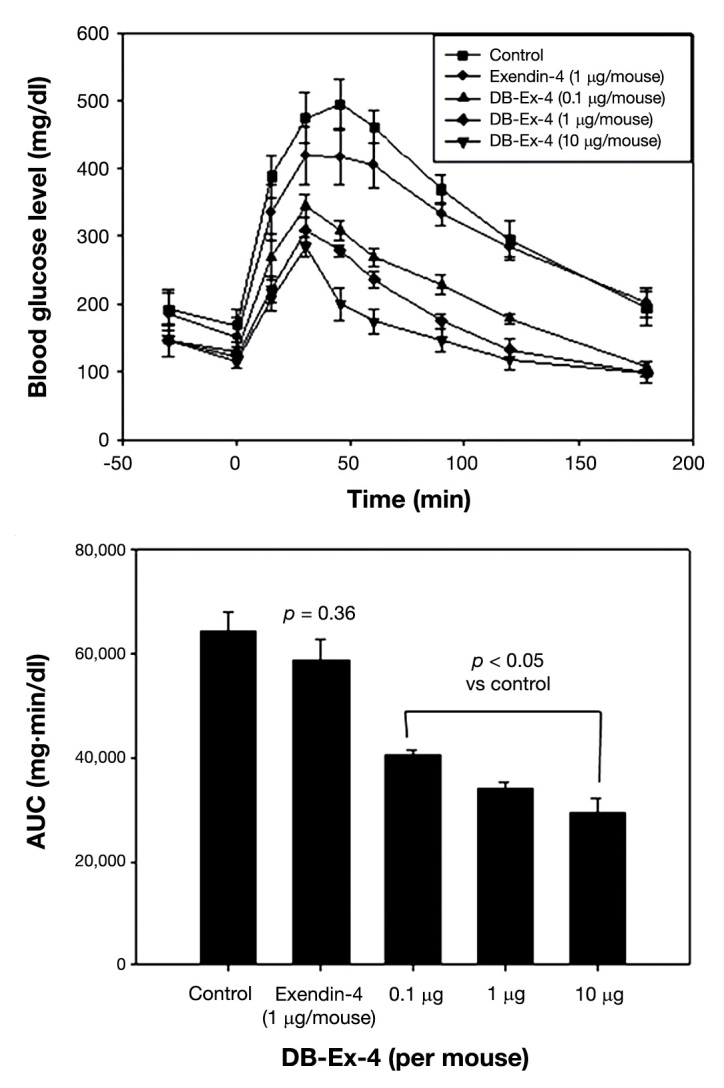
Oral hypoglycemic efficacies of exendin-4 and DB-Ex-4 in diabetic mice. Reprinted with permission from Journal of Controlled Release.67
When comparing all pharmacokinetic profiles, it was possible to observe that, after intravenous injections in diabetic mice, GLP-1 and exenatide-4 plasma concentrations were rapidly reduced, reaching the basal level 3 h post-injection. After oral administration, GLP-1, exenatide-4, and bibiotinylated GLP-1 remained on basal levels, showing that no absorption occurred, which can be explained by the rapid action of DPP-4 as discussed earlier, while DB-Ex-4 showed characteristic absorption patterns since exenatide-4 has a longer half-life than GLP-1.48,67
Therefore, biotin as a bioconjugate can lead to intestinal absorption of peptides and is thus a good strategy to enhance peptide bioavailability after oral administration. However, it is not a sustained system in itself, because it is only able to increase the absorption of peptides but not to protect them from degradation. To protect from degradation, it is necessary to prevent DPP-4 action by using GLP-1 analogs such as exenatide-4 or improving the drug delivery system.
PEGylated Glucagon-Like Peptide-1
As previous studies have demonstrated that adding polyethylene glycol (PEG) to peptides or nanoparticles increase its enzyme resistance as well as lower its clearance rate,56,68–72 Chae and coauthors73 also tested the influence of PEG in biotinylated GLP-1 (DBP-GLP-1). They showed that DBP-GLP-1 had even better results in absorption and enzyme resistance than just biotinylation (its plasma concentration increased quickly 30 min after oral administration), with as good results as native GLP-1 in peptide bioactivity and decreased glucose concentration [area under the curve (AUC) 0–180 min for 3 h reduced by 24.5%; Figure 4].
Figure 4.
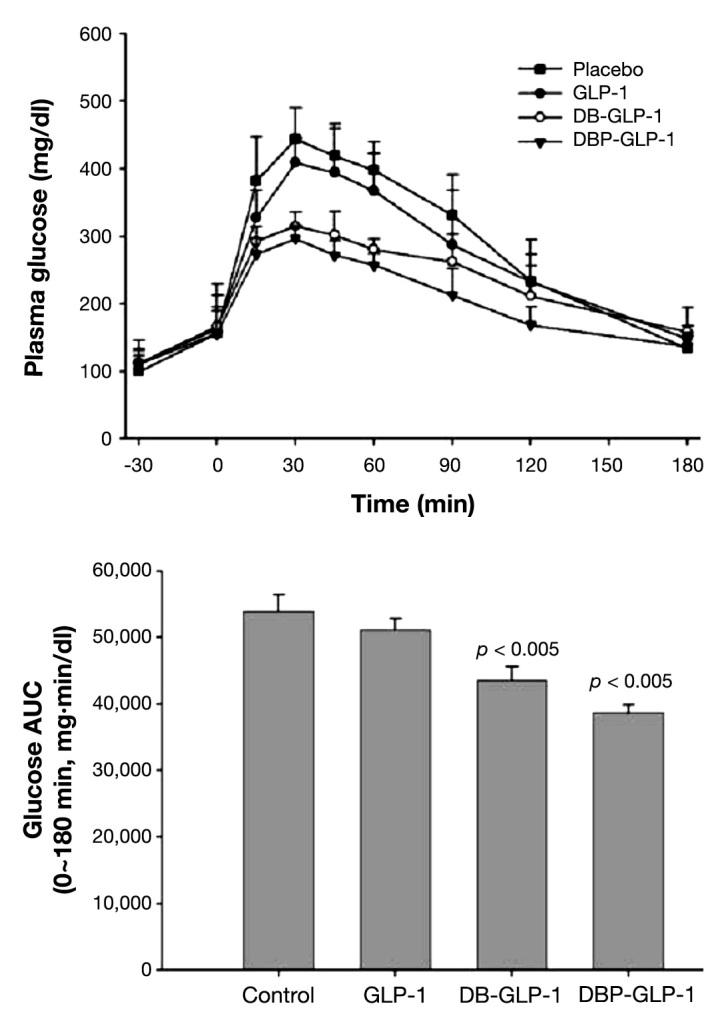
Glucose clearance kinetics after the oral administrations of GLP-1, DB-GLP-1, or DBP-GLP-1 prior to intraperitoneal glucose injections and glucose areas under the curve of the experimental groups (*p < .05). Reprinted with permission from Bioconjugating Chemistry.73
In view of these results, PEGylation can increase GLP-1 half-life without interfering with its biological effect and thus is a good approach to improve peptide bio-availability. Together with biotin, GLP-1 can be a good candidate for an oral antidiabetic drug.
Nanoparticles
Polymeric-Based Nanoparticles
Increasingly, nanotechnology plays an important role in the development of new forms of drug delivery. Several studies have been made using nanotechnology approaches in order to overcome the main biological obstacles to GLP-1 administration, namely, its short half-life.74–78 Although it is very important to prolong its time of action, it has yet to be administrated by parenteral routes.
Joseph and coauthors79 developed a d-ala2-GLP-1 (a GLP-1analog resistant to DPP-4) encapsulated into modified polylactide-co-glycolide (PLGA) nanoparticles that can be delivered orally because the peptides are entrapped in the nanoparticles and protected against the harsh environment of the GI tract. d-ala2-GLP-1 was released in therapeutic concentrations when administrated orally, decreasing glycemic response in diabetic mice (reduced AUC by 27% and 28% at 4 and 8 h tests, respectively). The authors also showed reduction in basal glycemia by 23% in 4 h and 35% in 8 h after treatment when compared with mice that had no treatment.
Thus the association of peptides/proteins to nanoparticles can also be a good alternative to continuous parenteral delivery.
pH-Sensitive Nanoparticles
Besides their nanoscale size, one of the greatest advantages in using polymeric nanoparticles as a drug delivery vehicle is the possibility of modifying its polymer structure and, thereafter, obtaining nanoparticles that are sensitive to influences such as pH80–82 and temperature.83 As with other peptides,84–87 exanetide-4 was also formulated in a pH-dependent nanoparticle carrier by Nguyen and coauthors.88 These nanoparticles were made of chitosan and poly(g-glutamic acid) coated with an enteric polymer that remained intact in low pH environments, protecting the nanoparticles from gastric content. This coating disintegrates with the increase of pH (above 5.5), and thus, when administrated orally, exenatide-4 nanoparticles are released in the small intestine only. Once there, they can open the TJs and, because of their pH-sensitivity, become unstable and disintegrate, making it possible for exenatide-4 to be transported via the paracellular pathway.88
Analyzing the pharmacokinetic and pharmacodynamic profiles after subcutaneous administration, it is possible to observe that the free form of exenatide-4 decreased shortly, with blood glucose levels returning to the basal level within time and increased insulin, reaching the maximum value 2 h after injection and returning to the basal level 3 h later. However, after oral administration, no alterations were observed in plasma concentrations, which was similar to results of capsules filled with emptynanoparticles. On the other hand, when administered orally, the capsule containing exendin-4 nanoparticles produced a slower but prolonged reduction in blood glucose levels, showing a maximum plasma concentration 5 hafter administration, and also increased the insulin levels in a slower but prolonged way (Figure 5). In this present case, the bioavailability of exenatide-4 encapsu-lated into nanoparticles was 14%, compared with exenatide-4 administrated subcutaneously, which is a significantly higher value compared with other examples in literature,88 a promising result considering the oral peptide administration in general. The longer onset of the hypoglycemic effect is a drawback and would be criticized to a rapid control of glycemia. Nevertheless, the advantages associated with the decrease in number of administrations and the constant plasmatic levels in the therapeutic window are beneficial in the long term.
Figure 5.
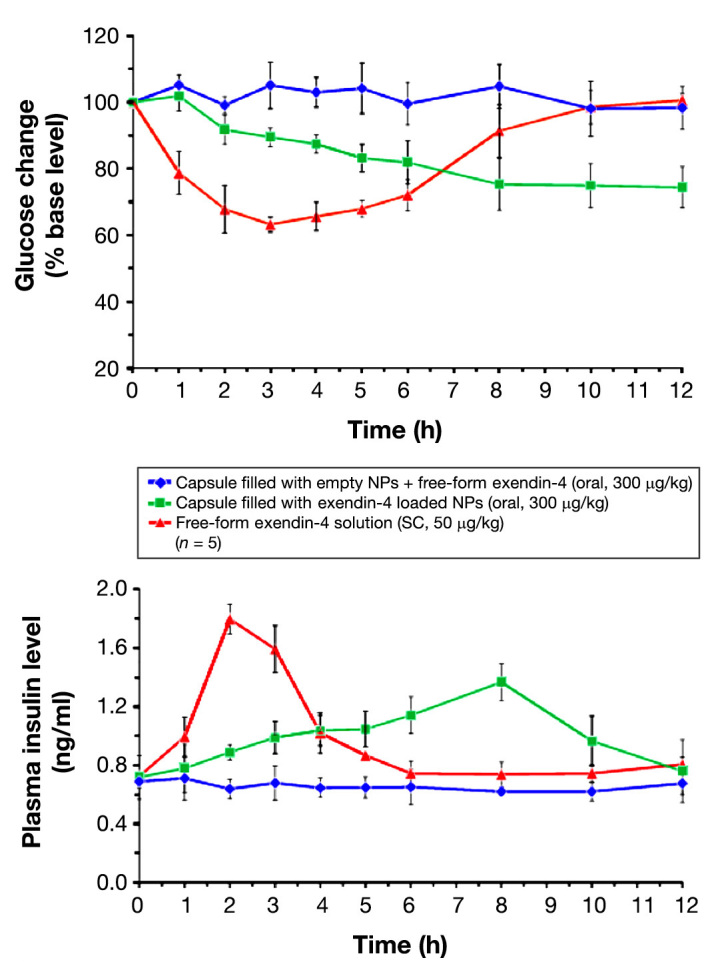
Blood glucose changes and plasma insulin levels versus time profiles of diabetic rats following the administration of different exendin-4 formulations. Reprinted with permission from Biomaterials.88 NP, nanoparticle; SC, subcutaneous
Nonpeptidic Receptor Agonist
Many efforts have been made to circumvent the need of administration of GLP-1 by parenteral routes. With this aim, the discovery of nonpeptide agonists has been studied in order to develop oral active pharmaceuticals; however, these attempts have generally been unsuccessful.
Chen and coauthors89 developed two substituted cyclo-butanes, S4P and Boc5 (Figure 6), which, like GLP-1, are GLP-1R specific and do not activate cells through glucagon or GLP-2 receptors (also a product of proglucagon gene). When tested, Boc5 behaved as a full GLP-1R agonist, whereas S4P behaved as a partial agonist. Boc5 amplified glucose-stimulated insulin secretion in isolated rat islets by a factor of up to 4.5 in comparison with controls. Oral administration of Boc5 dose-dependently inhibited food intake in mice, and a reduction in glycosylated hemoglobin to nondiabetic values was observed after daily injections of Boc5 into diabetic mice. Thus Boc5 behaved as a full GLP-1 mimetic both in vitro and in vivo. Other works reported similar research goals as described earlier, but none of the nonpeptide agonists were orally tested.90–92
Figure 6.
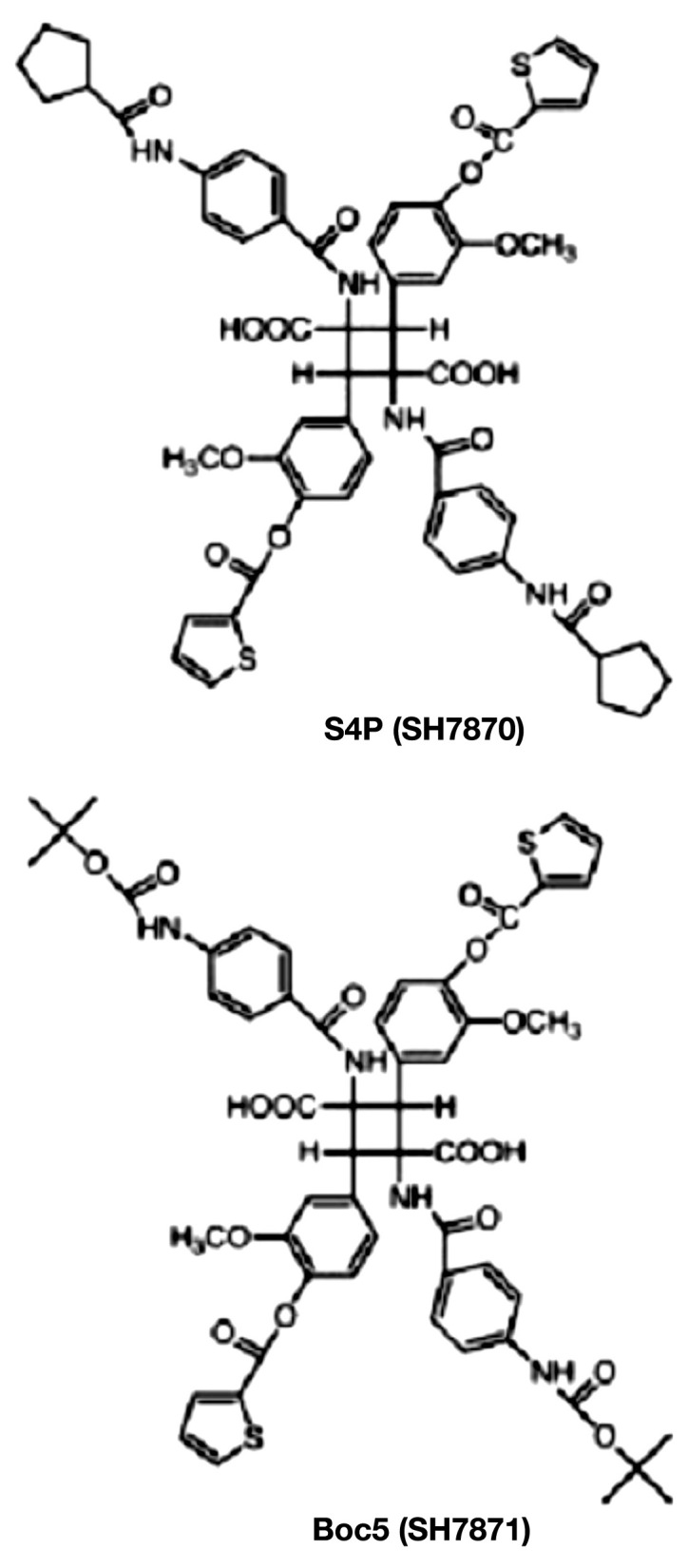
Chemical structure of substituted cyclobutanes S4P and Boc5. Reprinted with permission from Proceedings of the National Academy of Sciences of the United States of America.89
Regardless of whether nonpeptidic receptor agonist compounds may lead to the identification of orally active GLP-1 receptor agonists, further work is needed to improve the chemical stability and pharmacokinetic properties that enable their clinical development.
Advantages and Drawbacks Associated with Oral Glucagon-Like Peptide-1 and Glucagon-Like Peptide-1 Analog Delivery
Among the advantages for the patients who were previously discussed, such as more convenient and simple administration, comfortable way of treatment, increased patient compliance to the therapy, and therefore more clinical success, the costs of any oral formulation will also be an advantage for the pharmaceutical industry because it does not require specific conditions of sterility and other precautions against possible particle contamination.52
The biggest drawback of GLP-1-based therapy is the limited clinical experience with its use; it began only in 2005 and its analogs (e.g., oral GLP-1 and GLP-1 analog), delivered by parenteral routes started even later. So far, there are just a few studies in this field, and they are only in initial clinical trials phases. Glucagon-like peptide-1 acts only when plasma glucose levels are high and are excreted by renal clearance.8 Hence, it is possible to extrapolate that, when administrated orally, even if it is a high dose, GLP-1 will act only when glucose levels are high and possibly excrete if those levels are normal, not being toxic to the organism. Thus, the question still remains whether all its analogs or their carriers have some adverse effects.
Some side effects of exenatide have been reported sporadically, namely, nausea, vomiting, and cross immuno-reactivity, which decrease during the treatment, although these do not affect the therapy’s success. Like exenatide, liraglutide also causes these effects.15 However, not all GLP-1 analogs are associated with side effects. In 2010, a study using ORMD-0901 (a GLP-1 analog discussed later) administered enterically in pigs and dogs was performed in order to evaluate its safety and efficacy, and the study showed that this peptide was well tolerated in animals without any side effects.93
The carriers used in transporting GLP-1 and its analogs are considered excipients, being also eliminated by normal excretion pathways without presenting any adverse effect as reported by pharmaceutical companies.94–96
Further studies on detailed oral formulations are necessary to gain more knowledge on the advantages and drawbacks of oral GLP-1 and its analogs, namely, those related with intestinal mucosa toxicity over chronic administration.
Clinical Trials
With the continued increase of T2DM, it is imperative to satisfy the treatment needs of patients. Thus, many pharmaceutical companies are trying to develop long-acting GLP-1 analogs that can be orally administered.
Novo Nordisk developed an oral GLP-1 analog that is more resistant to enzymatic degradation (NN9924); it is in phase I clinical trial. This company established two different partnerships with two other pharmaceutical companies, Emisphere Technologies Inc. and Merrion Pharmaceuticals, which developed unique technologies, Eligen® and GIPET® (gastrointestinal permeation enhancement technology), respectively, which facilitate peptide absorption.94,95,97
Emisphere’s Eligen technology enables peptide/protein absorption without affecting biological properties or pharmacological activities. It uses a family of small carriers, in the case of GLP-1, SNAC (n-8-aminocaprylic acid), with low molecular weight that transport peptide/protein to the small intestine through weak and non-covalent interactions. They increase their lipophilicity, which enables the passive transcellular transport of drug molecules of all sizes through cell membranes instead of paracellular transport, and facilitate their passage across biological barriers, protecting them from chemical barriers. Once they are inside the cells, they dissociate, the peptide is free to reach the bloodstream, and the carrier is eliminated through normal excretion.54,94,98,99 This technology has already been used for oral delivery of other peptides such as insulin,98–100 and is not associated with side effects as demonstrated in clinical trials.54
Merrion’s GIPET technology consists of enteric-coated tablets targeting the duodenum, with peptide and patented absorption enhancers inside. Once it is in the intestinal lumen, the tablet dissolves, resulting in the release of the drug and the absorption enhancers, facilitating the peptide’s absorption across the epithelium and increasing bioavailability.95,101 These enhancers require high concen-trations for optimal enhancement, which leads to a transcellular perturbation due to surfactant properties, although this causes only a mild mucosal injury that is rapidly reversed without any major toxicity issues.54
Novo Nordisk, also Oramed Pharmaceuticals, developed a GLP-1 analog (ORMD-0901) that is presently in human clinical trials. This company exploited a delivery technique consisting of incorporation of adjuvants, pharmacopoeial registered protease inhibitors, and a carrier or excipient that protect the peptide from the chemical barriers. They increase paracellular permeability by chelating the calcium that is required to form intercellular junctions and promote its transportation across the epithelium to reach the bloodstream. These formulations were well tolerated in patients who experienced only mild GI events as side effects.54,96
Poxel is also developing an oral nonpeptidic GLP-1 analog. This project is not as advanced as the previous ones and is still in the lead optimization phase. Nevertheless, it has already shown positive effects in pathophysiological animal models.102
Conclusions
Glucagon-like peptide-1 and GLP-1 analogs have many advantages over existing therapies for the treatment of T2DM, including a superior ability to increase glucose-dependent insulin secretion and glucose-dependent glucagon suppression with consequent low risk of hypoglycemia. As oral delivery of proteins and peptides is still a great challenge for the modern pharmaceutical industry, oral delivery of GLP-1 and its analogs is a promising new scheme therapy.
Many studies have been conducted to overcome the main biological barriers of the body that prevent peptide and protein absorption through intestinal epithelium, but so far, the only available data from clinical trials cover a short time frame (2005-2012).
Many more in vivo and human studies are necessary until oral administration of GLP-1 and its analogs becomes fully acceptable for therapy, especially studies regarding safety in chronic administrations. In this review, there are very good indications that ongoing clinical trials will lead to new products in the near future.
Glossary
- (AUC)
area under the curve
- (DPP-4)
dipeptidyl peptidase-4
- (GI)
gastrointestinal
- (GLP-1)
glucagon-like peptide-1
- (GLP-1R)
glucagon-like peptide-1 receptor
- (PEG)
polyethylene glycol
- (PLGA)
polylactide-co-glycolide
- (T2DM)
type 2 diabetes mellitus
- (TJ)
tight junction
Funding
The authors acknowledge financial support from Fundação para a Ciência e a Tecnologia, Portugal (grants SFRH/BD/78127/2011 and SFRH/BPD/35996/2007), and the Academy of Finland (decision numbers 252215 and 256394).
References
- 1.Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract. 2010;87(1):4–14. doi: 10.1016/j.diabres.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 2.Tahrani AA, Piya MK, Kennedy A, Barnett AH. Glycaemic control in type 2 diabetes: targets and new therapies. Pharmacol Ther. 2010;125(2):328–361. doi: 10.1016/j.pharmthera.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 3.Nicholson G, Hall GM. Diabetes mellitus: new drugs for a new epidemic. Br J Anaesth. 2011;107(1):65–73. doi: 10.1093/bja/aer120. [DOI] [PubMed] [Google Scholar]
- 4.Arulmozhi DK, Portha B. GLP-1 based therapy for type 2 diabetes. Eur J Pharm Sci. 2006;28(1-2):96–108. doi: 10.1016/j.ejps.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 5.Sadrzadeh N, Glembourtt MJ, Stevenson CL. Peptide drug delivery strategies for the treatment of diabetes. J Pharm Sci. 2007;96(8):1925–1954. doi: 10.1002/jps.20848. [DOI] [PubMed] [Google Scholar]
- 6.Marre M, Penfornis A. GLP-1 receptor agonists today. Diabetes Res Clin Pract. 2011;93(3):317–327. doi: 10.1016/j.diabres.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 7.Nauck MA, Vardarli I, Deacon CF, Holst JJ, Meier JJ. Secretion of glucagon-like peptide-1 (GLP-1) in type 2 diabetes: what is up, what is down? Diabetologia. 2011;54(1):10–18. doi: 10.1007/s00125-010-1896-4. [DOI] [PubMed] [Google Scholar]
- 8.Baggio LL, Drucker DJ. Biology of Incretins: GLP-1 and GIP. Gastroenterology. 2007;132(6):2131–2157. doi: 10.1053/j.gastro.2007.03.054. [DOI] [PubMed] [Google Scholar]
- 9.Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. 2007;87(4):1409–1439. doi: 10.1152/physrev.00034.2006. [DOI] [PubMed] [Google Scholar]
- 10.Steinert RE, Poller B, Castelli MC, Friedman K, Huber AR, Drewe J, Beglinger C. Orally administered glucagon-like peptide-1 affects glucose homeostasis following an oral glucose tolerance test in healthy male subjects. Clin Pharmacol Ther. 2009;86(6):644–650. doi: 10.1038/clpt.2009.159. [DOI] [PubMed] [Google Scholar]
- 11.Steinert RE, Poller B, Castelli MC, Drewe J, Beglinger C. Oral administration of glucagon-like peptide 1 or peptide YY 3-36 affects food intake in healthy male subjects. Am J Clin Nutr. 2010;92(4):810–817. doi: 10.3945/ajcn.2010.29663. [DOI] [PubMed] [Google Scholar]
- 12.Takei I, Kasatani T. Future therapy of diabetes mellitus. Biomed Pharmacother. 2004;58(10):578–581. doi: 10.1016/j.biopha.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 13.Portha B, Tourrel-Cuzin C, Movassat J. Activation of the GLP-1 receptor signalling pathway: a relevant strategy to repair a deficient beta-cell mass. Exp Diabetes Res. 2011;2011:376509. doi: 10.1155/2011/376509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holst JJ, Christensen M, Lund A, de Heer J, Svendsen B, Kielgast U, Knop FK. Regulation of glucagon secretion by incretins. Diabetes Obes Metab. 2011;13(Suppl 1):89–94. doi: 10.1111/j.1463-1326.2011.01452.x. [DOI] [PubMed] [Google Scholar]
- 15.Ahrén B. GLP-1 for type 2 diabetes. Exp Cell Res. 2011;317(9):1239–1245. doi: 10.1016/j.yexcr.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 16.Perfetti R, Merkel P. Glucagon-like peptide-1: a major regulator of pancreatic beta-cell function. Eur J Endocrinol. 2000;143(6):717–725. doi: 10.1530/eje.0.1430717. [DOI] [PubMed] [Google Scholar]
- 17.Gallwitz B. New therapeutic strategies for the treatment of type 2 diabetes mellitus based on incretins. Rev Diabet Stud. 2005;2(2):61–69. doi: 10.1900/RDS.2005.2.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gutzwiller JP, Drewe J, Göke B, Schmidt H, Rohrer B, Lareida J, Beglinger C. Glucagon-like peptide-1 promotes satiety and reduces food intake in patients with diabetes mellitus type 2. Am J Physiol. 1999;276(5 Pt 2):R1541–4. doi: 10.1152/ajpregu.1999.276.5.R1541. [DOI] [PubMed] [Google Scholar]
- 19.Neumiller JJ. Clinical pharmacology of incretin therapies for type 2 diabetes mellitus: implications for treatment. Clin Ther. 2011;33(5):528–576. doi: 10.1016/j.clinthera.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 20.Davidson MH. Cardiovascular effects of glucagonlike peptide-1 agonists. Am J Cardiol. 2011;108(3 Suppl):33B–41B. doi: 10.1016/j.amjcard.2011.03.046. [DOI] [PubMed] [Google Scholar]
- 21.Cernea S, Raz I. Therapy in the early stage: incretins. Diabetes Care. 2011;34(Suppl 2):S264–71. doi: 10.2337/dc11-s223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vilsboll T, Krarup T, Madsbad S, Holst JJ. No reactive hypo-glycaemia in Type 2 diabetic patients after subcutaneous administration of GLP-1 and intravenous glucose. Diabet Med. 2001;18(2):144–149. doi: 10.1046/j.1464-5491.2001.00424.x. [DOI] [PubMed] [Google Scholar]
- 23.Holst JJ, Deacon C, Toft-Nielsen MB, Bjerre-Knudsen L. On the treatment of diabetes mellitus with glucagon-like peptide-1. Ann N Y Acad Sci. 1998;865:336–343. doi: 10.1111/j.1749-6632.1998.tb11193.x. [DOI] [PubMed] [Google Scholar]
- 24.Unger J. Clinical efficacy of GLP-1 agonists and their place in the diabetes treatment algorithm. J Am Osteopath Assoc. 2011;111(2 Suppl 1):eS2–9. [PubMed] [Google Scholar]
- 25.Unger JR, Parkin CG. Glucagon-like peptide-1 (GLP-1) receptor agonists: differentiating the new medications. Diabetes Ther. 2011;2(1):29–39. doi: 10.1007/s13300-010-0013-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garber AJ. Long-acting glucagon-like peptide 1 receptor agonists: a review of their efficacy and tolerability. Diabetes Care. 2011;34(Suppl 2):S279–84. doi: 10.2337/dc11-s231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, Zheng X, Tang L, Xu W, Gong M. GLP-1 analogs containing disulfide bond exhibited prolonged half-life in vivo than GLP-1. Peptides. 2011;32(6):1303–1312. doi: 10.1016/j.peptides.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 28.Deacon CF. Potential of liraglutide in the treatment of patients with type 2 diabetes. Vasc Health Risk Manag. 2009;5(1):199–211. doi: 10.2147/vhrm.s4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalra S, Kalra B, Kumar A. GLP-1 analogs: newer molecules, newer uses. Recent Pat Endocr Metab Immune Drug Discov. 2009;3(2):129–134. [Google Scholar]
- 30.Chen MC, Sonaje K, Chen KJ, Sung HW. A review of the prospects for polymeric nanoparticle platforms in oral insulin delivery. Biomaterials. 2011;32(36):9826–9838. doi: 10.1016/j.biomaterials.2011.08.087. [DOI] [PubMed] [Google Scholar]
- 31.Beglinger C, Poller B, Arbit E, Ganzoni C, Gass S, Gomez-Orellana I, Drewe J. Pharmacokinetics and pharmacodynamic effects of oral GLP-1 and PYY3-36: a proof-of-concept study in healthy subjects. Clin Pharmacol Ther. 2008;84(4):468–474. doi: 10.1038/clpt.2008.35. [DOI] [PubMed] [Google Scholar]
- 32.Cao X, Gibbs ST, Fang L, Miller HA, Landowski CP, Shin HC, Lennernas H, Zhong Y, Amidon GL, Yu LX, Sun D. Why is it challenging to predict intestinal drug absorption and oral bio-availability in human using rat model. Pharm Res. 2006;23(8):1675–1686. doi: 10.1007/s11095-006-9041-2. [DOI] [PubMed] [Google Scholar]
- 33.Hamman JH, Demana PH, Olivier EI. Targeting receptors, transporters and site of absorption to improve oral drug delivery. Drug Target Insights. 2007;2:71–81. [PMC free article] [PubMed] [Google Scholar]
- 34.Mahato RI, Narang AS, Thoma L, Miller DD. Emerging trends in oral delivery of peptide and protein drugs. Crit Rev Ther Drug Carrier Syst. 2003;20(2-3):153–214. doi: 10.1615/critrevtherdrugcarriersyst.v20.i23.30. [DOI] [PubMed] [Google Scholar]
- 35.Le Ferrec E, Chesne C, Artusson P, Brayden D, Fabre G, Gires P, Guillou F, Rousset M, Rubas W, Scarino ML. In vitro models of the intestinal barrier. The report and recommendations of ECVAM Workshop 46. European Centre for the Validation of Alternative methods. Altern Lab Anim. 2001;29(6):649–668. doi: 10.1177/026119290102900604. [DOI] [PubMed] [Google Scholar]
- 36.Krol S, Ellis-Behnke R, Marchetti P. Nanomedicine for treatment of diabetes in an aging population: state-of-the-art and future developments. Maturitas. 2012;73(1):61–67. doi: 10.1016/j.maturitas.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 37.Thompson C, Ibie C. The oral delivery of proteins using inter-polymer polyelectrolyte complexes. Ther Deliv. 2011;2(12):1611–1631. doi: 10.4155/tde.11.131. [DOI] [PubMed] [Google Scholar]
- 38.Lee HJ. Protein drug oral delivery: the recent progress. Arch Pharm Res. 2002;25(5):572–584. doi: 10.1007/BF02976925. [DOI] [PubMed] [Google Scholar]
- 39.Lai SK, Wang YY, Hanes J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv Drug Deliv Rev. 2009;61(2):158–171. doi: 10.1016/j.addr.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ensign LM, Cone R, Hanes J. Oral drug delivery with polymeric nanoparticles: the gastrointestinal mucus barriers. Adv Drug Deliv Rev. 2011;64(6):557–570. doi: 10.1016/j.addr.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Atuma C, Strugala V, Allen A, Holm L. The adherent gastro-intestinal mucus gel layer: thickness and physical state in vivo. Am J Physiol Gastrointest Liver Physiol. 2001;280(5):G922–9. doi: 10.1152/ajpgi.2001.280.5.G922. [DOI] [PubMed] [Google Scholar]
- 42.Johansson ME, Larsson JM, Hansson GC. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4659–4665. doi: 10.1073/pnas.1006451107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jin Y, Song Y, Zhu X, Zhou D, Chen C, Zhang Z, Huang Y. Goblet cell-targeting nanoparticles for oral insulin delivery and the influence of mucus on insulin transport. Biomaterials. 2012;33(5):1573–1582. doi: 10.1016/j.biomaterials.2011.10.075. [DOI] [PubMed] [Google Scholar]
- 44.Des Rieux A, Fievez V, Theate I, Mast J, Preat V, Schneider YJ. An improved in vitro model of human intestinal follicle-associated epithelium to study nanoparticle transport by M cells. Eur J Pharm Sci. 2007;30(5):380–391. doi: 10.1016/j.ejps.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 45.Des Rieux A, Fievez V, Momtaz M, Detrembleur C, Alonso-Sande M, Van Gelder J, Cauvin A, Schneider YJ, Préat V. Helodermin-loaded nanoparticles: characterization and transport across an in vitro model of the follicle-associated epithelium. J Control Release. 2007;118(3):294–302. doi: 10.1016/j.jconrel.2006.12.023. [DOI] [PubMed] [Google Scholar]
- 46.Fearn RA, Hirst BH. Predicting oral drug absorption and hepatobiliary clearance: human intestinal and hepatic in vitro cell models. Environ Toxicol Pharmacol. 2006;21(2):168–178. doi: 10.1016/j.etap.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 47.Griffin BT, O’Driscoll CM. Opportunities and challenges for oral delivery of hydrophobic versus hydrophilic peptide and protein-like drugs using lipid-based technologies. Ther Deliv. 2011;2(12):1633–1653. doi: 10.4155/tde.11.128. [DOI] [PubMed] [Google Scholar]
- 48.Youn YS, Chae SY, Lee S, Kwon MJ, Shin HJ, Lee KC. Improved peroral delivery of glucagon-like peptide-1 by site-specific biotin modification: design, preparation, and biological evaluation. Eur J Pharm Biopharm. 2008;68(3):667–675. doi: 10.1016/j.ejpb.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 49.Salamat-Miller N, Johnston TP. Current strategies used to enhance the paracellular transport of therapeutic polypeptides across the intestinal epithelium. Int J Pharm. 2005;294(1-2):201–216. doi: 10.1016/j.ijpharm.2005.01.022. [DOI] [PubMed] [Google Scholar]
- 50.Qiu Y, Chen Y, Zhang GG, Liu L, Porter WR, editors. Developing solid oral dosage forms: pharmaceutical theory and practice. Burlington: Academic Press; 2009. [Google Scholar]
- 51.Assimakopoulos SF, Papageorgiou I, Charonis A. Enterocytes’ tight junctions: From molecules to diseases. World J Gastrointest Pathophysiol. 2011;2(6):123–137. doi: 10.4291/wjgp.v2.i6.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khafagy el-S, Morishita M, Onuki Y, Takayama K. Current challenges in non-invasive insulin delivery systems: a comparative review. Adv Drug Deliv Rev. 2007;59(15):1521–1546. doi: 10.1016/j.addr.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 53.Park K, Kwon IC, Park K. Oral protein delivery: current status and future prospect. React Funct Polym. 2011;71(3):280–287. [Google Scholar]
- 54.Maher S, Brayden DJ. Overcoming poor permeability: translating permeation enhancers for oral peptide delivery. Drug Discov Today. 2012;9(2):e113–9. doi: 10.1016/j.ddtec.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 55.Morishita M, Peppas NA. Is the oral route possible for peptide and protein drug delivery? Drug Discov Today. 2006;11(19-20):905–910. doi: 10.1016/j.drudis.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 56.Fonte P, Andrade F, Araujo F, Andrade C, Neves JD, Sarmento B. Chitosan-coated solid lipid nanoparticles for insulin delivery. Methods Enzymol. 2012;508:295–314. doi: 10.1016/B978-0-12-391860-4.00015-X. [DOI] [PubMed] [Google Scholar]
- 57.Deli MA. Potential use of tight junction modulators to reversibly open membranous barriers and improve drug delivery. Biochim Biophys Acta. 2009;1788(4):892–910. doi: 10.1016/j.bbamem.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 58.Cano-Cebrian MJ, Zornoza T, Granero L, Polache A. Intestinal absorption enhancement via the paracellular route by fatty acids, chitosans and others: a target for drug delivery. Curr Drug Deliv. 2005;2(1):9–22. doi: 10.2174/1567201052772834. [DOI] [PubMed] [Google Scholar]
- 59.Liang JF, Yang VC. Insulin-cell penetrating peptide hybrids with improved intestinal absorption efficiency. Biochem Biophys Res Commun. 2005;335(3):734–738. doi: 10.1016/j.bbrc.2005.07.142. [DOI] [PubMed] [Google Scholar]
- 60.Chalasani KB, Russell-Jones GJ, Jain AK, Diwan PV, Jain SK. Effective oral delivery of insulin in animal models using vitamin B12-coated dextran nanoparticles. J Control Release. 2007;122(2):141–150. doi: 10.1016/j.jconrel.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 61.Francis MF, Cristea M, Winnik FM. Exploiting the vitamin B12 pathway to enhance oral drug delivery via polymeric micelles. Biomacromolecules. 2005;6(5):2462–2467. doi: 10.1021/bm0503165. [DOI] [PubMed] [Google Scholar]
- 62.Kim SK, Lee EH, Vaishali B, Lee S, Lee YK, Kim CY, Moon HT, Byun Y. Tricaprylin microemulsion for oral delivery of low molecular weight heparin conjugates. J Control Release. 2005;105(1-2):32–42. doi: 10.1016/j.jconrel.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 63.Said HM. Intestinal absorption of water-soluble vitamins in health and disease. Biochem J. 2011;437(3):357–372. doi: 10.1042/BJ20110326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reidling JC, Nabokina SM, Said HM. Molecular mechanisms involved in the adaptive regulation of human intestinal biotin uptake: A study of the hSMVT system. Am J Physiol Gastrointest Liver Physiol. 2007;292(1):G275–81. doi: 10.1152/ajpgi.00327.2006. [DOI] [PubMed] [Google Scholar]
- 65.Ramanathan S, Pooyan S, Stein S, Prasad PD, Wang J, Leibowitz MJ, Ganapathy V, Sinko PJ. Targeting the sodium-dependent multivitamin transporter (SMVT) for improving the oral absorption properties of a retro-inverso Tat nonapeptide. Pharm Res. 2001;18(7):950–956. doi: 10.1023/a:1010932126662. [DOI] [PubMed] [Google Scholar]
- 66.Said HM. Cell and molecular aspects of human intestinal biotin absorption. J Nutr. 2009;139(1):158–162. doi: 10.3945/jn.108.092023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jin CH, Chae SY, Son S, Kim TH, Um KA, Youn YS, Lee S, Lee KC. A new orally available glucagon-like peptide-1 receptor agonist, biotinylated exendin-4, displays improved hypoglycemic effects in db/db mice. J Control Release. 2009;133(3):172–177. doi: 10.1016/j.jconrel.2008.09.091. [DOI] [PubMed] [Google Scholar]
- 68.Lee S, Youn YS, Lee SH, Byun Y, Lee KC. PEGylated glucagon-like peptide-1 displays preserved effects on insulin release in isolated pancreatic islets and improved biological activity in db/db mice. Diabetologia. 2006;49(7):1608–1611. doi: 10.1007/s00125-006-0234-3. [DOI] [PubMed] [Google Scholar]
- 69.Lee SH, Lee S, Youn YS, Na DH, Chae SY, Byun Y, Lee KC. Synthesis, characterization, and pharmacokinetic studies of PEGylated glucagon-like peptide-1. Bioconjug Chem. 2005;16(2):377–382. doi: 10.1021/bc049735+. [DOI] [PubMed] [Google Scholar]
- 70.Youn YS, Chae SY, Lee S, Jeon JE, Shin HG, Lee KC. Evaluation of therapeutic potentials of site-specific PEGylated glucagon-like peptide-1 isomers as a type 2 anti-diabetic treatment: insulinotropic activity, glucose-stabilizing capability, and proteolytic stability. Biochem Pharmacol. 2007;73(1):84–93. doi: 10.1016/j.bcp.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 71.Youn YS, Jeon JE, Chae SY, Lee S, Lee KC. PEGylation improves the hypoglycaemic efficacy of intranasally administered glucagon-like peptide-1 in type 2 diabetic db/db mice. Diabetes Obes Metab. 2008;10(4):343–346. doi: 10.1111/j.1463-1326.2007.00823.x. [DOI] [PubMed] [Google Scholar]
- 72.Chae SY, Chun YG, Lee S, Jin CH, Lee ES, Lee KC, Youn YS. Pharmacokinetic and pharmacodynamic evaluation of site-specific PEGylated glucagon-like peptide-1 analogs as flexible postprandial-glucose controllers. J Pharm Sci. 2009;98(4):1556–1567. doi: 10.1002/jps.21532. [DOI] [PubMed] [Google Scholar]
- 73.Chae SY, Jin CH, Shin HJ, Youn YS, Lee S, Lee KC. Preparation, characterization, and application of biotinylated and biotin-PEGylated glucagon-like peptide-1 analogues for enhanced oral delivery. Bioconjug Chem. 2008;19(1):334–341. doi: 10.1021/bc700292v. [DOI] [PubMed] [Google Scholar]
- 74.Hanato J, Kuriyama K, Mizumoto T, Debari K, Hatanaka J, Onoue S, Yamada S. Liposomal formulations of glucagon-like peptide-1: Improved bioavailability and anti-diabetic effect. Int J Pharm. 2009;382(1-2):111–116. doi: 10.1016/j.ijpharm.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 75.Gao Z, Tang Y, Chen J, Bai R, Zhang Q, Hou Y, Lu Y, Bai G. A novel DPP-IV-resistant analog of glucagon-like peptide-1 (GLP-1): KGLP-1 alone or in combination with long-acting PLGA micro-spheres. Peptides. 2009;30(10):1874–1881. doi: 10.1016/j.peptides.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 76.Kwak HH, Shim WS, Son MK, Kim YJ, Kim TH, Youn HJ, Kang SH, Shim CK. Efficacy of a new sustained-release microsphere formulation of exenatide, DA-3091, in Zucker diabetic fatty (ZDF) rats. Eur J Pharm Sci. 2010;40(2):103–109. doi: 10.1016/j.ejps.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 77.Choi S, Baudys M, Kim SW. Control of blood glucose by novel GLP-1 delivery using biodegradable triblock copolymer of PLGA-PEG-PLGA in type 2 diabetic rats. Pharm Res. 2004;21(5):827–831. doi: 10.1023/b:pham.0000026435.27086.94. [DOI] [PubMed] [Google Scholar]
- 78.Yin D, Lu Y, Zhang H, Zhang G, Zou H, Sun D, Zhong Y. Preparation of glucagon-like peptide-1 loaded PLGA microspheres: characterizations, release studies and bioactivities in vitro/in vivo. Chem Pharm Bull (Tokyo). 2008;56(2):156–161. doi: 10.1248/cpb.56.156. [DOI] [PubMed] [Google Scholar]
- 79.Joseph JW, Kalitsky J, St-Pierre S, Brubaker PL. Oral delivery of glucagon-like peptide-1 in a modified polymer preparation normalizes basal glycaemia in diabetic db/db mice. Diabetologia. 2000;43(10):1319–1328. doi: 10.1007/s001250051529. [DOI] [PubMed] [Google Scholar]
- 80.Cai G, Jiang H. pH-sensitive nanoparticles self-assembled from a novel class of biodegradable amphiphilic copolymers based on chitosan. J Mater Sci Mater Med. 2009;20(6):1315–1320. doi: 10.1007/s10856-008-3689-6. [DOI] [PubMed] [Google Scholar]
- 81.Sonaje K, Lin KJ, Wang JJ, Mi FL, Chen CT, Juang JH, Sung HW. Self-assembled pH-sensitive nanoparticles: a platform for oral delivery of protein drugs. Adv Funct Mater. 2010;20(21):3695–3700. [Google Scholar]
- 82.Makhlof A, Tozuka Y, Takeuchi H. Design and evaluation of novel pH-sensitive chitosan nanoparticles for oral insulin delivery. Eur J Pharm Sci. 2011;42(5):445–451. doi: 10.1016/j.ejps.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 83.Soga O, van Nostrum CF, Ramzi A, Visser T, Soulimani F, Frederik PM, Bomans PH, Hennink WE. Physicochemical characterization of degradable thermosensitive polymeric micelles. Langmuir. 2004;20(21):9388–9395. doi: 10.1021/la048354h. [DOI] [PubMed] [Google Scholar]
- 84.Sung HW, Sonaje K, Liao ZX, Hsu LW, Chuang EY. pH-responsive nanoparticles shelled with chitosan for oral delivery of insulin: from mechanism to therapeutic applications. Acc Chem Res. 2012;45(4):619–629. doi: 10.1021/ar200234q. [DOI] [PubMed] [Google Scholar]
- 85.He P, Tang Z, Lin L, Deng M, Pang X, Zhuang X, Chen X. Novel biodegradable and pH-sensitive poly(ester amide) microspheres for oral insulin delivery. Macromol Biosci. 2012;12(4):547–556. doi: 10.1002/mabi.201100358. [DOI] [PubMed] [Google Scholar]
- 86.Wang XQ, Dai JD, Chen Z, Zhang T, Xia GM, Nagai T, Zhang Q. Bioavailability and pharmacokinetics of cyclosporine A-loaded pH-sensitive nanoparticles for oral administration. J Control Release. 2004;97(3):421–429. doi: 10.1016/j.jconrel.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 87.Chellampillai B, Pawar AP. Improved bioavailability of orally administered andrographolide from pH-sensitive nanoparticles. Eur J Drug Metab Pharmacokinet. 2011;35(3-4):123–129. doi: 10.1007/s13318-010-0016-7. [DOI] [PubMed] [Google Scholar]
- 88.Nguyen HN, Wey SP, Juang JH, Sonaje K, Ho YC, Chuang EY, Hsu CW, Yen TC, Lin KJ, Sung HW. The glucose-lowering potential of exendin-4 orally delivered via a pH-sensitive nanoparticle vehicle and effects on subsequent insulin secretion in vivo. Biomaterials. 2011;32(10):2673–2682. doi: 10.1016/j.biomaterials.2010.12.044. [DOI] [PubMed] [Google Scholar]
- 89.Chen D, Liao J, Li N, Zhou C, Liu Q, Wang G, Zhang R, Zhang S, Lin L, Chen K, Xie X, Nan F, Young AA, Wang MW. A nonpeptidic agonist of glucagon-like peptide 1 receptors with efficacy in diabetic db/db mice. Proc Natl Acad Sci U S A. 2007;104(3):943–948. doi: 10.1073/pnas.0610173104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Knudsen LB, Kiel D, Teng M, Behrens C, Bhumralkar D, Kodra JT, Holst JJ, Jeppesen CB, Johnson MD, de Jong JC, Jorgensen AS, Kercher T, Kostrowicki J, Madsen P, Olesen PH, Petersen JS, Poulsen F, Sidelmann UG, Sturis J, Truesdale L, May J, Lau J. Small-molecule agonists for the glucagon-like peptide 1 receptor. Proc Natl Acad Sci U S A. 2007;104(3):937–942. doi: 10.1073/pnas.0605701104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li Y, Xu W, Tang L, Gong M, Zhang J. A novel GLP-1 analog exhibits potent utility in the treatment of type 2 diabetes with an extended half-life and efficient glucose clearance in vivo. Peptides. 2011;32(7):1408–1414. doi: 10.1016/j.peptides.2011.05.026. [DOI] [PubMed] [Google Scholar]
- 92.Teng M, Johnson MD, Thomas C, Kiel D, Lakis JN, Kercher T, Aytes S, Kostrowicki J, Bhumralkar D, Truesdale L, May J, Sidelman U, Kodra JT, Jørgensen AS, Olesen PH, de Jong JC, Madsen P, Behrens C, Pettersson I, Knudsen LB, Holst JJ, Lau J. Small molecule ago-allosteric modulators of the human glucagon-like peptide-1 (hGLP-1) receptor. Bioorg Med Chem Lett. 2007;17(19):5472–5478. doi: 10.1016/j.bmcl.2007.06.086. [DOI] [PubMed] [Google Scholar]
- 93.Eldor R, Kidron M, Greenberg-Shushlav Y, Arbit E. Novel glucagon-like peptide-1 analog delivered orally reduces postprandial glucose excursions in porcine and canine models. J Diabetes Sci Technol. 2010;4(6):1516–1523. doi: 10.1177/193229681000400629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Emisphere. www.emisphere.com.
- 95. Merrion. www.merrionpharma.com.
- 96. Oramed. www.oramed.com.
- 97. Novo Nordisk. www.novonordisk.com.
- 98.Malkov D, Angelo R, Wang HZ, Flanders E, Tang H, Gomez-Orellana I. Oral delivery of insulin with the eligen technology: mechanistic studies. Curr Drug Deliv. 2005;2(2):191–197. doi: 10.2174/1567201053586001. [DOI] [PubMed] [Google Scholar]
- 99.Hoffman A, Qadri B. Eligen insulin--a system for the oral delivery of insulin for diabetes. IDrugs. 2008;11(6):433–441. [PubMed] [Google Scholar]
- 100. Diabetes in Control. www.diabetesincontrol.com.
- 101.Leonard TW, Lynch J, McKenna MJ, Brayden DJ. Promoting absorption of drugs in humans using medium-chain fatty acid-based solid dosage forms: GIPET. Expert Opin Drug Deliv. 2006;3(5):685–692. doi: 10.1517/17425247.3.5.685. [DOI] [PubMed] [Google Scholar]
- 102. Poxel. www.poxel.com.