

SUMMARY
Rhomboid proteases regulate key cellular pathways, but their biochemical mechanism including how water is made available to the membrane-immersed active site remains ambiguous. We performed four prolonged molecular dynamics simulations initiated from both gate-open and gate-closed states of Escherichia coli rhomboid GlpG in a phospholipid bilayer. GlpG was notably stable in both gating states, experiencing similar tilt and local membrane thinning, with no observable gating transitions, highlighting that gating is rate-limiting. Analysis of dynamics revealed rapid loss of crystallographic waters from the active site, but retention of a water cluster within a site formed by His141, Ser181, Ser185 and/or Gln189. Experimental interrogation of 14 engineered mutants revealed an essential role for at least Gln189 and Ser185 in catalysis with no effect on structural stability. Our studies indicate that spontaneous water supply to the intra-membrane active site of rhomboid proteases is rare, but its availability is ensured by an unanticipated active site element, the water-retention site.
Keywords: rhomboid protease, molecular dynamics simulation, lipid bilayer, water retention site, transmembrane helix, intramembrane proteolysis
INTRODUCTION
Intramembrane proteases are a class of enzymes that reside immersed within cellular membranes, where they catalyze the hydrolysis of peptide bonds (Erez et al., 2009; Urban, 2010; Wolfe, 2009). These enzymes are present in all forms of life, and play central roles in growth factor signaling, metabolic homeostasis, and mitochondrial dynamics including apoptosis (Brown et al., 2000; Selkoe and Wolfe, 2007; Urban, 2006). Recent investigations have also implicated these enzymes in a multitude of diverse microbial pathogens (Urban, 2009). Intramembrane proteases bear little or no sequence resemblance to soluble proteases but are thought to employ similar hydrolytic mechanisms. The obvious biochemical discrepancy has been how intramem-brane proteolysis can be accommodated considering that water, essential in peptide bond hydrolysis, is in short supply within the membrane.
Among the three mechanistic classes of intramembrane proteases, the rhomboid serine protease family is so far the best characterized through both biochemical investigation using pure enzyme reconstitution assays and a series of high-resolution crystal structures (as reviewed in (Urban, 2010)). Structural advances in particular have transformed how hydrolysis within the membrane is considered (Ben-Shem et al., 2007; Lemieux et al., 2007; Wang et al., 2006; Wu et al., 2006). For the first time, both the catalytic apparatus, and route of water entry, became visible. Crystal structures of the Escherichia coli rhomboid intramembrane protease GlpG revealed the catalytic residues S201 on TM4 and H254 on TM6 form a hydrogen-bonded catalytic dyad. These residues lie at the center of a compact, helical-bundle core domain comprised of six characteristic hydrophobic transmembrane helices (TM1–TM6) connected by five loops (L1–L5). Unlike other TMs, the TM4 central helix is very short and ends abruptly at the catalytic serine in the middle of the molecule, which provides space for a cavity that opens to the extracellular environment (Koide et al., 2007). Water molecules decorate the structures within this hydrophilic cavity, but this microenvironment remains segregated from membrane lipid laterally by trans-membrane helices. This architecture suggested that water enters the active site through the large, overlying cavity, but raised the question of how substrates enter the active site from the membrane.
Comparison of the various GlpG structures solved in different detergents and space groups revealed an amazing congruity overall, but suggested two different conformations of GlpG exist (Ben-Shem et al., 2007; Lemieux et al., 2007; Wang et al., 2006; Wu et al., 2006). Enzyme activity analyses have defined these differences as functionally important for substrate gating (Baker et al., 2007). The 2IC8 structure revealed a compact molecule with the catalytic apparatus completely enclosed (Wang et al., 2006). While in 2IC8 the L5 Cap clamps down on the active site, both the L5 Cap as well as the underlying TM5 were found to adopt significantly different conformations in the 2NRF as well as the 2IRV structures (Ben-Shem et al., 2007; Wu et al., 2006). The TM5 helix in 2NRF (molecule A) and 2IRV (molecule B) is titled further away from the rest of the helices with its L5 also uncovering the active site from above. It was therefore hypothesized that the 2IC8 structure is GlpG in the closed state, while 2NRF is GlpG in the open state. Enzymatic analyses revealed that mutation of residues on TM5, but not on the L5 Cap, resulted in a dramatic increase in enzyme activity, suggesting that TM5 forms the rate-limiting gate for substrate access from the membrane to the active site (Baker et al., 2007). The enhancement of enzyme activity as high as 10-fold was observed both in vitro with purified enzyme and in living bacterial cells (Urban and Baker, 2008).
Despite the wealth of structural information, the dynamic function of GlpG cannot be extrapolated from static crystal structures alone. In this light, computational simulations provide a means to study enzyme dynamics, and recent molecular dynamics (MD) simulations with one GlpG structure for 34 ns have provided an initial view of its properties (Bondar et al., 2009). Since a complete understanding GlpG dynamics in the lipid environment requires analysis of different conformers over extended periods of time, we performed a series of 110 ns MD simulations with GlpG in both the closed and open conformations as starting points. The prolonged simulations unexpectedly identified a pocket next to the catalytic serine as a region for water retention. Experimental analysis of 14 engineered GlpG mutants in living cells and purified components in vitro indicate that water retention is essential for ensuring catalytic efficiency.
RESULTS
GlpG Dynamics and Gating Transitions
We carried out four 110 ns molecule dynamics simulations on the E. coli rhomboid protease GlpG in a palmitoyl oleoyl phosphatidylethanolamine (POPE) lipid bilayer, the major lipid of the E. coli membrane. Simulations GlpG1 and GplG2 start from the enzyme in the closed state (2IC8) while GlpG3 and GlpG4 initiate from GlpG in the open state (2IRV molecule B). The overall structure of GlpG is quite dynamic but nevertheless stable in all four trajectories with Cα root-mean-squared deviation (RMSD) around 2 Å and all transmembrane helices (TMs) remainintact (Fig. 1a and Movie S1). However, the six TMs differ in their structural flexibility, with TM5 having the largest Cα RMSD value (Fig. 1b). This indicates that the position of TM5 is quite flexible, which is consistent with the experimental finding that TM5 is part of the substrate gate. For loops, the Cα RMSD values for L4 and L5 are also all quite large (Fig. S1).
Figure 1. GlpG dynamics in a bilayer.

(A) Image of gate-open GlpG in a bilayer during a si-mulation. Right panel shows Cα root mean-square deviation (RMSD) with respect to the corresponding crystal structure along four 110 ns MD simulation trajectories (link to Movie S1). Tra-jectories GlpG1 and GlpG2 start from a crystal structure in the closed state (2IC8), and simula-tions GlpG3 and GlpG4 employ a crystal structure in the open state (2IRV molecule B) as the initial model. (B) Cα root mean-square deviation (RMSD) of TM helices in the open (GlpG3) and closed GlpG2) states. (C) The sum of minimal distances between residues H150 and G240, L148 and M247, L148 and S248 along four 110 ns MD simulations was used to quantify possible gating transitions. Right panel shows the sum distribution of minimal distances observed in MD simulations, with the diamonds indicating corresponding values in crystal structures. See also Figures S1, S2 and S3, and Movie S1.
The mechanism of gate-opening and closing remains a key unaddressed question for all intramembrane proteases (Erez et al., 2009; Urban and Shi, 2008). The minimal distances between H150 on TM2 and G240 on TM5, L148 on TM2 and M247 on L5, L148 and S248 on L5 show large differences between open and close states. The sum of three distances ranges from 25 Å to 35 Å for simulations of the closed state, while spans from 42 Å to 56 Å were observed for the open state (Fig. 1c). Nevertheless, the peak values throughout the simulations centered around the experimental values, indicating no obvious transition from one state to the other. To determine whether there are any subtle transitions staring to occur, we fitted every trajectory to the crystal structure in the opposite state (Fig. S2 and S3). The overall drift of GlpG from the opposite state is also stable during the simulation. Just like in the crystal structures, the major difference between the two states exists in TM5 and L5 and maintains throughout all simulation, while the RMSDs for other helices and loops are all quite small and stable. Therefore, while GlpG is more flexible in the open than in the close state, GlpG remains globally stable in both states, and there is no transitions between two states in the time scale of 110 ns.
GlpG Position in a Lipid Bilayer
Simulations further provide an opportunity to explore how GlpG is situated in a lipid environment, and whether the orientation changes between the open and closed states. The arrangement of a protein with respect to the membrane can be defined by its shift along the bilayer normal, the tilt angle, and the thickness of its membrane-spanning region (Lomize et al., 2006). In all four simulations, GlpG remains in the center of the bilayer relative to the bilayer normal (Fig. S4), but the tilt angle fluctuates between 13°–30° over the last 80 ns of the simulation (Fig. 2a). Interestingly, no differences were evident between the gate-open and closed forms.
Figure 2. GlpG position in a bilayer.

(A) Changes in tilt angle of GlpG relative to the membrane normal during the MD simulations. (B) The bilayer hydrophobic core thickness as a function of distance circumferentially outwards from GlpG estimated over the last 80 ns of simulations. The x-axis denotes the radial distance moving away from GlpG versus the hydrophobic core thickness of the bilayer (distance between carbon 2 of the upper and lower leaflet lipid). The discrete points represent the first 6 rings of lipids surrounding GlpG. Closed state of simulations GlpG1 (A), GlpG2 (B), open state of simulations GlpG3 (C) and GlpG4 (D). (C) Selected MD snapshot of the location of the active site with respect to the lipid surface. Gold spheres denote phosphate atoms of the phospholipid headgroups. See also Figure S4.
Since bilayer structure can be significantly perturbed by protein, we computed the average hydrophobic bilayer thickness moving outwards circumferentially from GlpG over the last 80ns for all four simulations (Fig. 2b). The distance of a lipid molecule from the protein was calculated as the minimum distance between carbon atom 5 of the lipid and any Cα atom of the protein. The bilayer hydrophobic thickness was calculated as the distance between carbon 2 of the lipid molecules of the two monolayers (Tieleman et al., 1998), which corresponds roughly to the hydrophobic interior of the bilayer. For the pure POPE bilayer, the calculated average bilayer hydrophobic thickness was 34.68 ± 0.15 Å over the last 5 ns trajectory in our simulation. Due to the hydrophobic mismatch between GlpG and POPE bilayer, the first three rings of lipids around GlpG were affected and showed a decrease in thickness to ~30 Å, whereas the next three rings remained close to the POPE bulk thickness. The results are very consistent among the four different simulations, with no obvious differences between the gate-open and closed forms, and are further consistent with the 34 ns simulations of gate-open GlpG with Charmm force field (Bondar et al., 2009).
The hydrophobic thickness of GlpG estimated in all simulations differs significantly from the 20 Å value that was initially suggested based on crystal water positions in the detergent-solubilized GlpG (Wang et al., 2007). To understand the discrepancy, we performed a short MD simulation using crystal structure 3B45 from which water occupancy was used to derive the GlpG hydrophobic belt (Wang et al., 2007). The simulation revealed most of the protein-bound crystal water molecules were quickly released to the bulk water, suggesting caution should be exercised in estimating the hydrophobic thickness based on the presence of crystal water molecules. Therefore, as all simulations agree, the hydrophobic belt of GlpG should be considered to be 30 Å (Bondar et al., 2009).
To examine further the importance of lipid environment, we carried out an MD simulation of GlpG in a water box without lipids using the same starting structures. Under these conditions, the Cα RMSD quickly becomes larger than 3.0 Å, and helix structures become unstable, emphasizing that a hydrophobic environment plays an integral role in the structural stability of GlpG.
Analysis of Water Dynamics Identifies a Retention Site
The catalytic residues of GlpG consists of a hydrogen-bonded Ser-His pair contributed by the N terminus of TM4 and the upper portion of TM6, respectively. In our simulations, the average distance from the oxygen of the catalytic S201 to the lipid surface of the first annular ring of lipids is 11–12 Å. Therefore, the active site is located about 10 Å under the bilayer surface, and our simulations support that the enzyme reaction occurs within the intramembrane environment rather than at the membrane surface (Fig. 2c).
A key question is how water molecules are supplied to the active site residues during catalysis. Although a large cavity above the catalytic serine opens to the aqueous environment, we found that most water molecules positioned near the active site in crystal structures quickly diffused out to the bulk water in all simulations of GlpG in a bilayer. This key point is illustrated in Movie S2. Interestingly, we observed three water molecules remained resident in a hydrophilic cavity adjacent to the catalytic residue S201 in all four 110 ns MD simulations (Fig. 3a). This unexpected observation raised the possibility that water, which is essential for peptide bond hydrolysis, might not arrive randomly during catalysis, but rather might be provided by polar residues enriching water molecules next to the catalytic serine.
Figure 3. Water retention site in GlpG.
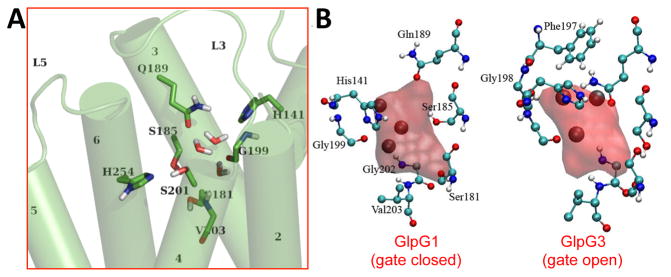
(A) Selected MD snapshot of a water cluster in a representative conformation from the MD Movies (link to Movies S2 and S3). (B) Mean water density of the triple water cluster and interaction of surrounding residues. The water positions in the x-ray structural models are in black spheres. The waters observed in the x-ray structures (black spheres) are within the Connolly surface, except one, but this water is very near the red area (see Fig. S5). The mean water densities are compatible with the water positions in the crystal structure models. See also Figure S5, and Movies S2 and S3.
The water retention site is located between TM3 and TM4, under L3 and a short non-TM helix in L1, and very near the catalytic dyad (Fig. 3a). The water molecules formed hydrogen bonds mainly with H141 on a short non-TM helix in the L1 loop, side chains of S181 and S185 on TM3, backbones of G202 and V203 on TM4, and the carbonyl group of G199 on L3. V203 forms the bottom for the water retention site by interacting with other hydrophobic residues from surrounding TM helices. Three water molecules inside the cavity sometimes form a single file of hydrogen-bonded molecules, and are stabilized by forming hydrogen bonds with the surrounding polar residues and protein backbones.
Because internal waters display anisotropic fluctuations during our simulations, a thorough description required a study of water density. We followed the method proposed by Kandt et. al. (Kandt et al., 2004). All snapshots were superimposed to a reference structure at 30 ns. A cubic spatial grid with an edge length of 1 Å was used to calculate the number of water oxygens per sub-cube. The evaluations were conducted on the last 80ns of each simulation to ensure that the system had equilibrated. Connolly surfaces were computed for the cells exceeding the cutoff 0.015H2O/Å 3 and a probe radius of 1.4 Å was used. A typical structure of the water cluster within GlpG (Fig. 3a) revealed that the water cluster mainly remained at the bottom of the cavity. The Connolly surfaces with the hydrogen bonded residues, taken from snapshots at 30ns, showed residues held the waters in a certain region by hydrogen bonds (Fig. 3b). Interestingly, Q189 is not within the Connolly surface of the water, which is consistent with the analysis of hydrogen bonds, and suggests that Q189 is involved in water conduction rather than retention directly.
In order to gain a more quantitative sense of the water retention, we measured the number and time that each H-bond formed by each side chain (Table 1 and S1). During four MD simulations, the average number of H-bonds formed between the three water molecules and GlpG is around five. In particular, the sidechain hydroxyl of S185, backbone carbonyl of G199, and backbone NH of V203 frequently formed H-bonds with water molecules, each of which were engaged for longer than half the time in all 4 simulations (Table 1). This revealed H-bonding with these individual sites totaling >50ns, which is remarkably long relative to water molecule loss that took <0.1ns (see below). From these calculations, our results indicate that, through a series of localized but dynamic H-bonds, water molecules are globally stable in the water retention site for at least 100 ns.
Table 1.
H-bond interactions for water molecules in and around the water-retention site cavity (values indicate percentage of total simulation time for which a given H-bond was present. An interaction is recorded only if it was present for >20% in at least one simulation). see also Table s1
| Interaction | GlpG1 | GlpG2 | GlpG3 | GlpG4 |
|---|---|---|---|---|
| S181OGHG-Wat:O | 8.5 | 19.4 | 7.8 | 24.5 |
| S185OGHG-Wat:O | 75.5 | 92.0 | 47.2 | 48.3 |
| Q189NEHE-Wat:O | 5.8 | - | 69.5 | - |
| G202NH-Wat:O | 10.9 | 9.7 | 75.7 | 33.6 |
| V203NH-Wat:O | 74.9 | 73.4 | 61.8 | 34.4 |
| H141ND-Wat:H | 27.9 | 57.4 | 70.6 | 42.7 |
| S181OG-Wat:H | 40.3 | 57.0 | 2.7 | 29.3 |
| S185OG-Wat:H | 44.0 | 63.5 | 23.6 | 27.1 |
| Q189OE-Wat:H | 12.8 | - | 76.0 | - |
| F197O-Wat:H | 2.9 | - | 1.4 | 26.0 |
| G198O-Wat:H | 3.4 | - | 19.4 | 0.1 |
| G199O-Wat:H | 82.6 | 87.4 | 70.5 | 47.5 |
While water molecules are quite mobile and hydrogen bonds dynamic inside the retention site, the exchange of water molecule between the retention site and the bulk is a rare event. In fact, only in one simulation (GlpG3) did one bulk water molecule exchange with one water molecule in the retention site (between 77.78 and 77.84 ns, see Movie S3 whose corresponding simulation trajectory is 1 ns); the new water molecule then stayed in the retention area for the remainder of the simulation. During the water exchange process, both the incoming and outgoing water molecules form hydrogen bonds with Q189 for prolonged lengths of time. Thus, in simulations with GlpG3, water molecules formed two hydrogen bonds with the side-chain of Q189 at percentages of 69.5% and 76.0%, while in other simulations the internal water molecules rarely formed hydrogen bonds with Q189.
Water-Retaining Residues are Essential for Catalysis not Structural Stability
The simulations raised the unexpected possibility of a water retention pocket within the enzyme being required for catalysis, which we sought to test experimentally by focusing on the key residues highlighted by this model. In addition to backbone interactions (which we cannot modify by mutation), the simulations identified the side chains of H141, Q189, S181 and S185 functioning in water conduction and/or retention. We therefore made a series of mutants at each of these residues and examined their contribution to GlpG enzymatic activity both in living E. coli cells (Urban and Baker, 2008) and in vitro with pure protein (Urban and Wolfe, 2005). Since activity analysis alone cannot distinguish between a requirement for catalysis from a role in maintaining structural stability, we also assessed whether our mutants perturb GlpG thermostability in a quantitative static light scattering assay. We recently developed this method and found it to be the most sensitive and robust assay currently available for assessing the structural stability of membrane proteases (Baker and Urban, 2012).
The sidechains of Q189 and H141 lie outward from the active site, and could thus be involved in water conduction. Mutation of Q189 to alanine, threonine, valine or tyrosine, and H141 to phenylalanine, or tyrosine resulted in a complete block to enzymatic activity either in bacterial cells or in vitro (Fig. 4a and b). To assess the effect of these mutations on protein folding, we raised temperature from 25°C to 85°C and quantified static light scattering at 0.5°C intervals. Mutation of Q189 to either alanine or threonine resulted in thermostability of GlpG that was indistinguishable from wildtype (Fig. 4c), yet all Q189 mutants proved to have no detectable catalytic activity either in E. coli cells or in vitro with pure proteins. Even Q189T, which retains one hydroxyl group capable of hydrogen bonding, proved inactive, highlighting how sensitive catalysis is to Q189 changes (Fig. 4d). While mutation of H141 to valine or threonine perturbed structural stability of GlpG, decreasing the transition temperature by 11–12°C (Fig. 3c), both mutants retained some proteolytic activity, suggesting that this residue is less important than Q189 for catalysis. Conversely, the H141F mutation resulted in a protein with thermostability closer to wildtype (~6°C decrease in transition temperature), but had nearly undetectable proteolytic activity. As such, increasing the hydrophobicity at residue 141 maintains protein structure but abolishes catalytic activity.
Figure 4. Protease activity and stability of upper water-retaining residue mutants H141 and Q189.

(A) Protease activity was assessed in vivo by co-expressing GlpG with a tagged Drosophila Spitz substrate in E. coli cells for the indicated times, followed by western analysis. Arrow indicates cleaved band, and SA denotes use of a catalytically-inactive GlpG as a negative control. (B) In vitro activity analysis of pure GlpG variants with APP+Spi7-Flag substrate in both detergent micelles and reconstituted in E. coli lipids. (C) Thermostability analysis of GlpG variants in a differential static light scattering assay. Transition temperatures with standard deviations are shown. Note that Q189 mutants did not perturb the thermostability of GlpG. (D) Activity analysis of GlpG variants with a Spitz substrate (polyA) harboring 4 and 3 alanine residues preceding and following the cleavage site, respectively. Small residues in the substrate could not rescue the protease activity defect of Q189 mutants.
S181 and S185 form the lower surface of the water retention compartment, we therefore also investigated the activity of their mutants (Fig. 5a and b). Interestingly, substitution of S185 with either valine or threonine resulted in structural stability of GlpG that is not statistically different from wildtype (~2°C difference in transition temperature; Fig. 5c). However, the S185V mutant dramatically exhibited no enzymatic activity under any conditions (Fig. 5a and b). Conversely, substituting S185 with threonine, a residue similar in size to valine but with a hydrophilic hydroxyl group that would be expected to help in water retention, partially restored activity both in living cells and in vitro. Mutation of S181 to valine perturbed GlpG structural stability despite retaining proteolytic activity (Fig. 5c), suggesting that S181 is not as important for catalysis directly. Therefore, the dramatic decrease in activity with substitutions at S185 do not result from effects on protein structure, but rather a key role for this residue in catalysis.
Figure 5. Protease activity and stability of lower water-retaining residue mutants S181 and S185.

(A) Protease activity of GlpG variants in vivo with a tagged Drosophila Spitz substrate in E. coli cells was examined by co-expression for the indicated times, followed by western analysis. Arrow indicates cleaved band, and SA denotes a catalytically-inactive GlpG used as a negative control. (B) Protease activity of pure GlpG variants in vitro with APP+Spi7-Flag substrate examined both in detergent micelles and reconstituted in E. coli lipids. (C) Thermostability of GlpG variants quantified in a differential static light scattering assay. Transition temperatures with standard deviations are shown. S185V did not significantly perturb the thermostability of GlpG. (D) Activity analysis of GlpG variants with a Spitz substrate (polyA) harboring 7 alanine residues surrounding the cleavage site. The activity defect of S185V was not rescued by a substrate bearing small residues, while enhancing the processing of all active enzymes.
Although the Q189 and S185 mutants do not affect structural stability of GlpG, it remained possible that they abolish protease activity because substrates with large residues surrounding the cleavage site can no longer be accommodated within the protease active site. In fact, Spitz has Leu, Glu and Lys preceding the P1 Ala residue. We therefore mutated 8 Spitz residues surrounding the cleavage site all to alanine, a small residue. The cleavage of this Spitz-polyAla substrate was actually enhanced by wildtype GlpG, yet S185V and all examined Q189 mutations failed to show any detectable protease activity against Spitz-polyAla (Fig. 4d and 5d). Therefore, interfering with accommodating substrates into the active site is not the basis of the lost protease activity.
Taken together, our activity and protein stability experiments revealed that substitutions expected to decrease interaction with water at S185 and Q189, in particular, strongly decrease catalytic activity without perturbing protein structure or substrate position. These observations are consistent with water retention playing an important role in supporting intramembrane pro-teolysis by GlpG.
DISCUSSION
Intramembrane proteases function immersed within the membrane to catalyze hydrolysis of peptide bonds (Erez et al., 2009; Urban, 2010; Wolfe, 2009). A wealth of recent biochemical and structural studies have provided a conceptual framework for understanding how rhomboid pro-teases function in general, but major gaps remain in our knowledge of the specific events involved in catalysis (as reviewed in (Urban, 2010)). A major challenge lies in understanding GlpG dynamics that underlie catalysis within the membrane, since this information cannot be extrapolated accurately from static crystal structures. We used molecular dynamics of open and closed-form GlpG structures in prolonged simulations to explore in detail two poorly-understood events in intramembrane catalysis; substrate gating dynamics and water delivery for hydrolysis.
Defining in detail how an intramembrane protease is positioned in the lipid bilayer is fundamental to understanding its mechanism. All but one (Vinothkumar, 2011) of the nearly dozen available GlpG structures were crystallized in detergent, providing little experimental information regarding how GlpG is positioned in the lipid bilayer (Bondar et al., 2009). All four of our simulations revealed GlpG within the membrane retains its overall shape completely unperturbed. Conversely, in all simulations the POPE bilayer adjusts its local thickness around GlpG due to hydrophobic mismatch, but the thickness of the first ring of lipids remains ~30 Å and not 20 Å as proposed from examining the crystal structure (Wang et al., 2007). Overall, the tilt and local membrane thinning immerse the gate region deeper, and the catalytic residues remain clearly beneath the membrane surface, further indicating that proteolysis is intramembrane (Bondar et al., 2009).
In an exciting recent advance, the first crystal structure of GlpG in a bicelle lipid environment has been solved (Vinothkumar, 2011). While this structure revealed several discrete interactions between GlpG and annular lipids, the overall shape or location of the membrane could not be visualized, limiting the information with respect to the position of GlpG within the membrane. Remarkably, however, the overall structure of GlpG in detergent versus in bicelle lipids was nearly identical, suggesting that the membrane does not change the overall structure of GlpG, which is also evident in our simulations.
Understanding the GlpG catalytic cycle also requires defining its conformational transitions and general dynamics in the membrane. For all gate-open and gate-closed 110ns simulations, we found GlpG to be globally stable, but dynamic in its motions. TM5, in particular, was the most dynamic among its six transmembrane helices, which is consistent with the proposal that TM5/L5 act as the substrate gate (Baker et al., 2007; Urban and Baker, 2008; Wu et al., 2006). Importantly, while it has been questioned whether the tilted TM5 in the open state is an artifact of crystal packing, our finding that the Cα RMSD value of TM5 in all simulations is strikingly comparable between the two states indicates that the tilted TM5 is stable and not a crystal-packing artifact. Moreover, in the gate-open state, the interaction between the hydrophilic protein interior and the hydrocarbon region of the lipid bilayer is compensated by the interaction between the protein and the head-group of the lipids, further suggesting that the gate-open form is stable.
Although our 110 ns simulations of both open and closed states of GlpG are over three times longer than those previously attempted for gate-open GlpG alone, our inability to detect gating transitions show that gating transitions are indeed a rare event, thereby reinforcing the enzymatic finding that gating is the rate-limiting step in intramembrane proteolysis. Nevertheless, it should be noted that there are some unavoidable limitations in a simulation study of this nature: although the simulation time in the order of 110 ns is comparable to other state-of-the-art simulations of membrane proteins (Bond and Sansom, 2003; Grossfield et al., 2007; Holyoake and Sansom, 2007), it is still too short to explore its conformation space fully; the accuracy of the molecular mechanical force field is nevertheless limited and no polarization effect has been considered; the protein has been simulated in a simple POPE lipid bilayer rather than the complex lipid environment in the E. coli inner membrane. Lastly, our analyses were conducted with GlpG as the only protein in the simulations, and no other bystander proteins or potential substrates. It is possible that the influence of other proteins could affect GlpG, directly or indirectly, to facilitate gating transitions. This remains a key avenue that merits future investigation.
Since water molecules were observed near the active site in all crystal structures, it has been assumed that water is readily available for intramembrane proteolysis (Ben-Shem et al., 2007; Lemieux et al., 2007; Wang et al., 2006; Wu et al., 2006). Completely unexpected was our observation that most crystal waters within the active site were rapidly lost, while a small clusters were specifically retained in a specialized area notably adjacent to the catalytic serine. In all four MD simulations, three water molecules always reside in this water retention site, forming hydrogen bonds with three backbone carbonyls and/or the side chains of H141, S181, S185 and Q189. Our mutational studies provide compelling evidence that at least the side chains of Q189 and S185 are important for catalysis, since their mutation abolished enzyme activity while leaving the structural stability of GlpG unperturbed. In fact, the light scattering assay that we have developed is able to detect very subtle changes in GlpG structure that are undetectable by other methods (R. Baker and S. Urban, 2012). As such, the identical thermal transition temperature of Q189 and S185 mutants are particularly informative, and suggest that water retention is essential for efficient intramembrane proteolysis. The fact that S181 and H141 mutants significantly perturb structural stability yet retain much higher protease activity than Q189 or S185 further emphasizes the importance of Q189 and S185 in catalysis. Moreover, multiple backbone NH and carbonyl groups also notably participate in water retention, and as such, it is not surprising that some side chain mutants (S181A, S185A, H141T) have only subtle effects until the mutations substantially increase hydrophobicity (e.g. S185V and H141F).
In summary, our combined computational and experimental studies suggest the existence of an internal water retention site in GlpG that ensures catalytic residues are supplied with water during catalysis. Why might this site be important if the catalytic serine is thought to be exposed to the aqueous phase? Water accessibility may in fact be restricted, but previous studies could not distinguish between bulk water accessibility to the active site versus internal ‘stored’ water, and as such our observations are fully consistent with prior experiments but offer a new interpretation. A second and key distinction lies in the nature of the active site at the time of catalysis. All structural and biochemical labeling studies of GlpG have been performed without the substrate bound. It is likely that the incoming substrate transmembrane segment exacerbates water loss by virtue of its hydrophobic transmembrane nature. As such, our investigations have provided an intriguing and unexpected insight into the function of membrane-immersed proteases; guaranteeing water supply to the catalytic residues at the time of hydrolysis is indeed a deliberate strategy for ensuring catalysis proceeds efficiently. In fact, recent work suggests that catalysis by soluble serine proteases actually requires water desolvation from the active site (Shokhen et al., 2008). Water retention at a specialized site could thus even be more widely applicable to general proteases, especially those that process hydrophobic substrates, although this awaits further analysis. In this regard, further studies of rhomboid proteases may provide new insights into proteolytic mechanisms in general.
EXPERIMENTAL PROCEDURES
Computational Methods
The 2IC8 GlpG structure (closed state) and molecule B of the 2IRV structures (open state) were used as two starting models for GlpG. The proteins were inserted into the pre-equilibrated POPE bilayer system using the ProtSqueeze algorithm (Yesylevskyy, 2007), and equilibration simulations were performed during which restraints on the protein atoms were gradually released. For each state of GlpG, we performed two simulations different from each other in the equilibration stage. Finally, 110 ns production run for each simulation was conducted. All simulations presented here were conducted using the GROMACS 3.3.1 MD simulation package (Van Der Spoel et al., 2005). The Berger et. al. force field (Berger et al., 1997) was used for the POPE molecules, the Gromos ffgmx force field for the protein (van Gunsteren and Berendsen, 1987), and the simple point charge (SPC) water potential31 for water molecules. Electrostatics were calculated using particle mesh Ewald (PME) with a 10 Å cutoff for the real space calculation.
DNA Constructs
All mutations were introduced by Quikchange site directed mutagenesis (Stratagene, La Jolla, USA) into GlpG in pGEX-6P-1 (GE Healthcare, Uppsala, Sweden) and GFP-Spitz-Flag in pET27b(+) (Novagen, Madison, USA), as described previously, and verified by DNA sequencing.
In vivo Proteolysis Assay in E. coli Cells
GlpG and Spitz were coexpressed in C43(DE3) cells grown in LB under ampicillin (100μg/ml) and kanamycin (50μg/ml) selection as described previously. Cells were harvested 4 and 6 hours after co-induction with 250 μM IPTG and resuspended in reducing Lamelli buffer. Protein lysates were resolved on 4–20% Tris-glycine SDS-PAGE gels and subjected to western analysis using infrared fluorescence scanning (LiCor Biosciences, Lincoln, USA).
In Vitro Proteolysis Assays
GlpG rhomboid proteins and the APP+Spi7-Flag substrate were expressed in E. coli cultures and purified using glutathione and immunoaffinity chromatography as described previously. Protein yields were quantified using infrared fluorescence scanning (LiCor Biosciences, Lincoln, USA) of Coomassie-stained SDS PAGE gels. Protease activity of GlpG and its mutants was assayed in a final volume of 20 μl containing 50 mM Tris (pH 7.5) and 150 mM NaCl with 0.1% do-decyl-b-D-maltoside (DDM) or 1 mg/ml E. coli lipid extract (Avanti Polar Lipids, Alabaster, USA) at 37°C for 1 hour. Reactions were resolved on 10% NuPAGE (Invitrogen, Carlsbad, USA) gels and detected by anti-Flag western analysis using infrared fluorescence.
GlpG Thermostability Analysis
Pure GlpG proteins were subjected to thermostability analysis in clear-bottom 384 well plates in a StarGazer instrument (Harbinger Biotech, Toronto, Canada). Temperature was raised from 25°C to 85°C at 1°C per minute, and light scattering from a ~620 nm LED source was quantified every 0.5°C and plotted. The transition temperature of each well was derived from fitting to a Boltzmann curve using StarGazer software. Each mutant was analyzed at least four times, yielding standard deviations of less than 1°C.
Supplementary Material
Figure S1: Cα RMSD value versus time for the loops in closed state of simulations GlpG1 (A), GlpG2 (B), open state of simulations GlpG3 (C) and GlpG4 (D).
Figure S2: Cα RMSD value versus time for the helices in closed state of simulations GlpG1 fitted to TM1-TM4+TM6 of open state (A), GlpG2 (B), open state of simulations GlpG3 fitted to closed state (C) and GlpG4 (D).
Figure S3: Cα RMSD value versus time for the loops in closed state of simulations GlpG1 fitted to TM1-TM4+TM6 of open state (A), GlpG2 (B), open state of simulations GlpG3 fitted to closed state (C) and GlpG4 (D).
Figure S4: MD trajectory of the GlpG center of mass along the Z axis relative to the center of mass of the lipid bilayer. Closed state of simulations GlpG1 (A), GlpG2 (B), open state of simulations GlpG3 (C) and GlpG4 (D).
Figure S5: A mean water density of the triple water cluster and interaction of surrounding residues. The water positions in the x-ray structural models are in black spheres. Closed state of simulation GlpG2 (left) and open state of simulation GlpG4 (right).
Table S1: H-bond interactions at the active site (The values indicate percentage of total simulation time for which a given H-bond was present. An interaction is recorded only if it was present for >20% in at least one simulation).
Molecular dynamics simulation of GlpG in a POPE bilayer. The catalytic serine and histidine are shown in space-fill.
Water dynamics during a MD simulation, starting from waters observed in the crystal structure. Note that the majority of crystallographic waters rapidly escaped, with no bulk waters entering to take their place. In contrast, several water molecules were retained near the catalytic center in a localized site (the ‘water retention’ site). The catalytic serine and histidine are shown in stick format. GlpG is simulated in a POPE bilayer, but phospholipids were removed from the movie for the sake of clarity.
Water dynamics in the active site of GlpG during a MD simulation. Note that the waters are dynamic, but constantly interacting with GlpG (side chains are shown in stick format). Only one water molecule (in blue and white) entered from bulk and only in this one of the four simulations. This water molecule remained at the active site due to its interactions with GlpG residues of the ‘water retention’ site.
Acknowledgments
We are grateful for the support from National Science Foundation (CHE-CAREER- 0448156 to YZ), the National Institute of Health (GM079223 to YZ; AI066025 to SU), and the Howard Hughes Medical Institute (SU). Special thanks to NYU-ITS and NCSA for providing computational resources. SU is a Fellow of the David and Lucile Packard Foundation.
Abbreviations used
- POPE
Palmitoyl oleoyl phosphatidylethanolamine
- TM
transmembrane
- MD
molecular dynamics
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baker RP, Urban S. Architectural and thermodynamic principles underlying intramembrane protease function. Nature Chem Biol. 2012 doi: 10.1038/nchembio.1021. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RP, Young K, Feng L, Shi Y, Urban S. Enzymatic analysis of a rhomboid intramembrane protease implicates transmembrane helix 5 as the lateral substrate gate. Proc Natl Acad Sci U S A. 2007;104:8257–8262. doi: 10.1073/pnas.0700814104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shem A, Fass D, Bibi E. Structural basis for intramembrane proteolysis by rhomboid serine proteases. Proc Natl Acad Sci U S A. 2007;104:462–466. doi: 10.1073/pnas.0609773104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger O, Edholm O, Jahnig F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys J. 1997;72:2002–2013. doi: 10.1016/S0006-3495(97)78845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond PJ, Sansom MS. Membrane protein dynamics versus environment: simulations of OmpA in a micelle and in a bilayer. J Mol Biol. 2003;329:1035–1053. doi: 10.1016/s0022-2836(03)00408-x. [DOI] [PubMed] [Google Scholar]
- Bondar AN, del Val C, White SH. Rhomboid protease dynamics and lipid interactions. Structure. 2009;17:395–405. doi: 10.1016/j.str.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Ye J, Rawson RB, Goldstein JL. Regulated intramembrane pro-teolysis: a control mechanism conserved from bacteria to humans. Cell. 2000;100:391–398. doi: 10.1016/s0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- Erez E, Fass D, Bibi E. How intramembrane proteases bury hydrolytic reactions in the membrane. Nature. 2009;459:371–378. doi: 10.1038/nature08146. [DOI] [PubMed] [Google Scholar]
- Grossfield A, Feller SE, Pitman MC. Convergence of molecular dynamics simulations of membrane proteins. Proteins. 2007;67:31–40. doi: 10.1002/prot.21308. [DOI] [PubMed] [Google Scholar]
- Holyoake J, Sansom MS. Conformational change in an MFS protein: MD simulations of LacY. Structure. 2007;15:873–884. doi: 10.1016/j.str.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Kandt C, Schlitter J, Gerwert K. Dynamics of water molecules in the bacterior-hodopsin trimer in explicit lipid/water environment. Biophys J. 2004;86:705–717. doi: 10.1016/S0006-3495(04)74149-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide K, Maegawa S, Ito K, Akiyama Y. Environment of the active site region of RseP, an Escherichia coli regulated intramembrane proteolysis protease, assessed by site-directed cysteine alkylation. J Biol Chem. 2007;282:4553–4560. doi: 10.1074/jbc.M607339200. [DOI] [PubMed] [Google Scholar]
- Lemieux MJ, Fischer SJ, Cherney MM, Bateman KS, James MN. The crystal structure of the rhomboid peptidase from Haemophilus influenzae provides insight into intramembrane proteolysis. Proc Natl Acad Sci U S A. 2007;104:750–754. doi: 10.1073/pnas.0609981104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomize AL, Pogozheva ID, Lomize MA, Mosberg HI. Positioning of proteins in membranes: a computational approach. Protein Sci. 2006;15:1318–1333. doi: 10.1110/ps.062126106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ, Wolfe MS. Presenilin: running with scissors in the membrane. Cell. 2007;131:215–221. doi: 10.1016/j.cell.2007.10.012. [DOI] [PubMed] [Google Scholar]
- Shokhen M, Khazanov N, Albeck A. Screening of the active site from water by the incoming ligand triggers catalysis and inhibition in serine proteases. Proteins. 2008;70:1578–1587. doi: 10.1002/prot.21727. [DOI] [PubMed] [Google Scholar]
- Tieleman DP, Forrest LR, Sansom MS, Berendsen HJ. Lipid properties and the orientation of aromatic residues in OmpF, influenza M2, and alamethicin systems: molecular dynamics simulations. Biochemistry (Mosc) 1998;37:17554–17561. doi: 10.1021/bi981802y. [DOI] [PubMed] [Google Scholar]
- Urban S. Rhomboid proteins: conserved membrane proteases with divergent biological functions. Genes Dev. 2006;20:3054–3068. doi: 10.1101/gad.1488606. [DOI] [PubMed] [Google Scholar]
- Urban S. Making the cut: central roles of intramembrane proteolysis in pathogenic mi-croorganisms. Nature Reviews Microbiology. 2009;7:411–423. doi: 10.1038/nrmicro2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban S. Taking the plunge: integrating structural, enzymatic and computational insights into a unified model for membrane-immersed rhomboid proteolysis. Biochem J. 2010;425:501–512. doi: 10.1042/BJ20090861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban S, Baker RP. In vivo analysis reveals substrate-gating mutants of a rhomboid intramembrane protease display increased activity in living cells. Biol Chem. 2008;389:1107–1115. doi: 10.1515/BC.2008.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban S, Shi Y. Core principles of intramembrane proteolysis: comparison of rhomboid and site-2 family proteases. Curr Opin Struct Biol. 2008;18:432–441. doi: 10.1016/j.sbi.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban S, Wolfe MS. Reconstitution of intramembrane proteolysis in vitro reveals that pure rhomboid is sufficient for catalysis and specificity. Proc Natl Acad Sci U S A. 2005;102:1883–1888. doi: 10.1073/pnas.0408306102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. Journal of computational chemistry. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- van Gunsteren WF, Berendsen HJ. Thermodynamic cycle integration by computer simulation as a tool for obtaining free energy differences in molecular chemistry. J Comput Aided Mol Des. 1987;1:171–176. doi: 10.1007/BF01676960. [DOI] [PubMed] [Google Scholar]
- van Gunsteren WF, Berendsen HJC. In: GROMOS-87 Manual. Biomos BV, editor. Groningen; The Netherlands: 1987. [Google Scholar]
- Vinothkumar KR. Structure of rhomboid protease in a lipid environment. J Mol Biol. 2011;407:232–247. doi: 10.1016/j.jmb.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Maegawa S, Akiyama Y, Ha Y. The role of L1 loop in the mechanism of rhomboid intramembrane protease GlpG. J Mol Biol. 2007;374:1104–1113. doi: 10.1016/j.jmb.2007.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang Y, Ha Y. Crystal structure of a rhomboid family intramembrane protease. Nature. 2006;444:179–180. doi: 10.1038/nature05255. [DOI] [PubMed] [Google Scholar]
- Wolfe MS. Intramembrane proteolysis. Chem Rev. 2009;109:1599–1612. doi: 10.1021/cr8004197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Yan N, Feng L, Oberstein A, Yan H, Baker RP, Gu L, Jeffrey PD, Urban S, Shi Y. Structural analysis of a rhomboid family intramembrane protease reveals a gating mechanism for substrate entry. Nature structural & molecular biology. 2006;13:1084–1091. doi: 10.1038/nsmb1179. [DOI] [PubMed] [Google Scholar]
- Yesylevskyy SO. ProtSqueeze: simple and effective automated tool for setting up membrane protein simulations. Journal of chemical information and modeling. 2007;47:1986–1994. doi: 10.1021/ci600553y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Cα RMSD value versus time for the loops in closed state of simulations GlpG1 (A), GlpG2 (B), open state of simulations GlpG3 (C) and GlpG4 (D).
Figure S2: Cα RMSD value versus time for the helices in closed state of simulations GlpG1 fitted to TM1-TM4+TM6 of open state (A), GlpG2 (B), open state of simulations GlpG3 fitted to closed state (C) and GlpG4 (D).
Figure S3: Cα RMSD value versus time for the loops in closed state of simulations GlpG1 fitted to TM1-TM4+TM6 of open state (A), GlpG2 (B), open state of simulations GlpG3 fitted to closed state (C) and GlpG4 (D).
Figure S4: MD trajectory of the GlpG center of mass along the Z axis relative to the center of mass of the lipid bilayer. Closed state of simulations GlpG1 (A), GlpG2 (B), open state of simulations GlpG3 (C) and GlpG4 (D).
Figure S5: A mean water density of the triple water cluster and interaction of surrounding residues. The water positions in the x-ray structural models are in black spheres. Closed state of simulation GlpG2 (left) and open state of simulation GlpG4 (right).
Table S1: H-bond interactions at the active site (The values indicate percentage of total simulation time for which a given H-bond was present. An interaction is recorded only if it was present for >20% in at least one simulation).
Molecular dynamics simulation of GlpG in a POPE bilayer. The catalytic serine and histidine are shown in space-fill.
Water dynamics during a MD simulation, starting from waters observed in the crystal structure. Note that the majority of crystallographic waters rapidly escaped, with no bulk waters entering to take their place. In contrast, several water molecules were retained near the catalytic center in a localized site (the ‘water retention’ site). The catalytic serine and histidine are shown in stick format. GlpG is simulated in a POPE bilayer, but phospholipids were removed from the movie for the sake of clarity.
Water dynamics in the active site of GlpG during a MD simulation. Note that the waters are dynamic, but constantly interacting with GlpG (side chains are shown in stick format). Only one water molecule (in blue and white) entered from bulk and only in this one of the four simulations. This water molecule remained at the active site due to its interactions with GlpG residues of the ‘water retention’ site.