

Abstract
Most anticancer drugs entering clinical trials fail to achieve approval from the US FDA. Drug development is hampered by the lack of preclinical models with therapeutic predictive value. Herein, we report the development and validation of a tumorgraft model of renal cell carcinoma (RCC) and its application to the evaluation of an experimental drug. Tumor samples from 94 patients were implanted in the kidney of mice without additives or disaggregation. Tumors from 35 patients formed tumorgrafts, and 16 stable lines were established. Samples from metastatic sites engrafted at high frequency, and stable engraftment of primary tumors in mice correlated with decreased patient survival suggesting that tumor growth in mice may reveal the acquisition by the tumor of an ability to thrive at distant sites and metastasize. Tumorgrafts retained the histology, gene expression, DNA copy number alterations, and over 90% of the protein-coding gene mutations of the corresponding tumors. As determined by the induction of hypercalcemia in tumorgraft-bearing mice, tumorgrafts were able to act on the host causing paraneoplastic syndromes. In studies simulating drug exposures in patients, RCC tumorgraft growth was inhibited by sunitinib and sirolimus (into which temsirolimus is converted in humans), but not by erlotinib, which was used as a control. Dovitinib, a drug in clinical development, showed greater activity than sunitinib and sirolimus. The routine incorporation of models recapitulating the molecular genetics and drug sensitivities of human tumors into preclinical programs has the potential to improve oncology drug development.
Keywords: xenograft, tumor graft, orthotopic, PK, NOD/SCID, kidney cancer, clear-cell renal cell carcinoma
INTRODUCTION
More than 80% of anticancer drugs administered to patients in clinical trials fail to reach FDA approval (1-3), twice the failure rate of drugs in other categories (1). Better paradigms and preclinical models are needed to reduce the toll on patient lives and resources.
Most preclinical studies evaluate drugs in tumor cell lines that have been passaged in culture for many years (i.e. NCI-60 panel) (4, 5). Although substantial contributions have been made with these cell lines, their value is diminished by new mutations acquired during the process of adaptation to growth in culture and subsequent expansion (6, 7). In addition, tumors formed by cell lines in mice tend to be poorly differentiated and likely dissimilar from the tumor from which the cell line was originally derived (6-9). These factors probably explain their limited utility in predicting drug responsiveness in patients.
Renal cell carcinoma (RCC) is especially well suited for the development of a tumorgraft model in which tumors derived from patients are implanted in mice. First, RCCs are typically large, providing access to abundant tumor material. Second, RCC is seldom treated with chemotherapy and thus the molecular genetics and behavior of the tumor is unlikely to be affected by prior exposure to DNA damaging agents. Third, RCCs implanted in mice preserve the histology and karyotype of patient tumors (10-16). Fourth, the implantation of tumors heterotopically in mice may affect tumor biology (17-19), and the site for orthotopic implantation of RCC, under the kidney capsule, is a privileged site for tumor growth (4). Finally, since RCC is largely treated with molecularly targeted medicines, a RCC tumorgraft model would permit testing the model against this most relevant class of drugs.
Advances in our understanding of the molecular genetics and biology of RCC have led to therapeutic developments and today, most patients with unresectable RCC are treated with angiogenesis inhibitors and inhibitors of mammalian target of rapamycin complex 1 (mTORC1) (20). RCC of clear-cell type (ccRCC) accounts for 70% of all RCC (21) and is characterized by inactivation of the tumor suppressor gene von Hippel-Lindau (VHL) (22). VHL inactivation results in constitutive activation of hypoxia-inducible factor (HIF) and consequent induction of vascular endothelial growth factor (VEGF) and platelet-derived growth factor β (PDGFβ) (23, 24). VEGF acts on VEGF receptor 2 (VEGFR2) on endothelial cells (25) and PDGFβ acts on PDGF receptor β (PDGFRβ) on pericytes, thereby promoting angiogenesis (26). These findings paved the way for the development of a VEGF neutralizing antibody, bevacizumab (27-30), and of several inhibitors targeting both VEGFR2 and PDGFRβ - sorafenib (31), sunitinib (32), pazopanib (33), and axitinib (34).
Similarities between two otherwise unrelated familial syndromes, von Hippel-Lindau, resulting from mutations in VHL, and tuberous sclerosis complex (TSC), resulting from mutations in the eponymic genes TSC1 and TSC2, led us to hypothesize that mTORC1, which is regulated by the TSC1 and TSC2 proteins, may be implicated in renal cancer (35). mTORC1 regulates cell growth and is constitutively activated in the majority of ccRCCs (36-39). It is negatively regulated by a complex formed by TSC1 and TSC2, and somatically acquired TSC1mutations occur in ~5% of sporadic ccRCCs (40). How mTORC1 is activated in most ccRCC remains unknown, but mTORC1 is allosterically inhibited by rapamycin (sirolimus) and two sirolimus analogs, temsirolimus (41) and everolimus (42), have been approved by the FDA.
Herein, we report the development and validation of an RCC tumorgraft model for the evaluation of molecularly targeted therapies.
RESULTS
Establishment of tumorgrafts
Between September 2009 and January 2011, 94 tumors were implanted in mice. Eligibility criteria were based on preoperative imaging studies and included tumors at least 5 cm in diameter, multifocal, bilateral or recurrent tumors, suspicion of invasion, and regional or distant metastasis. In a few instances, samples were implanted from metastatic sites (Table 1). Over 90% of the tumors were renal cell carcinomas and 75% were of clear-cell type. VHL mutations were detected in 87% of ccRCCs examined (Table 1). Over fifty percent of the RCC were of high grade, and sarcomatoid elements were found in approximately 10%.
Table 1.
Patient demographics and characteristics of tumors implanted
| T(ID) | Age | Sex | VHL | Source | Histology | Sarc. | Grade | Foc. | Size | pT | pN | TG-0 | TG≥2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 114 | 69 | M | mut | primary | clear-cell | Y | 4 | uni | 10 | 3 | 0 | Y | N |
| 2 | 115 | 65 | F | mut | primary | clear-cell | N | 4 | uni | 8 | 2 | x | Y | Y |
| 3 | 116 | 66 | M | mut | primary | clear-cell | Y | n/a | uni | 17 | 4 | 0 | N | N |
| 4 | 117 | 61 | M | n/a | primary | papillary | N | 3 | uni | 4 | 1 | x | N | N |
| 5 | 118 | 65 | F | mut | primary | clear-cell | N | 2 | uni | 2.5 | 1 | x | N | N |
| 6 | 119 | 79 | F | n/a | primary | clear-cell | N | 3 | uni | 5.7 | 1 | x | N | N |
| 7 | 120 | 62 | M | n/a | primary | clear-cell | N | 1 | uni | 2.9 | 1 | x | N | N |
| 8 | 121 | 78 | F | n/a | primary | papillary | N | 2 | uni | 3 | 1 | x | Y | Y |
| 9 | 122 | 68 | F | n/a | primary | chromophobe | N | 3 | uni | 25 | 3 | 0 | N | N |
| 10 | 123 | 56 | M | n/a | primary | papillary | N | 3 | multi | 3.1 | 1 | x | N | N |
| 11 | 124 | 63 | M | n/a | primary | clear-cell | N | 2 | uni | 3.2 | 1 | x | N | N |
| 12 | 125 | 62 | F | mut | primary | clear-cell | N | 2 | uni | 4.5 | 1 | x | Y | N |
| 13 | 126 | 53 | M | mut | primary | clear-cell | N | 2 | uni | 4.5 | 1 | x | N | N |
| 14 | 127 | 35 | M | mut | primary | clear-cell | N | 3 | uni | 6.2 | 3 | x | Y | Y |
| 15 | 128 | 71 | M | mut | primary | clear-cell | N | 3 | uni | 4 | 1 | x | N | N |
| 16 | 129 | 67 | M | n/a | met | clear-cell | N | n/a | n/a | 4 | n/a | n/a | N | N |
| 17 | 130 | 61 | F | mut | primary | clear-cell | N | 2 | uni | 6 | 3 | x | N | N |
| 18 | 131 | 49 | F | mut | primary | clear-cell | N | 2 | uni | 3 | 1 | 0 | Y | N |
| 19 | 132 | 57 | M | n/a | primary | papillary | N | 3 | uni | 7.9 | 2 | 0 | N | N |
| 20 | 133 | 58 | F | mut | primary | clear-cell | N | 2 | uni | 7.2 | 3 | 0 | N | N |
| 21 | 134 | 59 | F | n/a | primary | clear-cell | N | 2 | uni | 1.1 | 1 | x | N | N |
| 22 | 135 | 63 | M | n/a | primary | clear-cell | N | 1 | uni | 2.9 | 1 | x | N | N |
| 23 | 136 | 73 | F | wt | primary | clear-cell | N | 2 | uni | 8.5 | 2 | x | N | N |
| 24 | 137 | 77 | M | n/a | primary | papillary | N | 3 | uni | 2.2 | 1 | x | Y | N |
| 25 | 138 | 68 | F | n/a | primary | chromophobe | N | 2 | uni | 5.4 | 1 | x | N | N |
| 26 | 139 | 66 | M | n/a | primary | papillary | N | 2 | uni | 8.5 | 2 | x | Y | N |
| 27 | 140 | 52 | M | n/a | primary | papillary | N | 3 | multi | 6.2 | 1 | x | N | N |
| 28 | 141 | 51 | M | n/a | primary | chromophobe | N | 3 | uni | 5.7 | 1 | x | N | N |
| 29 | 142 | 56 | F | mut | primary | clear-cell | N | 3 | uni | 8.6 | 3 | x | Y | Y |
| 30 | 143 | 74 | F | wt | primary | clear-cell | Y | 4 | uni | 11 | 4 | x | Y | Y |
| 31 | 144 | 71 | M | mut | primary | clear-cell | N | 4 | uni | 7.5 | 2 | 1 | Y | Y* |
| 32 | 145 | 55 | F | mut | primary | clear-cell | N | 2 | uni | 7 | 1 | x | Y | N |
| 33 | 146 | 60 | F | mut | primary | clear-cell | N | 2 | uni | 4 | 1 | x | N | N |
| 34 | 147 | 59 | M | n/a | primary | liposarcoma | n/a | n/a | n/a | 14 | n/a | n/a | N | N |
| 35 | 148 | 64 | M | n/a | primary | papillary | N | 2 | uni | 6.5 | 1 | x | N | N |
| 36 | 149 | 56 | M | wt | primary | clear-cell | Y | 4 | uni | 11.5 | 3 | 0 | N | N |
| 37 | 150 | 50 | F | mut | primary | clear-cell | N | 3 | uni | 6.7 | 3 | x | Y | N |
| 38 | 151 | 74 | M | mut | primary | clear-cell | N | 2 | uni | 7.7 | 2 | x | Y | N |
| 39 | 152 | 25 | F | n/a | met | papillary | N | n/a | n/a | 4.5 | n/a | n/a | Y | Y |
| 40 | 153 | 62 | M | mut | primary | clear-cell | N | 2 | uni | 3.5 | 1 | x | Y | N |
| 41 | 154 | 62 | M | n/a | primary | oncocytoma | N | n/a | n/a | 4 | n/a | n/a | N | N |
| 42 | 155 | 64 | M | mut | primary | clear-cell | Y | 4 | uni | 10 | 3 | 0 | N | N |
| 43 | 156 | 65 | F | n/a | primary | papillary | N | 3 | uni | 11.5 | 2 | 0 | Y | Y |
| 44 | 157 | 72 | F | mut | primary | clear-cell | N | 2 | uni | 5.2 | 1 | x | N | N |
| 45 | 158 | 82 | F | mut | primary | clear-cell | Y | 3 | multi | 12.7 | 3 | x | Y | Y |
| 46 | 159 | 24 | M | n/a | primary | clear-cell | N | 2 | multi | 3 | 1 | x | N | N |
| 47 | 160 | 62 | M | mut | primary | clear-cell | N | 2 | uni | 4.2 | 1 | x | N | N |
| 48 | 161 | 36 | M | mut | primary | clear-cell | N | 2 | uni | 8.4 | 2 | x | Y | N |
| 49 | 162 | 56 | M | mut | primary | clear-cell | N | 3 | uni | 7.2 | 3 | x | Y | N |
| 50 | 163 | 52 | M | mut | primary | clear-cell | Y | 4 | uni | 4.5 | 1 | 0 | N | N |
| 51 | 164 | 75 | M | wt | primary | clear-cell | Y | 4 | uni | 11.3 | 4 | x | Y | Y |
| 52 | 165 | 42 | M | mut | met | clear-cell | N | 3 | n/a | 1.3 | n/a | n/a | Y | Y |
| 53 | 166 | 56 | M | mut | primary | clear-cell | Y | 4 | uni | 9 | 3 | 1 | Y | Y* |
| 54 | 167 | 74 | M | n/a | primary | clear-cell | N | 3 | uni | 4 | 1 | x | N | N |
| 55 | 168 | 53 | M | n/a | primary | unclassified | Y | 4 | uni | 8 | 3 | 1 | Y | Y |
| 56 | 169 | 52 | M | n/a | primary | unclassified | N | 4 | uni | 5.5 | 4 | 1 | Y | Y |
| 57 | 170 | 62 | F | mut | primary | clear-cell | N | 4 | uni | 8 | 3 | 0 | N | N |
| 58 | 171 | 62 | F | mut | primary | clear-cell | N | 2 | uni | 4.2 | 1 | x | N | N |
| 59 | 172 | 61 | F | n/a | primary | cystic nephr | N | n/a | n/a | 10.7 | n/a | n/a | N | N |
| 60 | 173 | 50 | M | mut | primary | clear-cell | N | 3 | multi | 12 | 3 | x | Y | N |
| 61 | 174 | 70 | F | n/a | primary | oncocytoma | N | n/a | n/a | 3.5 | n/a | n/a | N | N |
| 62 | 175 | 62 | F | mut | primary | clear-cell | N | 4 | uni | 11.5 | 2 | 0 | N | N |
| 63 | 176 | 71 | M | n/a | primary | papillary | N | 3 | uni | 11.5 | 3 | 1 | N | N |
| 64 | 177 | 56 | F | n/a | primary | clear-cell | N | 2 | uni | 3.2 | 1 | x | Y | N |
| 65 | 178 | 51 | M | n/a | primary | papillary | N | 2 | uni | 5.5 | 1 | x | Y | N |
| 66 | 179 | 60 | M | n/a | primary | clear-cell | N | 3 | uni | 7.5 | 2 | x | N | N |
| 67 | 180 | 56 | M | mut | met | clear-cell | N | n/a | n/a | 3.5 | n/a | n/a | Y | Y |
| 68 | 181 | 71 | M | n/a | primary | clear-cell | N | 3 | uni | 12 | 3 | 0 | N | N |
| 69 | 182 | 65 | M | n/a | primary | clear-cell | N | 3 | uni | 6 | 3 | 1 | N | N |
| 70 | 183 | 56 | M | mut | primary | clear-cell | N | 2 | uni | 4.3 | 1 | x | N | N |
| 71 | 184 | 49 | M | mut | primary | clear-cell | N | 3 | uni | 10 | 3 | 0 | N | N |
| 72 | 185 | 78 | F | n/a | primary | oncocytoma | N | n/a | n/a | 6 | n/a | n/a | Y | N |
| 73 | 186 | 51 | M | n/a | primary | papillary | N | 3 | uni | 9 | 2 | x | N | N |
| 74 | 187 | 45 | M | n/a | primary | unclassified | N | 3 | multi | 18 | 3 | 1 | N | N |
| 75 | 188 | 54 | F | n/a | primary | clear-cell | N | 2 | uni | 2.6 | 1 | x | N | N |
| 76 | 189 | 76 | M | n/a | primary | oncocytoma | N | n/a | n/a | 4 | n/a | n/a | N | N |
| 77 | 190 | 63 | M | n/a | primary | papillary | N | 3 | uni | 7.5 | 2 | x | N | N |
| 78 | 191 | 83 | M | mut | primary | clear-cell | N | 3 | uni | 9.6 | 3 | x | Y | N |
| 79 | 192 | 63 | F | mut | primary | clear-cell | N | 2 | uni | 6.8 | 1 | 0 | Y | N |
| 80 | 193 | 70 | M | mut | primary | clear-cell | N | 3 | uni | 12 | 2 | 0 | N | N |
| 81 | 194 | 56 | F | mut | primary | clear-cell | N | 3 | uni | 4.5 | 3 | x | N | N |
| 82 | 195 | 71 | F | mut | primary | clear-cell | N | 3 | uni | 5.3 | 3 | x | N | N |
| 83 | 196 | 57 | F | n/a | primary | chromophobe | N | 2 | uni | 11.5 | 2 | 0 | N | N |
| 84 | 197 | 60 | M | mut | primary | clear-cell | N | 3 | uni | 8.5 | 2 | x | N | N |
| 85 | 198 | 51 | F | n/a | primary | clear-cell | N | 2 | uni | 4.5 | 1 | 0 | N | N |
| 86 | 199 | 50 | M | wt | primary | clear-cell | N | 3 | uni | 13 | 3 | 0 | Y | N |
| 87 | 200 | 59 | F | n/a | primary | oncocytoma | N | n/a | n/a | 3.5 | n/a | n/a | N | N |
| 88 | 201 | 60 | M | n/a | primary | clear-cell | N | 2 | multi | 5.7 | 1 | 0 | N | N |
| 89 | 202 | 66 | M | mut | primary | clear-cell | N | 3 | uni | 13.3 | 3 | 0 | N | N |
| 90 | 203 | 65 | F | n/a | primary | clear-cell | N | 2 | uni | 3.5 | 1 | x | Y | N |
| 91 | 204 | 61 | F | wt | primary | clear-cell | N | 2 | uni | 3.5 | 1 | x | N | N |
| 92 | 205 | 47 | F | mut | primary | clear-cell | N | 3 | uni | 5.1 | 1 | x | N | N |
| 93 | 206 | 72 | M | n/a | met | clear-cell | N | n/a | n/a | 1.1 | n/a | n/a | Y | Y |
| 94 | 207 | 49 | F | n/a | primary | angiomyolipoma | N | n/a | n/a | 4.5 | n/a | n/a | N | N |
TG, Tumorgraft; VHL gene sequence (mut, mutation, germline and somatic; wt, wild-type); Sarc., sarcomatoid differentiation; Grade, Fuhrman nuclear grade; Size (cm); pT, pathologic T stage; pN pathologic N stage; TG-0, histologically confirmed tumor in recipient mouse cohort; TG≥2, histologically confirmed tumor passaged more than twice in mice; n/a, not applicable or assessed; uni, unifocal; multi, multifocal; Bold, tumorgraft lines giving rise to stable lines (TG≥2);
Tumor noted to induce hypercalcemia in tumorgraft.
Tumor fragments (2-3 mm in diameter) were implanted orthotopically, under the renal capsule, into 5 NOD/SCID mice (Fig. 1A). To preserve tumor architecture and minimize confounding factors, samples were implanted without disaggregation or additives. Tumorgrafts could be visualized by MRI (Fig. 1B) and ultrasound (Fig. 1C), but palpation was typically sufficient for follow-up. When tumors reached ~10 mm in diameter, they were passaged. Passage occurred earlier if the mouse became sick or was getting old. At initial passage, tumor diameters ranged from approximately 4 to >10 mm (Fig. 1D). As determined histologically by examining recipient mice (tumorgraft cohort 0, TGc0), 37% of grafts formed viable tumors in mice (Table 1). The average latency period from the day of implantation in mice until passage into the first cohort (TGc1) was highly variable, ranging from 1 to 8 months. The time was generally shorter for tumors with high nuclear grade or sarcomatoid elements and became shorter with sequential passage. At each passage, samples were fixed for histological analyses and, when sufficient material was available, samples were frozen for permanent storage (in DMSO) and separately for molecular studies (Fig. 1A).
Fig. 1.

Establishment of tumorgrafts. (A) Schema illustrating overall disposition of patient tumor samples: fragments are implanted orthotopically into 5 cohort 0 mice (TGc0), frozen without (F) and with DMSO (D), and fixed in formalin and paraffin embedded (FFPE). When tumorgrafts reach ~ 10 mm in diameter, they are passaged into cohort 1 mice (TGc1). Following 1-2 passages, tumorgrafts are implanted for subcutaneous (s.c.) growth evaluation and subsequently for drug trials. Tumors from mice not passaged are processed and preserved. (B) MRI of orthotopic tumorgraft-bearing kidney in a mouse. (C) Renal ultrasound of tumorgraft. (D) Macroscopic images of engrafted tumor and contralateral kidney. (E) Representative H&E sections of patient tumor and corresponding tumorgrafts of increasing passages [TG(I), TG(II) and TG(III)]. Scale bar: 100μm.
Orthotopic tumorgrafts resemble patient tumors histologically
Detailed analyses by a clinical pathologist specialized in genitourinary tumors (P.K.) showed that tumorgrafts retained not only the histology, but also fine histological features of the corresponding tumors in patients. Even within a specific histological type, architectural and cytological characteristics were preserved in the respective tumorgrafts (Fig. 1E). Overall, tumorgrafts maintained the architectural growth pattern, as well as cystic components, the presence of areas of necrosis and hemorrhage, sarcomatoid differentiation, cytological and nuclear features, Fuhrman nuclear grade, and the presence of some inflammatory cells (Table S1). In contrast, lymphocytic infiltrates were not preserved, which was expected because lymphocyte development is disrupted in NOD/SCID mice. Histological features were maintained despite serial passaging (Fig. 1E and Table S1).
Predictors of stable tumor engraftment in mice
From 16 different patients, tumorgraft lines were passaged at least twice in mice (TGc≥2), and these are referred to as stable lines. Sequential tumor growth in mice correlated with implantation from a metastatic site, sarcomatoid differentiation, high Fuhrman grade, pathologic tumor stage (a function of size and invasiveness), and the presence of regional lymph node or distant metastasis (Table 2). The same factors predicted engraftment when the analysis was limited to ccRCC (Table 2). The following did not predict for tumorgraft development in mice: histology, VHL mutation, focality, and tumor size. Notably, the engraftment rate of tumors implanted from metastases was considerably higher than that of primary tumors (80% vs. 14%; p=0.0028). Higher rates of engraftment were also observed with samples implanted from primary tumors of patients with distant metastases (60% vs. 8%; p=0.0014). These data suggested that the ability of tumors to grow serially in mice correlated with their ability to seed distant sites and metastasize.
Table 2.
Predictors of stable tumor engraftment
| All histologies |
clear-cell RCC |
|||
|---|---|---|---|---|
| Engrafted/Total |
(p)
|
Engrafted/Total |
(p)
|
|
| RCC histology | 0.15 | |||
| clear cell | 11/65 | |||
| papillary | 3/14 | |||
| chromophobe | 0/4 | |||
| oncocytoma | 0/5 | |||
| unclassified | 2/3 | |||
| VHL mutation (germline or somatic) | 0.59 | |||
| Mutant | 8/42 | |||
| Wild type | 2/6 | |||
| Tumor implanted from metastases | 4/5 | 0.0028 | 3/4 | 0.014 |
| Sarcomatoid differentiation | 5/10 | 0.012 | 4/9 | 0.038 |
| Fuhrman nuclear grade | 0.0008 | 0.002 | ||
| 1 | 0/2 | 0/2 | ||
| 2 | 1/32 | 0/26 | ||
| 3 | 5/34 | 4/22 | ||
| 4 | 7/13 | 5/11 | ||
| Focality | 1.000 | 0.52 | ||
| unifocal | 11/74 | 7/57 | ||
| multifocal | 1/7 | 1/4 | ||
| Size (cm) | 0.58 | 0.30 | ||
| ≤4 | 4/27 | 3/19 | ||
| >4-7 | 3/28 | 1/19 | ||
| >7-10 | 5/21 | 4/16 | ||
| >10 | 4/18 | 3/11 | ||
| Pathologic tumor stage | 0.0014 | 0.0026 | ||
| T1 | 1/37 | 0/28 | ||
| T2 | 3/15 | 2/9 | ||
| T3 | 5/25 | 4/21 | ||
| T4 | 3/4 | 2/3 | ||
| Pathologic lymph node stage | 0.0077 | 0.016 | ||
| N0 | 1/21 | 0/17 | ||
| N1 | 4/7 | 2/3 | ||
| Metastasis at presentation | 6/10 | 0.0014 | 4/7 | 0.013 |
Stable engraftment in mice is associated with poor survival in patients
Because stable engraftment correlated with metastases, we hypothesized that engraftment may unveil the acquisition of metastatic potential by the primary tumor. Due to short follow-up times and relatively small numbers, the study was not powered to definitively address this question. Nevertheless, we asked whether a correlation existed between engraftment in mice and outcomes in patients presenting with localized disease. Patients whose tumors engrafted in mice had a shorter survival, likely secondary to the development of metastases (Fig. 2). Similar results were observed when the analysis was limited to ccRCC (Fig. 2). Thus, stable engraftment in mice seems to predict for poor outcomes in patients.
Fig. 2.
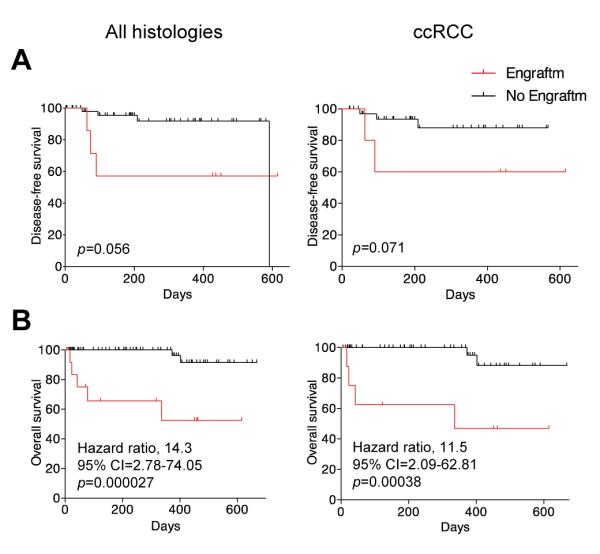
Evaluation of patient outcomes according to stable engraftment of primary tumors from patients with localized disease. (A) Kaplan-Meier analysis of disease-free survival according to stable engraftment for all histologies or ccRCC. (B) Kaplan-Meier analysis of overall survival according to engraftment status for all histologies or ccRCC. Cross indicates censored data.
Tumorgrafts retain the gene expression pattern of the original tumor
To ascertain the extent to which RCC tumorgrafts maintained the characteristics of the tumor from which they were derived, we performed gene expression analyses. Because recipient mice are immunodeficient, contributions to the global gene expression signature in tumors from infiltrating lymphocytes would be absent in tumorgrafts. In addition, as the stroma in the tumor is largely replaced by murine cells (43), human stromal transcripts may also be underrepresented in tumorgrafts. To account for these differences, transcripts upregulated in tumors by comparison to tumorgrafts (q<0.05 and fold change >1.5 fold) were subtracted (Fig. 3A). Pathway analyses on the subtracted probes showed that, as expected, they belonged largely to pathways implicated in immune-mediated processes such as antigen presentation, dendritic cell maturation, and NK signaling (Fig. 3A; Table S2).
Fig. 3.

Gene expression analyses of tumors and tumorgrafts. (A) Principal component analysis of tumor, paired normal renal cortex (for a subset of tumors), and tumorgraft samples before and after subtraction of probes upregulated in tumors vs. tumorgrafts. List of top Ingenuity Pathways corresponding to transcripts upregulated in tumors over tumorgrafts with a FDR q<0.05 and a fold change greater than 1.5. (B) Unsupervised hierarchical clustering of samples according to gene expression pattern after subtraction. Each tumor/tumorgraft clade is color-coded and includes patient tumor sample (T) and the corresponding tumorgrafts (numbers reflect mouse tumorgraft cohort). (C) Heatmap of tumor-specific gene expression changes after subtraction of immune/stromal signature including the top 25 up- and down-regulated probesets in tumors compared to adjacent normal parenchyma (ranked by q value). N, normal; T, tumor; TG, tumorgraft; FDR q, false discovery rate-corrected p value; FC, fold change.
To determine whether tumorgrafts retained the gene expression pattern of the original tumor, we performed unsupervised hierarchical clustering analyses. 21 out of 29 (72%) tumorgrafts clustered together with the corresponding tumor (Fig. 3B). In addition, clustering was maintained for cohort 7 and 8 tumorgrafts (Fig. 3B). Thus, the degree of similarity between most tumorgrafts and the corresponding patient tumors was greater than across tumors from different patients.
The approach we undertook also provides a means to separate gene expression signatures of tumor cells from those arising from non-neoplastic cells (Fig. 3C; Tables S3 and S4). Not unexpectedly, reducing the influence from non-neoplastic cells significantly affected the ranking of pathways deregulated in renal cancer and implicated some novel pathways, such as RAN signaling (Table S4).
Tumorgrafts preserve the DNA copy number alterations of tumors
We evaluated DNA copy number alterations (CNAs) in tumorgrafts ranging from primary tumors in recipient mice (TGc0) to cohort 8 (TGc8). CNAs in tumorgrafts were characteristic of RCC, including chromosome 3p loss and less frequent deletions of chromosome 14 and 9p (44-46). Tumorgrafts largely retained the pattern of CNAs of the corresponding tumor irrespective of passage (Fig. 4A). To evaluate the extent to which CNAs in tumorgrafts resembled those in the tumor from which they were derived, unsupervised hierarchical clustering analyses were performed. 20 out of 27 (74%) tumorgrafts clustered with the corresponding tumor (Fig. 4B).
Fig. 4.

DNA copy number alterations in tumors and tumorgrafts. (A) Representation of DNA copy number alterations in patient tumors (T) and corresponding tumorgrafts from the indicated cohorts. Red and blue denote amplifications and deletions, respectively. (B) Unsupervised hierarchical clustering of samples according to paired DNA copy number analyses. Each tumor/tumorgraft clade is color-coded and includes patient tumors (T) or metastasis (M) and corresponding tumorgrafts (numbers, which reflect cohort). (C) Paired copy number and allele-specific copy number analyses of a patient tumor (T), metastasis (M) and metastasis-derived tumorgrafts in the recipient mouse (mTGc0) and cohort 1 (mTGc1).
In one case, samples were available from both a primary tumor as well as a metastasis that was used to generate a tumorgraft line. The metastasis had acquired some alterations that were not found in the primary tumor, including losses in chromosome 1 and 8 (Fig. 4C). The presence of unique alterations in the metastasis allowed us to evaluate the degree of tumorgraft resemblance. CNAs in the tumorgrafts were indistinguishable from those in the metastasis and differed from the primary tumor (Fig. 4C). Thus, even within a patient, tumorgrafts appear to retain the specific signature of the tumor or metastasis from which they were generated.
Point mutations and indels are preserved in tumorgrafts
We performed whole genome (or exome) sequencing in 7 tumors (47) and examined the preservation of mutations in tumorgrafts. A total of 134 somatically-acquired point mutations or indels in protein-coding genes were examined across several tumorgraft cohorts (for specific mutations refer to ref 47). Ninety-two percent of mutations detected were retained in the tumorgrafts and the number did not significantly change in later passages (Table 3). VHL mutations were uniformly retained in tumorgrafts across different cohorts (Table 4), which was expected, as VHL mutations occur early during ccRCC development (48).
Table 3.
Mutations retained in tumorgraft cohorts
| Retained Mutations |
||||
|---|---|---|---|---|
| ID | # Mut Evaluated | TG(I) | TG(II) | TG(III) |
| TG22 | 48 | 45 | n/a | n/a |
| TG144 | 22 | 20 | 20 | 19 |
| TG166 | 21 | 21 | 21 | 21 |
| TG180 | 13 | 13 | 13 | n/a |
| TG164 | 12 | 11 | 11 | n/a |
| TG142 | 11 | 8 | 8 | n/a |
| TG127 | 7 | 5 | 5 | n/a |
|
| ||||
| Total | 134 | 123 (92%) | 78 (91%) | 40 (93%) |
|
|
||||
| Overall | 241/263 (92%) | |||
n/a, not assessed; TG, tumorgrafts of increasing passages.
Table 4.
VHL mutations in patient tumors and tumorgrafts
| Retained mutations |
||||
|---|---|---|---|---|
| ID | VHL mutation (Tumor) | TG(I) | TG(II) | TG(III) |
| TG22 | c.472C>G,p.L158V | + | n/a | n/a |
| TG127 | c.232_233delAA | + | + | n/a |
| TG142 | c.525_533delCAGGAGACT | + | + | n/a |
| TG144 | c.506T>C,p.L169P | + | + | + |
| TG166 | c.224_226delTCT | + | + | + |
| TG183 | c.414_421delATCTCTCA | + | + | n/a |
n/a, not assessed; TG, tumorgrafts of increasing passages.
Bidirectional Sanger sequencing in tumorgrafts was performed for 618 amplicons and we estimate that 247,200 bp of tumorgraft DNA was sequenced. Only one point mutation was confidently identified in a tumorgraft that was not detected in the primary tumor (Table 5). This mutation was in the TSC1 gene, which we recently reported to be somatically inactivated in ccRCCs (40), and the corresponding patient’s tumor had a different TSC1 mutation. Very deep sequencing of two independent samples (~2 million reads/sample) showed that the mutation pre-existed in the patient tumor at a frequency of 0.3% (Table S5). Consistent with genome-wide studies in other tumor types (49), these data suggest that the acquisition of new mutations by RCC tumorgrafts is a rare event.
Table 5.
Point mutations or indels in tumorgrafts but not patient tumors
| Total amplicons sequenced in tumorgrafts | 618 |
| Average number of bp per amplicon | 400 |
| Approximate number of bp sequenced | 247,200 |
| Mutations in tumorgrafts but not in patient tumors | 1‡ |
mutation detected in corresponding tumor by very deep sequencing.
Development of paraneoplastic hypercalcemia in tumorgraft-bearing mice
We observed that some tumorgraft-bearing mice became ill. As tumorgrafts grew, the mice became progressively less active and more hunched over, started losing weight, and eventually became moribund. The illness occurred only with specific tumorgraft lines (e.g. TG144 and TG166), suggesting that the tumor was directly responsible. The symptoms were somewhat reminiscent of untreated hypercalcemia in humans, so we considered whether tumorgrafts may be inducing paraneoplastic hypercalcemia in mice. A review of the medical records of the corresponding patients showed that they had presented with elevated calcium (Ca) levels and that hypercalcemia resolved after tumor resection. To determine whether TG144 and TG166 tumorgraft-bearing mice similarly developed paraneoplastic hypercalcemia, we measured Ca levels. Whereas in control tumorgraft-bearing mice serum Ca levels were within the normal range (8.5-10.5 mg/dL), Ca levels reached >15 mg/dL in both TG144 and TG166 mice (Fig. 5). Thus, as evidenced by hypercalcemia, paraneoplastic syndromes may develop in tumorgraft-bearing mice.
Fig. 5.
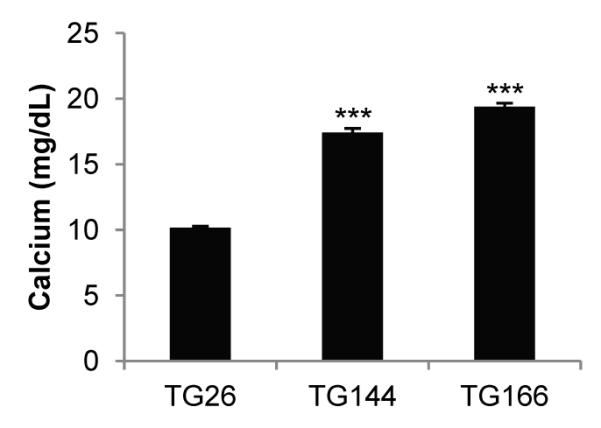
Calcium levels in tumorgraft-bearing mice. Serum calcium levels in mice implanted with tumors from patients with paraneoplastic hypercalcemia, TG144 (n=3) and TG166 (n=3), compared to tumorgraft-bearing mice from a patient without paraneoplastic hypercalcemia (TG26; n=5). Data are means + SE; ***, p<0.001.
Mimicking sunitinib and sirolimus exposures of RCC patients in NOD/SCID mice
Arguably, the most critical aspect in the evaluation of a tumor model is whether it reproduces the drug responsiveness of tumors in patients. To determine whether RCC tumorgrafts retained the sensitivity of RCC in the clinic, we tested their sensitivity to an inhibitor of angiogenesis, sunitinib, and an mTORC1 inhibitor, sirolimus. We used sirolimus instead of temsirolimus, because temsirolimus is largely a sirolimus prodrug; after temsirolimus administration 75% of circulating drug in humans is sirolimus (50, 51). In fact, we previously reported the treatment of a RCC patient with sirolimus (before temsirolimus became commercially available) (52). As a control, we sought to identify a small molecule kinase inhibitor that had been tested against RCC and was inactive. We settled on the EGFR kinase inhibitor, erlotinib, which is approved for non-small cell lung cancer (NSCLC) treatment. A randomized phase II trial of erlotinib in combination with bevacizumab failed to show improved outcomes by comparison to bevacizumab alone in patients with ccRCC (53).
Because the results of drug trials can be affected by differences in drug metabolism across species (7), pharmacokinetic (PK) studies were performed in mice to identify a regimen that mimicked human exposures. We sought to identify drug regimens balancing sustained therapeutic levels (above the Cmin in humans) without excessive peak (Cmax) and total exposures (AUClast). As differences may exist in drug metabolism across mouse strains, NOD/SCID mice were used.
Sirolimus, at 0.5 mg/kg intraperitoneally every 48 h resulted in trough levels within the therapeutic range in humans (5–15 ng/mL) (54, 55). However, this resulted in peak and overall exposures that were 2-3 fold higher than in humans (Table 6).
Table 6.
Pharmacokinetic analysis of drug regimens in mice with reference to human studies
| Drug | Species | Dose | Route | Schedule | PK data at |
Analyte | AUClast (ng•hr/mL) |
Cmax (ng/mL) |
tmax (h) |
Terminal t1/2 (h) |
Cmin (ng/mL) |
Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sirolimus | Mouse | 0.5 mg/kg | IP | q48h | 48h | Sirolimus | 4,276 ± 329 | 983 ± 98 | 1 | 13.6 | 14.9 ± 2.0 | |
|
| ||||||||||||
| Temsirolimus | Human | 25 mg | IV | qwk | D7 or D28 | Temsirolimus | 1,580 ± 270 | 595 ± 102 | 0.5 | 12.8 | Atkins et al., 2004 * | |
| Sirolimus | 3,810 ± 2,210 | 66 ± 35 | 1 | 48.8 | ||||||||
| Sum | 5,860 ± 2,340 | |||||||||||
|
| ||||||||||||
| Everolimus | Human | 10 mg | PO | qd | Everolimus | 514 ± 231 | 61 ± 17 | 13.2 ± 7.2 | Awada et al., 2008 * | |||
|
| ||||||||||||
|
Sunitinib
Malate |
Mouse | 10 mg/kg | G | q12h | 12h (D1) | Sunitinib | 449.8 ± 41.1 | 108.5 ± 5.5 | 2 | 2 | 4.2 ± 0.1 | |
|
Desethyl
Sunitinib |
174.4 ± 24.0 | 37.8 ± 0.2 | 2 | 2.6 | 3.0 ± 0.1 | |||||||
| Sum | 624.2 | 7.2 | ||||||||||
|
| ||||||||||||
| Sunitinib Malate |
Human | 50 mg | PO | qd (D1-28) | 24h (D1) | Sunitinib | 420 + 210 | 27.7 ± 14.1 | 5 | Faivre et al., 2006* | ||
| Desethyl Sunitinib | 63.6 ± 33.7 | 4.1 ± 2.2 | 5 | |||||||||
| Sum | 483.6 | |||||||||||
|
| ||||||||||||
| Sunitinib Malate |
Human | 50 mg | PO | qd (D1-28) | 24h (D28) | Sunitinib | 1,296 ± 609 | 72.2 ± 31.0 | 8.5 | 44.0 ± 26.0 | Faivre et al., 2006* | |
| Desethyl Sunitinib | 592 ± 391 | 33.7 ± 24.6 | 6.5 | 18.8 ± 8.5 | ||||||||
| Sum | 1,888 | 62.8 | ||||||||||
|
| ||||||||||||
| Erlotinib | Mouse | 12.5 mg/kg | G | q12h | 12h (D1) | Erlotinib | 27,042 ± 2,569 | 3,513 ± 271 | 3 | 3.1 | 521 ± 169 | |
|
Desmethyl
erlotinib |
3,970 ± 330 | 526 ± 77 | 3 | 2.5 | 50 ± 23 | |||||||
|
| ||||||||||||
| Erlotinib | Human | 150 mg | PO | qd | 24h (D1) | Erlotinib | 11,860 ± 5,010 | 872 ± 399 | 3 | 385 ± 213 | Raizer et al., 2010 * | |
| Desmethyl erlotinib |
835 ± 479 | 68 ± 45 | 3.6 | 25 ± 18 | ||||||||
|
| ||||||||||||
| Erlotinib | Human | 150 mg | PO | qd | 24h (D28) | Erlotinib | 43,760 ± 22,560 | 2,528 ± 1,187 | 24.2 | 1,473 ± 877 |
Tan et al., 2004 * | |
|
| ||||||||||||
| Dovitinib | Mouse | 30 mg/kg | G | qd | 24h (D1) | Dovitinib | 6078 ± 710 | 373 ± 14.6 | 6 | 5.4 | 41.2 ± 9.3 | |
| Dovitinib | Human | 500 mg | PO | 5 d on/2 d off |
24h (D1) | Dovitinib | 2200-8251 | 180-487 | Angevin, et al 2009 | |||
|
| ||||||||||||
| Dovitinib | Human | 500 mg | PO | qd | 24h (D1) | Dovitinib | 3734 ± 2115 | 223 ± 128 | Kim, et al 2011* | |||
| (D15) | Dovitinib | 4340 ± 3775 | 267 ± 178 | 21 | ||||||||
Pharmacokinetic parameters (mean ± SE) based on noncompartmental analysis using WinNonlin (Pharsight Corp.).
Mean ± SD; IP, intraperitoneal; IV, intravenous; PO, orally; G, gavage; qwk, weekly; qd, daily.
Sunitinib is metabolized to desethyl sunitinib, which is active, and PK studies were performed to evaluate both sunitinib and its metabolite. Whereas on day 1 the metabolite represented 13% of the total circulating drug in humans, it made up 38% in mice (Table 6). The half-life of sunitinib was much shorter in mice, and mice were dosed every 12 h. The administration of sunitinib 10 mg/kg by gavage every 12 h resulted in peak exposures that were slightly higher than in humans and overall exposures (parent compound + metabolite) that were within the range of exposures between D1-28 in humans (Table 6). While sunitinib levels in humans build up over time, troughs in mice are likely to be lower than in humans. Nevertheless, given the every 12 h dosing, and the overall drug exposures, this was considered acceptable.
In humans, erlotinib is metabolized to O-desmethyl-erlotinib, which is active. The ratios of parent compound to metabolite were similar in humans and mice (Table 6). Whereas the half-life of erlotinib in humans is 24 h (56), it was 3 h in mice, and we dosed mice every 12 h. As erlotinib was used as a negative control, it was preferable to err on the side of overdosing. Erlotinib at 12.5 mg/kg by gavage every 12 h resulted in trough levels within the human range (56, 57), although, this resulted in higher peak and overall exposures (Table 6).
Tumorgrafts reproduce the drug responsiveness of RCC
The use of orthotopic tumorgrafts to study drug responsiveness is hampered by the need for frequent imaging to monitor tumor growth during drug trials. Because measurements would be significantly easier for subcutaneous (s.c.) tumors, we evaluated the growth of orthotopically growing tumorgrafts in the subcutaneous space. Only ~65% of the tumorgraft lines growing orthotopically grew subcutaneously within a manageable timeframe. Despite the heterotopic location, RCC sometimes metastasizes to the subcutaneous space in humans and the histological characteristics of the tumorgrafts were preserved when grown subcutaneously (Fig. 6A). In addition, as determined in one tumorgraft line (TG164), drug effects did not differ whether the tumor was implanted subcutaneously or orthotopically, and similar anti-tumor responses were observed in earlier and later passage tumors (Fig. S1).
Fig. 6.

Effect of sunitinib, sirolimus, and erlotinib on ccRCC tumorgrafts. (A) H&E sections comparing orthotopic tumorgrafts (ortho) vs. tumorgrafts implanted subcutaneously (s.c..) for the 8 ccRCC tumorgraft lines evaluated for drug responsiveness. (B) Tumorgraft volumes from 8 tumorgraft lines treated, starting at day 0, with vehicle (n=32), erlotinib (n=25), sunitinib (n=32), and sirolimus (n=33). Graph includes 1,482 volume measurements (vehicle, n=385; erlotinib, n=304, sunitinib, n=380, and sirolimus, n=413). Trend lines were generated using a second order polynomial quadratic regression analysis. p values by comparison to vehicle control were determined using a linear mixed model. (C) Tumor volumes of lung adenocarcinoma cell line-derived xenografts from mice treated with erlotinib (n=3) or vehicle (n=3). Data are means + SE. (D) Representative macroscopic tumorgraft images from several tumors from different lines at the end of trial. (E) Immunohistochemistry of tumorgrafts from mice treated as indicated and evaluated for the mTORC1 pathway effector marker phospho-S6. Scale bar: 100 μm.
Eight ccRCC tumorgraft lines that grew subcutaneously were evaluated in drug trials (Fig. 6A). For each trial, ~20 mice were implanted with ~64 mm3 tumor fragments, and 2-4 weeks after implantation, tumor volume measurements begun. Tumor growth rates varied considerably among the 8 tumorgraft lines, and drug administration started when average tumor size reached approximately 250-300 mm3. To avoid biases from excessively weighting any particular dimension, tumor volumes were calculated according to the formula l × w × h, where l is maximal length, w is maximal width perpendicular to l, and h is maximal height. Tumorgraft-bearing mice of similar characteristics (tumor volume, tumor growth rate, and mouse weight) were distributed evenly across treatment arms. Arms were kept balanced, and statistical analyses at the completion of drug trials showed no biases at the start.
For each drug trial, 3-5 mice of each tumorgraft line were allocated to one of four treatment groups: sirolimus, sunitinib, erlotinib, and vehicle. Drug trials were carried out for ~28 days. During the trial, tumor measurements were taken twice weekly. Mice were weighted weekly, and drug administration was adjusted accordingly.
A total of 122 mice, from 8 different tumorgraft lines, were evaluated in drug trials (Fig. 6B; see also Fig. S2). Using ccRCC vehicle-treated mice as a reference, erlotinib treatment had no statistically significant effect on tumorgraft growth (Fig. 6B). However, the same erlotinib regimen had a profound effect on a NSCLC cell line-derived xenograft used as a control (Fig. 6C). In contrast, ccRCC tumorgraft growth was substantially inhibited by treatment with sunitinib (p<0.0001) (Fig. 6B and D). Likewise, sirolimus inhibited tumorgraft growth (p<0.0001) (Fig. 6B and D).
Taken together, these data show that tumorgrafts reproduce the sensitivity to sunitinib and (tem)sirolimus of RCC observed in the clinic.
Pharmacodynamic studies in tumorgrafts show mTORC1 pathway inhibition
Next we evaluated the effects of sirolimus and sunitinib on mTORC1 pathway activity in tumor cells. For this analysis, we used an antibody that recognizes phospho-S6240/244 (p-S6240/244), a faithful marker of mTORC1 activity (58). p-S6240/244 immunohistochemistry was evaluated by a clinical pathologist (P.K.) who was masked to the treatment allocation. Scores were given based on both signal intensity and the percentage of positive cells. A decrease in p-S6240/244 was observed in sirolimus-treated tumorgrafts (p<0.0001; see also Fig. 6E). A more modest, but significant, reduction in p-S6240/244 was also observed after sunitinib treatment (p=0.021). By contrast, erlotinib had no effect on p-S6240/244 (p=0.62).
Dovitinib inhibits tumorgraft growth
Finally, we used our model to evaluate an investigational agent, dovitinib. Dovitinib, is a highly potent inhibitor of VEGFR1-3, PDGFRβ and FGFR1-3 (59) that is being evaluated in clinical trials, but its effectiveness against RCC is presently unknown. Pharmacokinetic studies indicated that 30 mg/kg daily by gavage resulted in peak and overall exposures in mice within the range reported in humans (Table 6).
Clinical trials were conducted comparing dovitinib to sunitinib and sirolimus in 4 ccRCC tumorgraft lines, including a ccRCC tumorgraft line that we had not previously evaluated (TG206). Dovitinib profoundly suppressed ccRCC tumorgraft growth (Fig. 7A, B and Fig. S3), resulting in a greater inhibition of tumor growth than both sunitinib and sirolimus (Fig. 7A-C). We also evaluated a papillary RCC tumorgraft line (TG121), in which dovitinib was also very active (Fig. S3). In addition, dovitinib inhibited the development of paraneoplastic hypercalcemia in mice (Fig. 7D). Finally, as indicated by changes in body weight, dovitinib was not particularly detrimental in most mice (Fig. S4). Overall, these results show that dovitinib was more effective against RCC tumorgrafts than sunitinib or sirolimus and that it is reasonably well tolerated.
Fig. 7.

Effect of dovitinib on RCC tumorgrafts. (A) Tumorgraft volumes of mice treated with vehicle (n=17), sunitinib (n=16), sirolimus (n=15), and dovitinib (n=15). A second order polynomial quadratic regression analysis was used to generate trend lines and a linear mixed model was used for the calculation of p values respect to vehicle control. (B) Macroscopic tumorgraft images from several tumors at the end of trial. (C) Tumorgraft volumes at the end of drug trial. (D) Serum calcium levels of TG144 at the end of the trial (n=3-4 per treatment arm). Data are means + SE. *, p<0.05; **, p<0.01.
DISCUSSION
RCC tumorgrafts in the mouse kidney reproduced the histology, gene expression, molecular genetics, and treatment responsiveness of RCC in patients. Our results with FDA approved drugs establish a proof-of-principle for the evaluation of molecularly targeted therapies for renal cancer in mice. We used this platform to evaluate an agent in clinical development, dovitinib, which shows remarkable activity against RCC.
Tumorgrafts reproduced not only the histology of the patient tumor, but also finer characteristics such as tumor architecture as well as cytological and nuclear features. In unsupervised hierarchical clustering analyses of gene expression and DNA copy number alterations, approximately 70% of tumorgrafts clustered together with the corresponding tumors from patients. Thus, there were greater similarities between tumorgrafts and their corresponding tumors than across tumors from different patients. Even within a patient, DNA copy number analyses showed that a metastasis-derived tumorgraft was more similar to the metastasis than the metastasis was to the primary tumor. Significantly, tumorgrafts preserved 92% of the somatically-acquired protein-coding gene mutations of patient tumors. In addition, these studies failed to reveal any confident point mutations or indels in the tumorgraft that could not be found in the original patient tumor. Overall, these results show that tumorgrafts in mice are faithful models of the corresponding tumors in humans.
RCC tumorgrafts retained the drug sensitivity of RCC in the clinic. We performed large, controlled experiments with clinically relevant drug regimens, giving exposures comparable to those in humans. The tumorgrafts retained the sensitivities to sunitinib and (tem)sirolimus observed in the clinic, and failed to respond to erlotinib, used as a control. These data establish the validity of tumorgrafts as a model for the evaluation of targeted therapies for this cancer and others potentially.
Two studies reported similar results (60, 61). They evaluated two ccRCC tumorgraft lines and, in one study, a single mouse from each line was treated per condition (61). In the other study, tumorgraft growth was inhibited by sorafenib (60), although it is not clear whether the sorafenib regimen was clinically relevant, and the specificity of the response could not be ascertained due to a lack of a control drug. In addition, the admixing of tumorgraft tissue with matrigel raises the possibility that sorafenib inhibition may have resulted, at least in part, from inhibition of the effects of ectopic growth factors in the extract. Nevertheless, these results are consistent with our data.
Dovitinib, an investigational agent in clinical development, more potently inhibited RCC tumorgraft growth than did sunitinib and sirolimus. Our results in a validated tumorgraft model lead us to predict that dovitinib will similarly be effective against renal cancer in humans. These data support the evaluation of dovitinib in randomized clinical trials of RCC patients by comparison to sunitinib and temsirolimus.
Tumorgraft engraftment may reflect metastatic potential. We found a correlation between stable engraftment in mice and poor survival. We hypothesize that tumorgraft growth in mice reflects the acquisition by the tumor of a capability to thrive at other sites, which is characteristic of metastases. Consistent with this notion, samples implanted from metastatic sites engrafted at higher frequencies than those from primary tumors. Thus, tumorgrafts may help dissect biological determinants of metastases.
Tumorgraft features were preserved despite serial passage in mice. In addition, while divergence is expected with passage in mice over time, late passage tumorgrafts did not appear to accumulate mutations. Finally, neither passage nor subcutaneous implantation seemingly affected tumorgraft responsiveness to drugs. Whereas the engraftment frequency in mice was 37%, stable tumorgraft lines were obtained in approximately half (17%). Several factors may account for the low rate of stable engraftment including residual NK function in NOD/SCID mice and tumorgraft loss due to infiltration by lymphoma cells arising spontaneously in aged NOD/SCID mice.
Inasmuch as the only cellular compartment that regenerates itself in tumorgrafts long-term is the neoplastic compartment (43), tumorgrafts may represent nearly pure populations of human tumor cells and this may have great utility. Tumorgrafts are instrumental, in fact, to accurately determine mutant allele frequencies in tumors (47) and, given the mutation heterogeneity in RCC (48, 62), they may aid in the identification of driver mutations. In addition, tumorgrafts provide a means to separate gene expression signatures arising from the tumor from those contributed by non-malignant immune and stromal cells. This could have great utility in parsing out immune cell signatures in RCCs and begin to shed light into an important but very difficult clinical problem, the elucidation of determinants of tumor responsiveness to high-dose IL-2.
Tumorgrafts have many other applications. Inasmuch as tumorgrafts retain the mutations found in patient tumors and preserve their drug responsiveness, they can be used to determine whether targeting a specific pathway disrupted by mutation results in anti-tumor effects. In exploring new targeted agents, tumorgrafts are instrumental to determine whether the target was successfully inhibited in the tumor, which is a challenge in patients. Tumorgrafts can also be used to prioritize drug combinations and to dissect difficult problems such as how resistance to anti-angiogenic agents develops. In addition, they provide an experimental system in which to investigate poorly understood aspects of tumor biology such as some paraneoplastic syndromes and cancer-induced cachexia. The exploitation of differences across species may render tumorgraft-bearing mice useful for the identification of tumor markers. In addition, tumorgrafts can be used to study unusual forms of RCC and recently, we derived a tumorgraft line from a papillary RCC of a patient with a de novo germline mutation in fumarate hydratase (63). Finally, tumorgrafts may be useful in the evaluation of new imaging modalities and to assess, for instance, the effects of HIF on metabolism (64).
In summary, this study establishes a proof-of-concept for the development and application of RCC tumorgrafts for the evaluation of molecularly targeted therapies. We propose that the routine incorporation of such models into the development of molecularly targeted drugs could advance preclinical drug evaluation programs and improve oncology drug development.
MATERIALS AND METHODS
Regulatory
Patients enrolled in the study provided written consent allowing the use of discarded surgical samples for research purposes and genetic studies on an Institutional Review Board approved protocol. Tumorgraft studies were based on a protocol approved by the University of Texas Southwestern Medical Center Institutional Animal Care and Use Committee.
Nomenclature and annotation
Patient tumor samples were designated as T, followed by the ID. Tumors growing in mice are referred to as tumorgrafts (designated as TG, followed by the ID). Tumorgrafts arising in the recipient mouse are designated as TGc0 (TG cohort 0). Subsequent tumorgraft passages are designated by TG followed by the cohort number (e.g. TGc1, TGc2, etc.). TG followed by a roman numeral in brackets indicates a specific sequence of tumorgrafts without specifying the cohort, e.g. TG(I), TG(II), TG(III), where (I) precedes (II) and so on. T22, T26, T79, T84 (and the respective TGs) correspond to ccRCCs obtained prior to the series described in Table 1.
Tumor samples (pT) were annotated based on the AJCC TNM classification and the edition corresponding to the date of surgery. However, based on the 7th edition, all lymph node metastases are referred to as pN1.
Tumorgraft sample processing and implantation
Eligibility criteria were based on preoperative imaging studies and included renal tumor samples greater than 5 cm, multiple, bilateral or recurrent tumors, suspicion of invasion, lymphadenopathy and distant metastasis. Any of these criteria were sufficient for inclusion. Patients were excluded if they were known to be positive for HBV, HCV or HIV. Samples were collected, placed on ice, and typically processed within 3 h. They were transferred to a sterile dish with PBS and cut into 8-27 mm3 fragments.
Typically, 4-6 week old male or female NOD/SCID mice were used for implantation. Mice were anesthetized by inhalation with an isofluorane vaporizer and 0.0015 mg buprenorphine was administered by intraperitoneal (IP) injection immediately after surgery, while the mice were still anesthetized, and within 24 hours after surgery. Mice were placed on a warming pad, fur was shaved, and area was sterilized using Betadine. A transverse incision was made posteriorly over the mid lumbar spine and the skin was bluntly dissected. A smaller ~1cm incision in the same direction was made of the body wall over the left flank and the kidney was exteriorized by gently applying pressure. A 2 mm longitudinal incision was made in the dorsal aspect of the kidney using spring-loaded scissors. A pocket was carefully created underneath avoiding damage to the parenchyma which would otherwise bleed and 2-3 samples were gently pushed inside. The kidney was eased back into the retroperitoneum, the body wall was sutured and the skin stapled.
Additional fragments were frozen at -80°C or placed in 10% DMSO in Hank’s buffered salt solution (HBSS), frozen at -80°C, and subsequently transferred within 7 days to liquid nitrogen. In addition, a sample was fixed in 10% buffered formalin acetate, paraffin-embedded, and stained with hematoxylin and eosin. Light microscopic evaluation of tissue sections was performed by a urological pathologist (W.K. or P.K.). Images were obtained using a Nikon Eclipse 80i microscope and NIS-Elements D 3.10 camera.
Mice were evaluated for tumor growth by physical exam typically twice weekly. When tumors reached ~10 mm in diameter, or mice became ill, they were anesthetized with isofluorane, exsanguinated by cardiac puncture, and tumors were processed as above and serially transplanted into subsequent cohorts of NOD/SCID mice.
Serum calcium was measured at the UT Southwestern Mouse Metabolic Phenotyping Core using a Vitros 250 Chemistry System (Johnson and Johnson).
Tissue processing for genomic studies
Flash-frozen samples preserved at -80°C were processed while on dry ice. Tumor content was inferred through pathological analyses of flanking sections oriented using pathology dyes (StatLab Medical Products). Tumor and tumorgraft samples were carefully selected to ensure >70% tumor cellularity and the absence of necrosis or hemorrhage. Normal benign renal cortex or peripheral blood was used as reference. DNA and RNA were simultaneously extracted from the same sample using AllPrep (Qiagen) and mirVana (Ambion) kits after homogenizing the tissues with an RNase-free pestle (VWR) and a QIAshredder column (Qiagen) as detailed in (47). RNA quality was inspected using an Agilent 2100 Bioanalyzer.
Gene expression analyses
RNA samples were labeled with biotin and hybridized to Affymetrix Human Genome U133 Plus 2.0 arrays by the UTSW Microarray Core using standard procedures. CEL intensity files were analyzed as previously described (65). Probesets with non-specific hybridization (10,588, 19%) were discarded. Differences in gene expression between tumors and tumorgrafts were assessed using t tests and a Benjamini and Hochberg false discovery rate (FDR) correction (66). Probesets with an FDR q<0.05 and upregulated at least 1.5 fold in tumors vs. tumorgrafts were subtracted (2,443 probesets, 4%). These probesets were analyzed using Ingenuity Pathways Analysis (IPA). Probesets were also analyzed using principal components and unsupervised hierarchical clustering with Euclidean dissimilarity and an average linkage method in Partek Genomics Suite.
Copy number analyses
DNA samples were hybridized to Affymetrix SNP Arrays 6.0 by the Genome Science Resource (Vanderbilt University) using standard procedures. The CEL intensity files were quartile normalized with Partek Genomics Suite adjusting for fragment length and probe sequence without background correction as detailed in (47). Briefly, paired copy numbers were calculated from intensities for all samples except for TG143, for which a reference copy number baseline from Partek was used, as paired normal was unavailable. Unsupervised hierarchical clustering for 1.8 million markers was computed using Euclidean dissimilarity and an average linkage method. Copy numbers were adjusted for local GC content and were segmented with the circular binary segmentation (CBS) algorithm (67) using the DNAcopy package of R/Bioconductor. Segmented copy numbers were displayed with the Integrative Genomics Viewer (Broad Institute). Genotypes were obtained from Affymetrix Genotyping Console 4.0 and used for calculating the allele-specific copy number of each sample in Partek Genomics Suite.
Mutation analyses
Bidirectional Sanger DNA sequencing was performed on genomic DNA from tumors and tumorgrafts by Beckman Coulter Genomics using proprietary primers. Point mutations and indels were identified in chromatograms with Mutation Surveyor v3.30 and v3.98. Mutations within seven nucleotides before or after an exon were considered to affect splicing sites. Mutations are fully detailed in (47). Deep sequencing of the TSC1 mutation was carried out on the Illumina platform by direct amplicon sequencing. TSC1 sequences were amplified using TSC1 primers (forward, 5′ CACATCATTGCTGTCTTTATTT; reverse, 5′ CCAACTCTGGACAACATTCTAT) and including common adaptor sequences (forward, 5′-ACACGACGCTCTTCCGATCT-TSC1; reverse, 5′-GACGTGTGCTCTTCCGATCT-TSC1). Adaptor sequences were utilized as priming sites in a second PCR reaction, where oligo sequences converted PCR products into ‘sequencer ready’ templates by the addition of sequences required for clustering, sequencing and indexing.
Pharmacokinetic analyses
PK analyses were performed using 6-8 week old male NOD/SCID mice. PK parameters were calculated in sparse sampling mode using the noncompartmental analysis tool of WinNonlin (Pharsight Corporation). Sirolimus (LC Laboratories) was dosed at 0.5 mg/kg IP. The compound was dissolved in 5% EtOH, 5% PEG400, 5% Tween 80 and 85% of a 5% dextrose [D5W] solution. Mice were sacrificed at varying times post dose by CO2 inhalation, whole blood was drawn using an EDTA coated syringe, 100 μl of whole blood were mixed with 50 μl of 0.2 M Zinc Sulfate and the mixture was precipitated with 150 μl of 10 mM ammonium acetate in methanol + 0.1% formic acid containing 60 ng/sample of N-benzylbenzamide internal standard (IS). After mixing and a 10 min incubation at RT, samples were cleared twice by centrifugation and subsequently evaluated by LC/MS/MS using an Applied Biosystems/MDS Sciex 3200-QTRAP coupled to a Shimadzu Prominence LC. Standards, prepared by spiking fresh whole blood from uninjected NOD/SCID mice with varying concentrations of sirolimus, were processed following the same procedure. Chromatography conditions were as follows. Buffer A consisted of 10 mM ammonium acetate in water + 0.1% formic acid and Buffer B consisted of 10 mM ammonium acetate in methanol + 0.1% formic acid. The column flow rate was 1.5 ml/min using an Agilent C18 XDB, 5 micron packing 50 × 4.6 mm size column. The gradient conditions were 0.01 – 1.0 min 100% A, 1.0 - 1.5 min gradient to 100% B, 1.5 - 3.0 min 100% B, 3.0 - 3.1 min gradient to 0% B, 3.1 - 4.1 100% A. Sirolimus was detected in MRM mode by following the precursor to fragment ion transition 931.3 to 864.4. N-benzylbenzamide (transition 212.1 to 91.1) was used as an internal standard. Instrument settings for sirolimus were: Dwell time 150 ms, DP 31 volts, EP 6.5 volts, CEP 40 volts, CE 29 volts, CXP 10 volts, CUR 30, CAD med, IS 4500 volts, TEM 400°C, GS1 60 psi, GS2 40 psi. A value 3-times above the signal obtained from blank whole blood was designated as the limit of detection (LOD). The limit of quantitation (LOQ) was defined as the lowest concentration at which back calculation yielded a concentration within 20% of theoretical and which was above the LOD. The LOQ for sirolimus was 1 ng/ml. In general, back calculation of points yielded values within 15% of theoretical over five orders of magnitude (1 ng/ml to 10000 ng/ml).
Sunitinib malate (Pfizer) was administered at 10 mg/kg by oral gavage in 0.2 ml. The compound was resuspended in 0.5% W/V carboxymethylcellulose (CMC) in D5W, pH 7.2. Various times post dose, animals were sacrificed by CO2 inhalation, blood was collected using an acid-citrate-dextrose coated syringe by cardiac puncture, and plasma was isolated by centrifugation. 100 μl of plasma was precipitated with 400 μl of acetonitrile containing 40 ng/sample N-benzylbenzamide internal standard (IS). After clearing by centrifugation, 450 μl supernatant was lyophilized and resuspended in 180 μl of 50:50 acetonitrile:water with 0.1% formic acid. Standards, prepared by spiking blank mouse plasma (Bioreclamation, Inc.) with varying concentrations of sunitinib malate and its active metabolite N-desethyl sunitinib, were processed following the same procedure used for samples. Chromatography conditions were as follows. Buffer A consisted of water + 0.1% formic acid and Buffer B consisted of acetonitrile + 0.1% formic acid. The column flow rate was 1.5 ml/min using an Agilent C18 XDB, 5 micron packing, 50 × 4.6 mm size column. The gradient conditions were 0.01- 1.5 min 100% A, 1.5 - 2.0 min gradient to 100% B, 2.0 - 3.5 min 100% B, 3.5 - 3.6 min gradient to 0% B, 3.6 - 5.0 min 100% A. Sunitinib was detected in MRM (multiple reaction monitoring) mode by following the precursor to fragment ion transition 399.1 to 283.2. Instrument settings for sunitinib were: Dwell time 150 ms, DP 41 volts, EP 4.5 volts, CEP 24 volts, CE 33 volts, CXP 4 volts, CUR 45, CAD med, IS 4500 volts, TEM 650°C, GS1 60 psi, GS2 45 psi. Desethyl sunitinib was detected in MRM (multiple reaction monitoring) mode by following the precursor to fragment ion transition 371.0 to 326.2. Instrument settings for N-desethyl sunitinib were: Dwell time 150 ms, DP 31 volts, EP 5 volts, CEP 16 volts, CE 21 volts, CXP 4 volts, CUR 45, CAD med, IS 4500 volts, TEM 650°C, GS1 60 psi, GS2 45 psi. A value 3x above the signal obtained from blank plasma was designated as LOD. The LOQ was defined as the lowest concentration at which back calculation yielded a concentration within 20% of theoretical and which was above the LOD. The LOQ for both sunitinib and desethyl sunitinib was 1 ng/ml. In general back calculation of points yielded values within 15% of theoretical over four orders of magnitude (1 ng/ml to 1000 ng/ml).
Erlotinib (LC Laboratories) was administered at 12.5 mg/kg by oral gavage in 0.2 ml. The compound was dissolved in 5% ethanol, 0.5% Tween 80, 94.5% of a 0.3% w/v CMC in 0.174 M sodium acetate/acetic acid, pH 4.0. Various times post dose, animals were sacrificed by CO2 inhalation, blood was collected using an acid-citrate-dextrose coated syringe by cardiac puncture, and plasma was isolated by centrifugation. 100 μl of plasma was precipitated with 200 μl of methanol containing 30 ng/sample of N-benzylbenzamide internal standard and 0.1% formic acid. After clearing by centrifugation, samples were analyzed by LC/MS/MS using an Applied Biosystems/MDS Sciex 3200-QTRAP coupled to a Shimadzu Prominence LC. Standards prepared by spiking blank mouse plasma (Bioreclamation, Inc.) with varying concentrations of erlotinib and O-desmethyl-erlotinib were processed following the same procedure used for samples. Chromatography conditions were as follows. Buffer A consisted of water + 0.1% formic acid and Buffer B consisted of methanol + 0.1% formic acid. The column flow rate was 1.5 ml/min using an Agilent C18 XDB, 5 micron packing, 50 × 4.6 mm size column. The gradient conditions were 0.01- 1.2 min 100% A, 1.2 - 2.5 min gradient to 100% B, 2.5 - 3.5 min 100% B, 3.5 - 3.6 min gradient to 0% B, 3.6 - 5.0 min 100% A. Erlotinib was detected in MRM (multiple reaction monitoring) mode by following the precursor to fragment ion transition 394.2 to 278.2. O-desmethyl-erlotinib was detected as the 380.1 to 278.1 transition. N-benzylbenzamide (transition 212.1 to 91.1) was used as an internal standard (IS). Instrument settings for erlotinib were: Dwell time 150 ms, DP 56 volts, EP 5 volts, CEP 18 volts, CE 45 volts, CXP 4 volts, CUR 45, CAD med, IS 4500 volts, TEM 650°C, GS1 60 psi, GS2 45 psi. Settings for desmethyl erlotinib were: Dwell time 150 ms, DP 56 volts, EP 5 volts, CEP 18 volts, CE 45 volts, CXP 4 volts, CUR 45, CAD med, IS 4500 volts, TEM 650°C, GS1 60 psi, GS2 45 psi. A value 3x above the signal obtained from blank plasma was designated as LOD. The LOQ was defined as the lowest concentration at which back calculation yielded a concentration within 20% of theoretical and which was above the LOD. The LOQ for both erlotinib and O-desmethyl-erlotinib was 0.5 ng/ml. In general back calculation of points yielded values within 15% of theoretical over four orders of magnitude (0.5 ng/ml to 1000 ng/ml).
Dovitinib (LC Laboratories) was administered at 30 mg/kg by oral gavage in 0.2 ml. The compound was dissolved in 20 mM lactic acid in D5W. Various times post dose, animals were sacrificed by CO2 inhalation, blood was collected using an acid-citrate-dextrose coated syringe by cardiac puncture, and plasma was isolated by centrifugation. 100 μl of plasma was precipitated with 200 μl of acetonitrile containing 80 ng/sample of N-benzylbenzamide internal standard. After clearing by centrifugation, 250 μl of supernatant was mixed with 250 μl dH20 containing 0.2% formic acid. Samples were analyzed by LC/MS/MS using an Applied Biosystems/MDS Sciex 3200-QTRAP coupled to a Shimadzu Prominence LC. Standards prepared by spiking blank mouse plasma (Bioreclamation, Inc.) with varying concentrations of dovitinib were processed following the same procedure used for samples. Chromatography conditions were as follows. Buffer A consisted of water + 0.1% formic acid and Buffer B consisted of methanol + 0.1% formic acid. The column flow rate was 1.5 ml/min using an Agilent C18 XDB, 5 micron packing, 50 × 4.6 mm size column. The gradient conditions were 0.01- 1.5 min 100% A, 1.5 – 2.0 min gradient to 100% B, 2.0 - 3.5 min 100% B, 3.5 - 3.6 min gradient to 0% B, 3.6 - 5.0 min 100% A. Dovitinib was detected in MRM (multiple reaction monitoring) mode by following the precursor to fragment ion transition 393.1 to 336.2. N-benzylbenzamide (transition 212.1 to 91.1) was used as an internal standard (IS). Instrument settings for dovitinib were: Dwell time 150 ms, DP 71 volts, EP 9 volts, CEP 18 volts, CE 39 volts, CXP 4 volts, CUR 45, CAD med, IS 4500 volts, TEM 650°C, GS1 60 psi, GS2 45 psi. A value 3x above the signal obtained from blank plasma was designated as LOD. The LOQ was defined as the lowest concentration at which back calculation yielded a concentration within 20% of theoretical and which was above the LOD. The LOQ for dovitinib was 5 ng/ml. In general back calculation of points yielded values within 15% of theoretical over four orders of magnitude (5 ng/ml to 5000 ng/ml).
Drug trials
Approximately 64 mm3 fragments of tissue from stably growing orthotopic tumorgrafts were implanted subcutaneously in 4-6 week old mice. When tumor volumes reached 250-300 mm3, mice were segregated into appropriate treatment groups (3-5 mice/group) based on (i) tumor volume, (ii) growth rate, and (iii) mouse weight. Erlotinib (LC Laboratories) was administered by oral gavage every 12 h at 12.5 mg/kg in 5% ethanol, 0.5% Tween 80, 94.5% of a 0.3% w/v CMC in 0.174M sodium acetate/acetic acid, pH 4.0. Sirolimus (LC Laboratories) was administered by IP injection every 48 h at 0.5 mg/kg in 5% ethanol, 5% PEG400, 5% Tween 80 and 85% D5W. Vehicle (5% ethanol, 5% PEG400, 5% Tween 80, 85% D5W) was administered by IP injection every 48 h. Sunitinib (LC Laboratories) was administered by oral gavage every 12 h at 10 mg/kg in 0.5% CMC in D5W. Dovitinib (LC Laboratories) was administered at 30 mg/kg by oral gavage in 20 mM lactic acid in D5W. Tumor dimensions were measured twice a week using a digital caliper and tumor volume was calculated by the formula: tumor volume = l x w x h, where l is the largest dimension of the tumor, w is the largest diameter perpendicular to l, and h is maximal height of the tumor. Weekly weights were taken and treatment dose was modified accordingly. Mice were typically sacrificed after 4 weeks of treatment, or earlier if they became sick (e.g. hypercalcemia), or if tumors became too large.
Immunohistochemistry
Paraffin-embedded sections were cut and stained with phospho-S6 antibody S240/244 (1:100) (Cell Signaling) as described (40). Immunostained slides were evaluated independently by two investigators (S.S. and P.K.) who were blinded to clinicopathologic data. Quantitation was performed by P.K. according to the following scale: 1+ (weak), 2+ (moderate), 3+ (strong), and 4+ (very strong) and percentage of tumor cells with cytoplasmic (p-S6) staining: no staining (0), 1-24% (1), 25-49% (2), 50-74% (3), and >75% (4). The product of staining intensity and the percentage of positive cells was used for statistical analysis.
Statistical Analyses
The association of specific pathological characteristics and tumor engraftment was evaluated using a Fisher exact test for categorical variables and a Student t test for continuous variables. Outcome analyses excluded the liposarcoma (TG147). Treatment effects on tumor growth were evaluated using a linear mixed model assuming a AR(1) covariance structure. For time-to-event outcome, a Log-Rank test was used. For deceased patients, when the date of demise was not available, the date of the last encounter was used. Reported p-values are two-sided and not adjusted for multiple comparisons unless otherwise indicated. Statistical analyses were performed using SAS 9.2 for Windows (SAS Institute Inc.) and SPSS Statistics 17.0. For statistical analyses of microarray data, please see the corresponding sections.
Supplementary Material
ACKNOWLEDGEMENTS
We thank all the patients who donated samples. We thank Aneesa Husain, Amulya Yadlapalli, and Anusha Purushotham for technical support, Dr. J. Minna for NSCLC tumor cell line, the UT Southwestern tissue resource staff for tissue procurement, Pfizer for providing sunitinib, and all the members of the Brugarolas lab for reviewing the manuscript. Funding: This work was supported by the following grants to J.B.: American Cancer Society Research Scholar Grant (115739), RO1CA129387 and Cancer Prevention and Research Institute of Texas (RP101075). N.S.W. and the PK studies were supported in part by P01CA095471 (PI, Steve McKnight). The Vevo770 ultrasound was purchased with 1S10RR02564801 and imaging was facilitated by the SAIRP supported in part by U24CA126608. The tissue management shared resource is supported in part by NCI (1P30CA142543). S.P.-L. was partially supported by a fellowship of excellence from Generalitat Valenciana (BPOSTDOC06/004). J.B. is a Virginia Murchison Linthicum Scholar in Medical Research at UT Southwestern. The content is solely the responsibility of the authors and does not represent official views from any of the granting agencies.
Footnotes
Author contributions: S.S. set up and maintained the tumorgraft lines and database, processed tissue for histology and reviewed the slides, was responsible for most of the drug trials and wrote the first draft of the manuscript. S.P.-L. processed, managed, and extracted nucleic acids from tissues, performed gene expression, DNA copy-number and mutational analyses, and the corresponding statistical analyses. H.Z. performed statistical analyses under the supervision of X-J.X. B.K. assisted with drug trials and immunohistochemistry. P.S. conducted dovitinib drug trials. A.P.-J. processed tissue and helped with tumorgraft colony maintenance. T.Y. helped with imaging studies. D.J.M. performed deep sequencing studies under the supervision of M.T.R. and D.R.B. J.G. consented patients and N.C.W. established procedures for tumorgraft processing. L.M. performed all PK analyses under the supervision of N.S.W. who developed protocols for drug preparation and administration. Y.L., G.V.R., A.S., V.M., and J.A.C., performed surgeries and provided tissue samples. P.K. prepared, stained, and analyzed, together with W.K., all histological sections, including immunohistochemical stains. J.B. conceived the study, directed the project, and wrote the manuscript with input from other authors.
Competing interests: The authors declare that they have no competing interests.
Gene expression and SNP arrays were deposited in ArrayExpress under accession IDs E-MTAB-1050 and E-MTAB-1051, respectively.
REFERENCES AND NOTES
- 1.Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3:711–715. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 2.Ocana A, Pandiella A, Siu LL, Tannock IF. Preclinical development of molecular-targeted agents for cancer. Nat Rev Clin Oncol. 2010;8:200–209. doi: 10.1038/nrclinonc.2010.194. [DOI] [PubMed] [Google Scholar]
- 3.Hait WN. Anticancer drug development: the grand challenges. Nat Rev Drug Discov. 2010;9:253–254. doi: 10.1038/nrd3144. [DOI] [PubMed] [Google Scholar]
- 4.Suggitt M, Bibby MC. 50 years of preclinical anticancer drug screening: empirical to target-driven approaches. Clin Cancer Res. 2005;11:971–981. [PubMed] [Google Scholar]
- 5.Sellers WR. A blueprint for advancing genetics-based cancer therapy. Cell. 2011;147:26–31. doi: 10.1016/j.cell.2011.09.016. [DOI] [PubMed] [Google Scholar]
- 6.Fiebig HH, Maier A, Burger AM. Clonogenic assay with established human tumour xenografts: correlation of in vitro to in vivo activity as a basis for anticancer drug discovery. Eur J Cancer. 2004;40:802–820. doi: 10.1016/j.ejca.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 7.Peterson JK, Houghton PJ. Integrating pharmacology and in vivo cancer models in preclinical and clinical drug development. Eur J Cancer. 2004;40:837–844. doi: 10.1016/j.ejca.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 8.Sausville EA, Burger AM. Contributions of human tumor xenografts to anticancer drug development. Cancer Res. 2006;66:3351–3354. doi: 10.1158/0008-5472.CAN-05-3627. [DOI] [PubMed] [Google Scholar]
- 9.Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 10.Grisanzio C, Seeley A, Chang M, Collins M, Di Napoli A, Cheng SC, Percy A, Beroukhim R, Signoretti S. Orthotopic xenografts of RCC retain histological, immunophenotypic and genetic features of tumours in patients. J Pathol. 2011;225:212–221. doi: 10.1002/path.2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Angevin E, Glukhova L, Pavon C, Chassevent A, Terrier-Lacombe MJ, Goguel AF, Bougaran J, Ardouin P, Court BH, Perrin JL, et al. Human renal cell carcinoma xenografts in SCID mice: tumorigenicity correlates with a poor clinical prognosis. Lab Invest. 1999;79:879–888. [PubMed] [Google Scholar]
- 12.Maurer-Schultze B, Bassukas ID, Boswald M, Harasim M. Cell proliferation in human tumours growing in nude mice: renal cell carcinomas, larynx and hypopharynx carcinomas. J Cancer Res Clin Oncol. 1992;118:255–268. doi: 10.1007/BF01208614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knofel WT, Otto U, Baisch H, Kloppel G. Stability of human renal cell carcinomas during long term serial transplantation into nude mice: histopathology, nuclear grade, mitotic rate, and DNA content in thirty tumors. Cancer Res. 1987;47:221–224. [PubMed] [Google Scholar]
- 14.Clayman RV, Figenshau RS, Bear A, Limas C. Transplantation of human renal carcinomas into athymic mice. Cancer Res. 1985;45:2650–2653. [PubMed] [Google Scholar]
- 15.Kopper L, Magyarosy E, Nagy P, Lapis K, Szamel I, Eckhardt S, Csata S, Wabrosch G, Repassy D. Renal cell carcinoma--xenotransplantation into immuno-suppressed mice. Oncology. 1984;41:19–24. doi: 10.1159/000225784. [DOI] [PubMed] [Google Scholar]
- 16.Baisch H, Otto U, Kloppel G. Long-term serial transplantation of 30 different human renal cell carcinomas into NMRI (nu/nu) mice: flow cytometric, histologic, and growth studies. J Natl Cancer Inst. 1986;76:269–276. [PubMed] [Google Scholar]
- 17.Talmadge JE, Singh RK, Fidler IJ, Raz A. Murine models to evaluate novel and conventional therapeutic strategies for cancer. Am J Pathol. 2007;170:793–804. doi: 10.2353/ajpath.2007.060929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoffman RM. Orthotopic metastatic mouse models for anticancer drug discovery and evaluation: a bridge to the clinic. Invest New Drugs. 1999;17:343–359. doi: 10.1023/a:1006326203858. [DOI] [PubMed] [Google Scholar]
- 19.Fidler IJ. Critical factors in the biology of human cancer metastasis: twenty-eighth G.H.A. Clowes memorial award lecture. Cancer Res. 1990;50:6130–6138. [PubMed] [Google Scholar]
- 20.Brugarolas J. Renal-cell carcinoma--molecular pathways and therapies. N Engl J Med. 2007;356:185–187. doi: 10.1056/NEJMe068263. [DOI] [PubMed] [Google Scholar]
- 21.Baldewijns MM, van Vlodrop IJ, Schouten LJ, Soetekouw PM, de Bruine AP, van Engeland M. Genetics and epigenetics of renal cell cancer. Biochim Biophys Acta. 2008;1785:133–155. doi: 10.1016/j.bbcan.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 22.Nickerson ML, Jaeger E, Shi Y, Durocher JA, Mahurkar S, Zaridze D, Matveev V, Janout V, Kollarova H, Bencko V, et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res. 2008;14:4726–4734. doi: 10.1158/1078-0432.CCR-07-4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaelin WG., Jr. The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer. 2008;8:865–873. doi: 10.1038/nrc2502. [DOI] [PubMed] [Google Scholar]
- 24.Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferrara N. VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer. 2002;2:795–803. doi: 10.1038/nrc909. [DOI] [PubMed] [Google Scholar]
- 26.Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 27.Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- 28.Yang JC, Haworth L, Sherry RM, Hwu P, Schwartzentruber DJ, Topalian SL, Steinberg SM, Chen HX, Rosenberg SA. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med. 2003;349:427–434. doi: 10.1056/NEJMoa021491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Escudier B, Bellmunt J, Negrier S, Bajetta E, Melichar B, Bracarda S, Ravaud A, Golding S, Jethwa S, Sneller V. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol. 2010;28:2144–2150. doi: 10.1200/JCO.2009.26.7849. [DOI] [PubMed] [Google Scholar]
- 30.Rini BI, Halabi S, Rosenberg JE, Stadler WM, Vaena DA, Archer L, Atkins JN, Picus J, Czaykowski P, Dutcher J, et al. Phase III trial of bevacizumab plus interferon alfa versus interferon alfa monotherapy in patients with metastatic renal cell carcinoma: final results of CALGB 90206. J Clin Oncol. 2010;28:2137–2143. doi: 10.1200/JCO.2009.26.5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125–134. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- 32.Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik C, Kim ST, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115–124. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 33.Sternberg CN, Davis ID, Mardiak J, Szczylik C, Lee E, Wagstaff J, Barrios CH, Salman P, Gladkov OA, Kavina A, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061–1068. doi: 10.1200/JCO.2009.23.9764. [DOI] [PubMed] [Google Scholar]
- 34.Rini BI, Escudier B, Tomczak P, Kaprin A, Szczylik C, Hutson TE, Michaelson MD, Gorbunova VA, Gore ME, Rusakov IG, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet. 2011;378:1931–1939. doi: 10.1016/S0140-6736(11)61613-9. [DOI] [PubMed] [Google Scholar]
- 35.Brugarolas JB, Vazquez F, Reddy A, Sellers WR, Kaelin WG., Jr. TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell. 2003;4:147–158. doi: 10.1016/s1535-6108(03)00187-9. [DOI] [PubMed] [Google Scholar]
- 36.Pantuck AJ, Seligson DB, Klatte T, Yu H, Leppert JT, Moore L, O’Toole T, Gibbons J, Belldegrun AS, Figlin RA. Prognostic relevance of the mTOR pathway in renal cell carcinoma: implications for molecular patient selection for targeted therapy. Cancer. 2007;109:2257–2267. doi: 10.1002/cncr.22677. [DOI] [PubMed] [Google Scholar]
- 37.Robb VA, Karbowniczek M, Klein-Szanto AJ, Henske EP. Activation of the mTOR signaling pathway in renal clear cell carcinoma. J Urol. 2007;177:346–352. doi: 10.1016/j.juro.2006.08.076. [DOI] [PubMed] [Google Scholar]
- 38.Abou Youssif T, Fahmy MA, Koumakpayi IH, Ayala F, Al Marzooqi S, Chen G, Tamboli P, Squire J, Tanguay S, Sircar K. The mammalian target of rapamycin pathway is widely activated without PTEN deletion in renal cell carcinoma metastases. Cancer. 2011;117:290–300. doi: 10.1002/cncr.25402. [DOI] [PubMed] [Google Scholar]
- 39.Lin F, Zhang PL, Yang XJ, Prichard JW, Lun M, Brown RE. Morphoproteomic and molecular concomitants of an overexpressed and activated mTOR pathway in renal cell carcinomas. Ann Clin Lab Sci. 2006;36:283–293. [PubMed] [Google Scholar]
- 40.Kucejova B, Peña-Llopis S, Yamasaki T, Sivanand S, Tran TA, Alexander S, Wolff NC, Lotan Y, Xie XJ, Kabbani W, et al. Interplay Between pVHL and mTORC1 Pathways in Clear-Cell Renal Cell Carcinoma. Mol Cancer Res. 2011;9:1255–1265. doi: 10.1158/1541-7786.MCR-11-0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, Staroslawska E, Sosman J, McDermott D, Bodrogi I, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271–2281. doi: 10.1056/NEJMoa066838. [DOI] [PubMed] [Google Scholar]
- 42.Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grunwald V, Thompson JA, Figlin RA, Hollaender N, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449–456. doi: 10.1016/S0140-6736(08)61039-9. [DOI] [PubMed] [Google Scholar]
- 43.Hahn SA, Seymour AB, Hoque AT, Schutte M, da Costa LT, Redston MS, Caldas C, Weinstein CL, Fischer A, Yeo CJ, et al. Allelotype of pancreatic adenocarcinoma using xenograft enrichment. Cancer Res. 1995;55:4670–4675. [PubMed] [Google Scholar]
- 44.Chen M, Ye Y, Yang H, Tamboli P, Matin S, Tannir NM, Wood CG, Gu J, Wu X. Genome-wide profiling of chromosomal alterations in renal cell carcinoma using high-density single nucleotide polymorphism arrays. Int J Cancer. 2009;125:2342–2348. doi: 10.1002/ijc.24642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dondeti VR, Wubbenhorst B, Lal P, Gordan JD, D’Andrea K, Attiyeh EF, Simon MC, Nathanson KL. Integrative genomic analyses of sporadic clear cell renal cell carcinoma define disease subtypes and potential new therapeutic targets. Cancer Res. 2012;72:112–121. doi: 10.1158/0008-5472.CAN-11-1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peña-Llopis S, Vega-Rubin-de-Celis S, Liao A, Leng N, Pavía-Jiménez A, Wang S, Yamasaki T, Zhrebker L, Sivanand S, Spence P, et al. Submitted. Novel Tumor Suppressor, BAP1, Sets Foundation for Molecular Genetic Classification of Renal Cell Carcinoma
- 48.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. New England Journal of Medicine. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW, Harris CC, McLellan MD, Fulton RS, Fulton LL, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Atkins MB, Hidalgo M, Stadler WM, Logan TF, Dutcher JP, Hudes GR, Park Y, Liou SH, Marshall B, Boni JP, et al. Randomized phase II study of multiple dose levels of CCI-779, a novel mammalian target of rapamycin kinase inhibitor, in patients with advanced refractory renal cell carcinoma. J Clin Oncol. 2004;22:909–918. doi: 10.1200/JCO.2004.08.185. [DOI] [PubMed] [Google Scholar]
- 51.Raymond E, Alexandre J, Faivre S, Vera K, Materman E, Boni J, Leister C, Korth-Bradley J, Hanauske A, Armand JP. Safety and pharmacokinetics of escalated doses of weekly intravenous infusion of CCI-779, a novel mTOR inhibitor, in patients with cancer. J Clin Oncol. 2004;22:2336–2347. doi: 10.1200/JCO.2004.08.116. [DOI] [PubMed] [Google Scholar]
- 52.Brugarolas J, Lotan Y, Watumull L, Kabbani W. Sirolimus in metatastic renal cell carcinoma. J Clin Oncol. 2008;26:3457–3460. doi: 10.1200/JCO.2008.16.4590. [DOI] [PubMed] [Google Scholar]
- 53.Bukowski RM, Kabbinavar FF, Figlin RA, Flaherty K, Srinivas S, Vaishampayan U, Drabkin HA, Dutcher J, Ryba S, Xia Q, et al. Randomized phase II study of erlotinib combined with bevacizumab compared with bevacizumab alone in metastatic renal cell cancer. J Clin Oncol. 2007;25:4536–4541. doi: 10.1200/JCO.2007.11.5154. [DOI] [PubMed] [Google Scholar]
- 54.Boni JP, Leister C, Bender G, Fitzpatrick V, Twine N, Stover J, Dorner A, Immermann F, Burczynski ME. Population pharmacokinetics of CCI-779: correlations to safety and pharmacogenomic responses in patients with advanced renal cancer. Clin Pharmacol Ther. 2005;77:76–89. doi: 10.1016/j.clpt.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 55.Awada A, Cardoso F, Fontaine C, Dirix L, De Greve J, Sotiriou C, Steinseifer J, Wouters C, Tanaka C, Zoellner U, et al. The oral mTOR inhibitor RAD001 (everolimus) in combination with letrozole in patients with advanced breast cancer: results of a phase I study with pharmacokinetics. Eur J Cancer. 2008;44:84–91. doi: 10.1016/j.ejca.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 56.Tan AR, Yang X, Hewitt SM, Berman A, Lepper ER, Sparreboom A, Parr AL, Figg WD, Chow C, Steinberg SM, et al. Evaluation of biologic end points and pharmacokinetics in patients with metastatic breast cancer after treatment with erlotinib, an epidermal growth factor receptor tyrosine kinase inhibitor. J Clin Oncol. 2004;22:3080–3090. doi: 10.1200/JCO.2004.08.189. [DOI] [PubMed] [Google Scholar]
- 57.Raizer JJ, Abrey LE, Lassman AB, Chang SM, Lamborn KR, Kuhn JG, Yung WK, Gilbert MR, Aldape KA, Wen PY, et al. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro Oncol. 2010;12:95–103. doi: 10.1093/neuonc/nop015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anjum R, Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat Rev Mol Cell Biol. 2008;9:747–758. doi: 10.1038/nrm2509. [DOI] [PubMed] [Google Scholar]
- 59.Trudel S, Li ZH, Wei E, Wiesmann M, Chang H, Chen C, Reece D, Heise C, Stewart AK. CHIR-258, a novel, multitargeted tyrosine kinase inhibitor for the potential treatment of t(4;14) multiple myeloma. Blood. 2005;105:2941–2948. doi: 10.1182/blood-2004-10-3913. [DOI] [PubMed] [Google Scholar]
- 60.Yuen JS, Sim MY, Siml HG, Chong TW, Lau WK, Cheng CW, Huynh H. Inhibition of angiogenic and non-angiogenic targets by sorafenib in renal cell carcinoma (RCC) in a RCC xenograft model. Br J Cancer. 2011;104:941–947. doi: 10.1038/bjc.2011.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Karam JA, Zhang XY, Tamboli P, Margulis V, Wang H, Abel EJ, Culp SH, Wood CG. Development and characterization of clinically relevant tumor models from patients with renal cell carcinoma. Eur Urol. 2011;59:619–628. doi: 10.1016/j.eururo.2010.11.043. [DOI] [PubMed] [Google Scholar]
- 62.Xu X, Hou Y, Yin X, Bao L, Tang A, Song L, Li F, Tsang S, Wu K, Wu H, et al. Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell. 2012;148:886–895. doi: 10.1016/j.cell.2012.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yamasaki T, Tran TA, Oz OK, Raj GV, Schwarz RE, Deberardinis RJ, Zhang X, Brugarolas J. Exploring a glycolytic inhibitor for the treatment of an FH-deficient type-2 papillary RCC. Nat Rev Urol. 2011;8:165–171. doi: 10.1038/nrurol.2010.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kucejova B, Sunny NE, Nguyen AD, Hallac R, Fu X, Peña-Llopis S, Mason RP, Deberardinis RJ, Xie XJ, Debose-Boyd R, et al. Uncoupling hypoxia signaling from oxygen sensing in the liver results in hypoketotic hypoglycemic death. Oncogene. 2011;30:2147–2160. doi: 10.1038/onc.2010.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peña-Llopis S, Vega-Rubin-de-Celis S, Schwartz JC, Wolff NC, Tran TA, Zou L, Xie XJ, Corey DR, Brugarolas J. Regulation of TFEB and V-ATPases by mTORC1. EMBO J. 2011;30:3242–3258. doi: 10.1038/emboj.2011.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B-Methodological. 1995;57:289–300. [Google Scholar]
- 67.Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.