

Abstract
Chagas disease is a Trypanosoma cruzi-induced zoonosis that has no natural cure. Local damage induced by the parasite and the immune response causes chronic heart and digestive lesions. Efforts to develop a therapeutic vaccine that boosts the immune response to completely clear the parasite are needed because there is no effective treatment for chronically infected patients. In an attempt to modify the host-parasite equilibrium to increase parasite destruction, we analyzed cardiopathy and the immune response in chronically infected mice that were challenged with live homologous parasites. Challenge with a single dose of parasite increased CD4+ and CD8+ T cell populations, gamma interferon (IFN-γ) production, and serum-specific IgG levels. However, subpatent parasitemias and cardiac tissue were not affected. Because of the short duration of the immune boost after a single challenge, we next evaluated the impact of four parasite doses, administered 3 weeks apart. At 1 to 2 months after the last dose, the numbers of CD4+ T cells and IFN-γ-producing CD4+ memory cells and the CD4+ T cell proliferative response to T. cruzi antigen were increased in the spleen. The frequency of IFN-γ-producing CD8+ memory cells in the blood was also increased. However, the sustained challenge did not favor TH1 development; rather, it induced an increase in serum-specific IgG1 levels and mixed TH1/TH2 cytokine production. Moreover, there were no significant changes in cardiac lesions and subpatent parasitemias. In conclusion, we believe that this study may help in elucidating the necessary elements for a successful therapeutic vaccine which may reduce cardiomyopathy in chronically infected human patients.
INTRODUCTION
Chagas disease is a Latin America zoonosis that is caused by the flagellate protozoan Trypanosoma cruzi (1). The most severe consequences of this disease occur in the chronic phase, with the establishment of digestive megasyndromes and/or myocarditis, with death due to arrhythmia or congestive heart failure eventually occurring in the last case. Importantly, no spontaneous cure occurs in either humans or experimental models.
In spite of the development of a specific immune response that allows control of the acute infection, total elimination of T. cruzi does not occur (2), inasmuch as a small number of parasites remain, leading the infection to a state of chronicity. The incomplete clearance of T. cruzi is strictly related to the strategies developed by the parasite to evade the immune system, such as escaping from the parasitophorous vacuole to the cytosol in mononuclear phagocytes (3), evading complement-dependent lysis (4, 5), interfering with the intracellular signaling pathway of infected structural cells (6), and hijacking transforming growth factor β (TGF-β) (7), or is due to the host deficiencies with respect to constraining the infection, such as the negative regulation of effector leukocytes that results in CD4+ T cell senescence and the loss of the effector activity of tissue-infiltrating CD8+ T cells (8, 9) and the immunodominance of an anti-T. cruzi CD8+ effector T cell response (10, 11).
In the previous decade, government efforts designed for housing amelioration and vector control have led to a sharp reduction in T. cruzi infections and almost complete elimination of new cases in countries of endemicity such as Uruguay, Chile, and Brazil (12). These policies have also been implemented in other American countries, and it is expected that the peridomiciliar form of the infection will be eradicated in the near future. Nevertheless, estimates indicate that 8 million people have already been infected and that 30 to 40% will suffer from the life-threatening complications of chronic infection. The options for chronic patients are limited because specific chemotherapy treatments do not result in the positive outcomes observed in patients in the acute phase of the disease (1, 13). Therefore, there is a great need to develop a therapeutic vaccine that can increase the ability of the immune system to eliminate T. cruzi infection.
On the basis of these considerations, we sought to enhance the specific immune response by challenging chronically infected mice with homologous parasites. Previously, we showed that mice that were chronically infected with the Y strain of T. cruzi presented a reduction in the subpatent parasitemia levels and enhanced anti-T. cruzi effector mechanisms when they were intravenously (i.v.) challenged with live homologous parasites (14). No effect of the parasite challenge on the heart could be analyzed in those experiments, since T. cruzi Y parasites rarely cause chronic cardiac inflammation. Therefore, to address the latter issue, we evaluated here the effects of homologous parasitic challenge on C3H/HePAS mice that were chronically infected with Sylvio X10/4 parasites, an experimental model that shows strong chronic cardiopathy with many characteristics that resemble the human disease (15). Two opposite nonexclusive effects could be anticipated after the challenge; on the one hand, part of the injected parasites could colonize the host cells and increase tissue damage; on the other hand, they could also boost the immune response and facilitate T. cruzi elimination. Nevertheless, neither a single dose of homologous parasites nor a sustained four-dose challenge was able to cause reproducible changes over the course of the chronic cardiac inflammation.
MATERIALS AND METHODS
Mice, parasites, and infection.
Six-to-8-week-old C3H/HePAS male and female mice were bred under specific, pathogen-free conditions at the Isogenic Mouse Facility of the Biomedical Sciences Institute of the University of São Paulo, São Paulo, Brazil. The T. cruzi Sylvio X10/4 clone (16) was maintained by in vitro infection of LLCMK2 cells. Mice were infected with 105 trypomastigotes by the intraperitoneal (i.p.) route. All experiments were carried out in accordance with the ethical guidelines for experiments with mice provided by the Animal Experimentation Brazilian College (COBEA), and the protocols were approved by the Health Animal Committee (CEEA) of the University of São Paulo.
T. cruzi antigen preparation.
Sylvio X10/4 T. cruzi parasites in LLCMK2 culture supernatant were washed in sterile phosphate-buffered saline (PBS) and subjected to 20 freeze-thaw cycles. The antigen concentration was adjusted to 1 × 108 parasites/ml, and the preparation was stored at −20°C.
Peripheral blood and spleen cell suspensions.
Blood mononuclear cells, isolated by Percoll gradient separation of heparinized blood obtained by cardiac puncture, and spleen cells, obtained from homogenized spleens, were maintained in RPMI 1640 supplemented with penicillin (100 U/ml), streptomycin (100 μg/ml), 2-mercaptoethanol (50 μM), l-glutamine (2 mM), sodium pyruvate (1 mM), and 3% heat-inactivated fetal calf serum (FCS). All supplements were purchased from Life Technologies. Cell number was estimated using a Neubauer chamber.
Cell phenotyping.
A total of 1 × 106 spleen and blood mononuclear cells from individual mice were stained with fluorescein isothiocyanate (FITC)-, phycoerythrin (PE)-, and Cy-chrome-labeled monoclonal antibodies (MAb) (BD Pharmingen) to CD4 (H129.19), CD8 (53-6.7), and B220 (RA3-6B2), respectively. Cells were analyzed by flow cytometry using a FACSCalibur flow cytometer and CELLQUEST software (Becton, Dickinson).
Intracellular staining of Foxp3.
Spleen cells (1 × 106) were surface stained with fluorochrome-labeled anti-CD4 and anti-CD25 (PC61) MAbs. The cells were permeabilized and intracellularly stained for FoxP3 according to the manufacturer guidelines (catalog no. 12-5773-82; eBioscience), washed, and analyzed by flow cytometry.
Intracellular staining of IFN-γ.
A total of 1 × 106 spleen cells from individual mice or mononuclear cell suspensions from pooled blood of mice in the same group were cultured with GolgiStop according to the manufacturer's guidelines (catalog no. 55471; BD Pharmingen) in the presence of 5 μg/ml of plate-bound, purified anti-CD3 MAb (145-2C11) or medium alone for 10 h at 37°C in a 5% CO2 atmosphere. After washing, cells were stained with FITC- or Cy-Chrome-labeled MAb to surface CD4 and CD8. Cells were fixed with Cytofix/Cytoperm buffer and incubated with a PE-labeled MAb to gamma interferon (IFN-γ) (XMG-1.2) diluted in Perm/Wash buffer. Subsequent analysis was performed by flow cytometry. All reagents were purchased from BD Pharmingen.
CFSE proliferation assay.
CD4+ and CD8+ T cell proliferation was measured as previously described (17). Briefly, 20 × 106 cells/ml in PBS–0.1% bovine serum albumin (BSA) were incubated with 5,6-carboxy fluorescein succinimidyl ester (CFSE; Molecular Probes) at a final concentration of 2.5 μM, for 20 min at 37°C. Cells (1 × 106) were then cultured in 96-well plates (Costar) with a T. cruzi antigen dose equivalent to 2.5 × 106 parasites, anti-CD3 (10 μg/ml)/anti-CD28 (2 μg/ml) MAb (BD Pharmingen), or medium alone for 96 h, at 37°C in a 5% CO2 atmosphere. After incubation, cells were stained with PE- or Cy-chrome-labeled MAb to CD4 and CD8 and analyzed by flow cytometry.
T. cruzi-specific ELISA.
Anti-T. cruzi antibodies were quantified by enzyme-linked immunosorbent assay (ELISA) as previously described (18). In brief, 96-well flat-bottom microtest plates (Costar) were coated overnight (4°C) with a T. cruzi parasite antigen dose equivalent to 5 × 106 parasites. Plates were saturated with 1% bovine serum albumin (BSA) for 1 h. After washing, 100 μl of mouse serum samples (diluted 800× to 12,000×) was added and left for 90 min at room temperature. Plates were incubated using goat anti-mouse IgG1 or IgG2a peroxidase-conjugated antibodies (Southern Biotechnology Associates) for 1 h. After washing, 100 μl of tetra-methyl-benzidine (TMB; Zymed) was added to each well, and the absorbance values were quantified after 15 min by a Spectra Max 190 spectrophotometer (Molecular Devices) with a 650-nm-wavelength filter.
Cytokine analysis in culture supernatants.
IFN-γ, tumor necrosis factor alpha (TNF-α), interleukin-2 (IL-2), IL-4, IL-5, and IL-10 levels in spleen cell culture supernatant after stimulation with a T. cruzi antigen dose equivalent to 2.5 × 106 parasites for 48 to 72 h were estimated by the use of a cytometric bead array (CBA) Th1/Th2 kit (BD Pharmingen) following manufacturer guidelines. Briefly, 50-μl aliquots of supernatant were added to a pool of capture beads specific for each cytokine. Subsequently, phycoerythrin-fluorochrome-labeled detection reagent was added to these mixtures. After a 2 h of incubation at room temperature in the dark, bead mixtures were washed, and the pellets were analyzed by flow cytometry. The cytokine concentration in the supernatant was determined by the mean fluorescence intensity in fluorescence detector 2 in relation to titration curves obtained for each cytokine.
Analysis of subpatent parasitemia.
T. cruzi parasites in the blood of individual chronically infected mice were detected by triplicate cultures of 5-μl blood aliquots in axenic liver infusion tryptose (LIT) medium (1 ml/well) incubated at 28°C. Cultures were screened for epimastigote growth twice a week during 1 month.
Histopathology.
Mice were deeply anesthetized and bled to death by cardiac puncture. The heart was collected, fixed in 10% formalin, and preserved in paraffin. Six 5-μm-thick nonconsecutive slides were stained with hematoxylin-eosin for microscopy. According to the intensity of infiltrates in different areas of the heart, the following scores were established: absent (0), discrete (1), moderate (2), intense (3), very intense (4), and severe (5).
Statistical analysis.
Statistical analysis was performed with a t test and 2-way analysis of variance (ANOVA), using Graph Pad Prism 4 software. Differences between two groups were statistically considered significant when the P value was < 0.05.
RESULTS
Single homologous challenge boosts the immune response but does not affect cardiac lesion.
Sylvio X10/4 is a low-virulence T. cruzi clone that yields no patent parasitemia in immunocompetent mice but induces low-level subpatent parasitemia that is revealed by amplification methods (19). Challenging Sylvio X10/4 chronically infected mice with a single i.v. dose of homologous parasites (5 × 106 trypomastigotes) increased the subpatent parasitemia, which peaked 1 h postchallenge (p.c.) and then declined to the baseline levels of chronic mice (data not shown). Ten days after the challenge, chronically infected mice presented increased frequencies and numbers of CD4+ and CD8+ cells, but not B (B220+) cells, in the spleen, subsiding by day 60 p.c., when these values were similar to those observed in unchallenged chronic mice (see Fig. S1A in the supplemental material). Of note, a transient increase in the frequency of CD4+ and CD8+ cells was also observed in the blood of challenged mice (see Fig. S1B in the supplemental material). Significant augmentations of the numbers of CD4+ and CD8+ splenocytes that were producing IFN-γ spontaneously or after anti-CD3/CD28 stimulus were also observed 10 days p.c., but not by day 60 p.c. (Fig. 1A). Likewise, a transient increase in the frequency of IFN-γ-producing CD4+ and CD8+ cells was observed in the blood, but, differently from the spleen, it was detected only after in vitro stimulation with anti-CD3/CD28 MAbs (see Fig. S1C in the supplemental material). As previously reported (20), IgG2a was the dominant subclass of serum antibody in chronic mice that were infected with Sylvio X10/4 T. cruzi even after the parasite challenge (Fig. 1B). However, enhanced levels of both IgG2a and IgG1 anti-T. cruzi serum antibodies were observed on day 10 p.c.; this effect was no longer observed by day 60 p.c. The kinetics of the anti-T. cruzi IgG1 and IgG2a antibody serum levels are shown in Fig. S1D in the supplemental material, where the challenge resulted in a brief alteration of antibody levels between days 7 and 30 p.c. In the experiment shown, the challenge-induced increase of IgG1 was more robust than that observed with IgG2a.
Fig 1.

Effect of a single challenge of chronically infected mice with homologous T. cruzi parasites. Sylvio X10/4-infected chronic mice that were subjected to i.v. challenge with a single dose of homologous parasites and unchallenged chronically infected mice were euthanized at 10 and 60 days p.c. (A) Numbers of spontaneous and anti-CD3/CD28-induced IFN-γ-producing CD4+ and CD8+ cells in the spleen; (B) anti-T. cruzi IgG2a and IgG1 antibody serum levels (the 1/12,000 dilution is shown) (O.D, optical density); (C) intensity of inflammatory infiltrates in the pericardium, endocardium, myocardium, and global heart tissue. Data represent the results determined with unchallenged (■) and challenged (□) chronically infected mice. *, P < 0.05; **, P < 0.01 (compared to unchallenged chronically infected mice). Representative experiments are shown.
Chronic mice that were challenged with a single dose of homologous parasites were then analyzed for the presence of cardiac lesions. It is known that, during chronic infection with Sylvio X10/4 parasites, C3H/HePAS mice develop an intense infiltration of mononuclear cells into the heart tissue that affects the pericardium, endocardium, and myocardium accompanied by the occasional presence of parasite nests (15). Challenging C3H/HePAS mice with homologous parasites did not modify the intensity of mononuclear cell infiltration in the heart after 10 or 60 days (Fig. 1C), which remained very high (nearly 4 points) according to our score.
Effect of sustained challenge with homologous parasites on the immune response of chronically infected mice.
We considered the possibility that continuous stimulation of the immune system was necessary to modulate the inflammatory response in the cardiac tissue because challenging chronically infected mice with a single parasite dose resulted in a brief boost in the immune response that did not affect the level of cardiac lesion (Fig. 1C). To test this possibility, Sylvio X10/4 chronically infected C3H/HePAS mice (infected for 3 to 4 months) were challenged with four doses of 5 × 106 parasites given at 3-week intervals. The first two doses were given i.v., and the other two were given i.p. The effects of these challenges were analyzed 1 to 2 months after the last dose. Sustained challenge increased the number of CD4+ cells but not the number of CD8+ cells in the spleen of chronically infected mice (Fig. 2A). In addition, while sustained challenge did not affect the spontaneous production of IFN-γ, it increased the number of IFN-γ-secreting CD4+ cells after in vitro stimulation with anti-CD3/anti-CD28 MAbs (Fig. 2B). No major changes were observed after sustained challenge in the frequencies of CD4+, CD8+, and B220+ cells in the bloodstream. However, challenged mice showed an increase in the frequency of CD8+ blood cells but not of CD4+ blood cells that produced IFN-γ after in vitro anti-CD3/CD28 stimulation (see Fig. S2 in the supplemental material).
Fig 2.
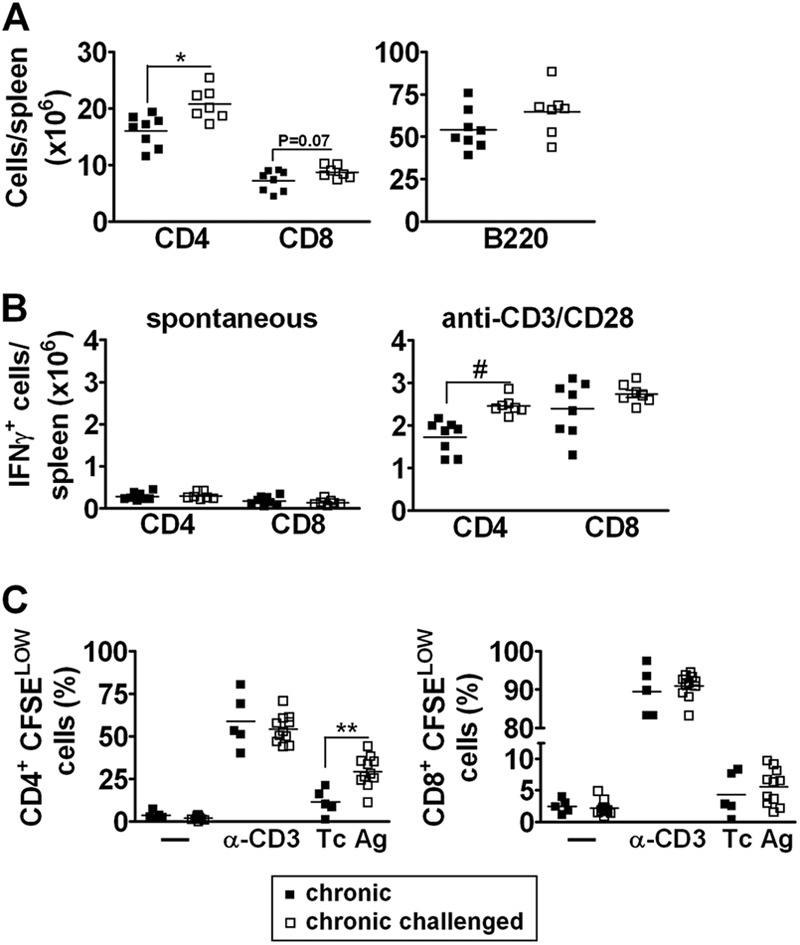
Analysis of CD4+ and CD8+ splenocytes in chronically infected mice that were subjected to sustained challenge with homologous T. cruzi parasite cells. Unchallenged mice, chronically infected mice, and chronically infected mice that were submitted to sustained challenge with homologous parasites were euthanized 1 to 2 months after the last challenge dose and the spleen cell suspensions analyzed for (A) total numbers of CD4+, CD8+, and B (B220+) cells, (B) numbers of CD4+ and CD8+ cells producing IFN-γ, either spontaneously or after in vitro stimulation for 12 h with anti-CD3/anti-CD28 MAbs, and (C) frequencies of proliferating (CFSELOW) CD4+ and CD8+ cells in response to stimulation with anti-CD3 (α-CD3)/anti-CD28 MAbs, T. cruzi antigen (Tc Ag), or medium alone (—). Data represent the results determined with unchallenged (■) and challenged (□) chronically infected mice. *, P < 0.05; *, P < 0.01; #, P < 0.001 (compared to unchallenged chronically infected mice). Representative experiments are shown.
The spleen cell response in chronically infected mice subjected to sustained antigenic challenge was further evaluated by measuring the T cell proliferative response after in vitro stimulation. Spleen cells taken from challenged and unchallenged mice were labeled with CFSE and cultured for 96 h in medium (spontaneous proliferation) or with anti-CD3/anti-CD28 MAbs or T. cruzi antigen. The frequency of proliferating cells was estimated by the percentage of CD4+ and CD8+ cells with reduced CFSE levels. Compared to those from unchallenged mice, CD4+ cells from challenged mice showed an increased proliferative response to T. cruzi antigen (Fig. 2C). However, the proliferative responses of CD8+ cells were not different between unchallenged and challenged mice. Overall, our results demonstrate that a sustained challenge with homologous parasites increased the number of parasite-specific CD4+ memory cells in the spleen and CD8+ memory cells in the blood. This reinforces the notion that challenge induces a boost of the specific immune response to infection.
Sustained challenge promotes an increased production of both TH1 and TH2 cytokines, IgG1 antibody levels, and regulatory T (TREG) cells.
Control of T. cruzi parasite infection requires a dominant TH1 response, characterized by the production of IFN-γ and a shift to IFN-γ-dependent IgG2a antibodies (21, 22). To indirectly evaluate the impact of a sustained parasite challenge on the effector cytokine pattern in chronically infected mice, we analyzed the specific IgG1 and IgG2a serum levels in unchallenged and challenged mice. Figure 3A shows that in challenged mice, the serum levels of anti-T. cruzi IgG1 antibodies were notably increased, while T. cruzi IgG2a antibody levels were increased in just one of the four experiments that were performed (data not shown).
Fig 3.
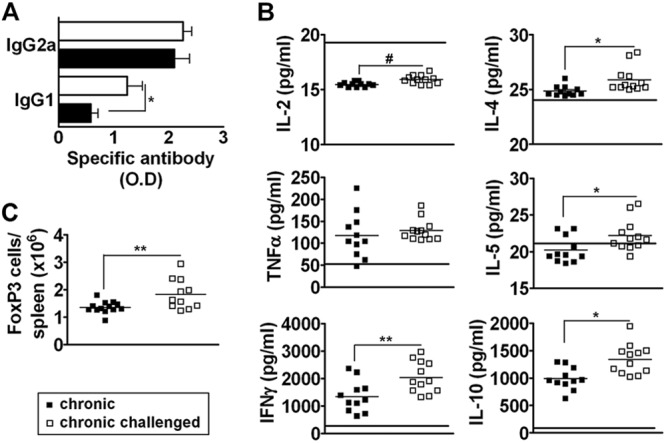
Analysis of functional polarization in chronically infected mice that were subjected to sustained challenge with homologous T. cruzi parasite cells. Unchallenged mice, chronically infected mice, and chronically infected mice that were submitted to sustained challenge with homologous parasites were bled and euthanized 1 to 2 months after the last challenge dose and analyzed for (A) T. cruzi-specific serum IgG2a and IgG1 antibody levels (O.D values, 1/12,000 dilution), (B) in vitro T. cruzi antigen-induced production of TH1 and TH2 cytokines by spleen cells, and (C) the numbers of CD4+ FoxP3+ cells in the spleen. Data represent the results determined with unchallenged (■) and challenged (□) chronically infected mice. In panel B, the horizontal bars represent the mean values of cytokine levels in T. cruzi antigen-stimulated spleen cell cultures from noninfected mice. *, P < 0.05; **, P < 0.01; #, P < 0.001 (compared to unchallenged chronically infected mice). Representative experiments are shown.
Because the switch to the IgG1 isotype is promoted by IL-4, a cytokine induced at low levels during T. cruzi infection, we wonder if a sustained challenge would alter the cytokine profile of chronically infected mice. Spleen cells from challenged and unchallenged mice were stimulated for 48 to 72 h with T. cruzi antigen, and the levels of cytokines were measured by the CBA. We observed that the sustained challenge promoted an increase in the production of IFN-γ and IL-2 by the splenocytes (Fig. 3B). However, IL-2 levels were lower in unchallenged or challenged mice than in the naïve ones (Fig. 3B, horizontal bars). A similar level of TNF-α production was observed in splenocyte cultures from challenged mice (Fig. 3B). Interestingly, sustained challenge promoted a significant increase in the production of the TH2 cytokines IL-10, IL-4, and IL-5. These results indicate that a sustained challenge boosts the immune response in a mixed TH1 and TH2 profile.
We also investigated whether regulatory cell subsets were expanded in challenged mice because both pro- and anti-inflammatory cytokines were induced by a sustained immunologic challenge. Spleen CD4+ cells from challenged and unchallenged mice were analyzed for the expression of Foxp3, a transcription factor that is characteristic of regulatory T (TREG) cells. Figure 3C shows that sustained challenge promoted an expansion of TREG cells, most of which displayed the IL-2 receptor alpha chain CD25 (data not shown). These results reinforce the notion that repeated stimulation with homologous parasites boosts the regulatory circuits.
Effects of sustained challenge on subpatent parasitemia and cardiopathy in chronically infected mice.
Our next step was to evaluate the effect of sustained challenge with homologous parasites on subpatent parasitemia and cardiopathy of Sylvio X10/4 chronically infected mice. Four independent experiments were performed with 34 challenged and 30 unchallenged mice (Fig. 4). Subpatent parasitemias were estimated by determining the percentage of positive cultures in LIT medium using 5-μl blood aliquots from each mouse. In two of the experiments, chronically infected mice that were subjected to sustained challenge displayed a trend toward a decrease in subpatent parasitemia. In the other two experiments, the opposite result was observed, that is, an increase in parasitemia levels or a tendency for increased parasitemia after the four rounds of homologous challenge (Fig. 4A). Similarly, the effects of a sustained challenge with homologous parasites on cardiac lesion were not consistent, since we observed a small decrease in global cardiopathy in two experiments, an augmentation of pericarditis and a tendency for higher global cardiopathy in a third experiment, and no major changes in the fourth experiment (Fig. 4B). Overall, our results indicate that sustained immunologic challenge does not consistently influence the inflammatory reaction in the heart of chronically infected mice. However, in spite of the variation observed in the results, a correlation was seen in the effects of the sustained challenge regarding the parasitemia and the cardiopathy. Thus, in the two experiments where the challenge decreased the cardiac compromise, a trend for lower parasitemia was observed. Additionally, in the experiment where the challenge enhanced the parasitemia, an increased level of pericarditis and a tendency for augmented global cardiopathy were evidenced.
Fig 4.

Effect of sustained challenge with homologous parasites on the levels of subpatent parasitemia and cardiac pathology in chronically infected mice. Unchallenged chronically infected mice and chronically infected mice that were submitted to sustained challenge with homologous Sylvio X10/4 parasites were euthanized 1 to 2 months after the last challenge dose to evaluate (A) the subpatent parasitemia levels by culture of blood aliquots from individual mice in LIT medium and (B) the intensity of pericarditis, endocarditis, myocarditis, and global heart inflammation. Data represent the results determined with unchallenged (■) and challenged (□) chronically infected mice. *, P < 0.05 compared to unchallenged chronically infected mice.
DISCUSSION
The persistence of T. cruzi in the heart is considered the main underlying factor behind chronic chagasic cardiomyopathy (23–25). In consequence, a great number of experimental studies pursued the goal of reducing the parasite load as a means of diminishing or eliminating the chronic cardiopathy (26) by applying different approaches, such as prophylactic or therapeutic vaccines based on recombinant T. cruzi proteins (27, 28), naked parasite DNA (29), or recombinant viral vectors carrying T. cruzi genes (30–32).
To investigate the requirements to diminish the cardiac lesion in chronically infected hosts, we evaluated the effect of homologous challenge on C3H/HePAS mice that were chronically infected with Sylvio X10/4 parasites, a model that yields strong chronic myocarditis (14). We hypothesized that if parasite challenge reduced the systemic and the cardiac tissue parasite load in chronically infected mice, it should result in the long term in decreased cardiac inflammation and damage. We first evaluated the effects of challenge with a single parasite dose and observed increased specific serum IgG levels as well as the spontaneous IFN-γ production by CD4+ and CD8+ cells in the spleen and blood. Nonetheless, these were transient boosts and were not noticeable 60 days after challenge. More importantly, we did not observe changes in the cardiopathy, with the intensity of mononuclear cell infiltrates seen in the cardiac tissue similar to that in unchallenged mice.
Bearing in mind that the immune-mediated destruction of parasites in the heart occurs preferentially following nest rupture and that the rupture of a significant number of nests is a process that takes a long period of time (33), we thought that the failure of single-dose challenge to modify the heart inflammation could be explained by the brevity of the boosted response. With the aim of inducing a long-lasting modification of the immune response, chronically infected mice were submitted to a sustained challenge with four parasite doses, and the effects on immune parameters were analyzed 1 to 2 months after the last dose. Sustained challenge promoted a significant increase in IFN-γ-producing memory CD4+ cells in the spleen and IFN-γ-producing memory CD8+ cells in the blood. It also increased the parasite-specific IgG. Meanwhile, its effects on cardiac pathology were not consistent. Moreover, the effects of the sustained challenge in the levels of systemic parasitism differed in the different experiments. Yet, a correlation was observed in the effect of sustained challenge upon blood parasitism and cardiac pathology, a result that suggests a connection between the two parameters.
Different reasons can be hypothesized to explain why sustained challenge did not modify the cardiopathy, among which we should not discard the remote possibility of increased heart tissue infection by the newly inoculated parasites. More likely, we should consider that the boosted immune response in challenged mice was quantitatively and/or qualitatively insufficient to significantly diminish the levels of parasites that were present in the bloodstream or in the tissues. A qualitative defect is suggested by the fact that the boosted immune response of challenged mice shifted away from the protective TH1 response in chronically infected mice. Thus, the increase in T. cruzi-specific serum antibody levels occurred mainly for IgG1, an IL-4-dependent isotype (34) that shows lower opsonizing and complement activation capacities compared to IgG2a. Moreover, the boosted response included increases in both TH1 (IFN-γ, IL-2) and TH2 (IL-4, IL-5) cytokines, anti-inflammatory molecules (IL-10), and TREG cell population. A shift away from the TH1 lineage suggests that the host did not identify the challenged inocula as a dangerous stimulus that could easily be eliminated (35, 36). Destruction of the challenge inocula might have occurred because the injected trypomastigotes bound to specific IgG and were subsequently killed by phagocytic cells in the liver, lung, spleen, or peritoneal cavity, and the signal generated in this process might have not been sufficient to fully activate the immune response. Supporting this interpretation, we have previously shown that, following i.v. challenge of Y strain-infected chronic mice with homologous parasites, the liver parenchyma is infiltrated by a high number of memory CD8+ cells, but very few CD8+ effector cells (19). Another reason that could contribute to the absence of a consistent modification of the cardiopathy after sustained challenge is that the boosted immune response would be restricted to the blood, lungs, liver, and lymphoid organs (19); therefore, its impact upon the local inflammatory response in the heart could have been negligible. Thus, even if the levels of T. cruzi-specific IgG in the heart tissue would be similar to those observed in the serum, the same might not have occurred regarding the inflammatory cells (effector T cells, monocytes, etc.), since an increased local signaling would be required for an increased cell recruitment.
Due to the absence of the effect of a single-dose or a sustained immunologic challenge upon heart infiltrates, we may hypothesize that boosting the parasite-specific immune response of chronically infected mice with T. cruzi extracts or purified or recombinant antigens would not be relevant in the treatment of the chronic cardiopathy because, as in the case of the homologous challenge, the boosted response would not find the requirements for reducing the parasite load in the tissues. We suggest that, for a therapeutic vaccine to be effective in reducing the effects of the chronic disease, it might require the continuous provision of danger signals, a means of inducing a selective, long-lasting boost in IFN-γ-producing and cytotoxic CD8+ cells, a requirement that could be met by immunization with viral vectors containing T. cruzi parasite sequences (32, 37, 38). In this respect, it is important to mention the recent observation of Vasconcelos et al. (38) that a vaccination with an adenovirus vector carrying the ASP-2 T. cruzi gene was able to overcome the qualitative deficits in the specific CD8+ response of T. cruzi-infected mice.
Currently, various therapeutic vaccine candidates have proven that a reduction in the systemic and local T. cruzi parasite load and a reduction in heart tissue inflammation are possible (31, 39, 40). Hence, these studies must be pursued, as they represent, alone or in combination with specific chemotherapy, the most promising candidates for treating the complications of the chronic human disease.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo) and CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico).
We extend our thanks to Rogério Silva do Nascimento, Meire Ioshie Hiyane, and Bernardo Paulo Albe for technical assistance.
Footnotes
Published ahead of print 12 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/CVI.00032-12.
REFERENCES
- 1. Lescure FX, Le Loup G, Freilij H, Develoux M, Paris L, Brutus L, Pialoux G. 2010. Chagas disease: changes in knowledge and management. Lancet Infect. Dis. 10:556–570 [DOI] [PubMed] [Google Scholar]
- 2. Dias JC, Dias E, Martins-Filho OA, Vitelli-Avelar D, Correia D, Lages E, Prata A. 2008. Further evidence of spontaneous cure in human Chagas disease. Rev. Soc. Bras. Med. Trop. 41:505–506 [DOI] [PubMed] [Google Scholar]
- 3. Nogueira N, Cohn Z. 1976. Trypanosoma cruzi: mechanism of entry and intracellular fate in mammalian cells. J. Exp. Med. 143:1402–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fischer E, Ouaissi MA, Velge P, Cornette J, Kazatchkine MD. 1988. Gp 58/68, a parasite component that contributes to the escape of the trypomastigote form of T. cruzi from damage by the human alternative complement pathway. Immunology 65:299–303 [PMC free article] [PubMed] [Google Scholar]
- 5. Kipnis TL, David JR, Alper CA, Sher A, da Silva WD. 1981. Enzymatic treatment transforms trypomastigotes of Trypanosoma cruzi into activators of alternative complement pathway and potentiates their uptake by macrophage. Proc. Natl. Acad. Sci. U. S. A. 78:602–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marinho CR, Nunez-Apaza LN, Bortoluci KR, Bombeiro AL, Bucci DZ, Grisotto MG, Sardinha LR, Jorquera CE, Lira S, Lima MR, Alvarez JM. 2009. Infection by the Sylvio X10/4 clone of Trypanosoma cruzi: relevance of a low-virulence model of Chagas' disease. Microbes Infect. 11:1037–1045 [DOI] [PubMed] [Google Scholar]
- 7. DosReis GA. 2011. Evasion of immune responses by Trypanosoma cruzi, the etiological agent of Chagas disease. Braz. J. Med. Biol. Res. 44:84–90 [DOI] [PubMed] [Google Scholar]
- 8. Albareda MC, Olivera GC, Laucella SA, Alvarez MG, Fernandez ER, Lococo B, Viotti R, Tarleton RL, Postan M. 2009. Chronic human infection with Trypanosoma cruzi drives CD4(+) T cells to immune senescence. J. Immunol. 183:4103–4108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leavey JK, Tarleton RL. 2003. Cutting edge: dysfunctional CD8(+) T cells reside in nonlymphoid tissues during chronic Trypanosoma cruzi infection. J. Immunol. 170:2264–2268 [DOI] [PubMed] [Google Scholar]
- 10. Martin DL, Weatherly DB, Laucella SA, Cabinian MA, Crim MT, Sullivan S, Heiges M, Craven SH, Rosenberg CS, Collins MH, Sette A, Postan M, Tarleton RL. 2006. CD8(+) T-cell responses to Trypanosoma cruzi are highly focused on strain-variant trans-sialidase epitopes. PLoS Pathog. 2:e77 doi:10.1371/journal.ppat.0020077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rodrigues MM, de Alencar BCG, Claser C, Tzelepis F. 2009. Immunodominance: a new hypothesis to explain parasite escape and host/parasite equilibrium leading to the chronic phase of Chagas' disease? Braz. J. Med. Biol. Res. 42:220–223 [DOI] [PubMed] [Google Scholar]
- 12. Schofield CJ, Jannin J, Salvatella R. 2006. The future of Chagas disease control. Trends Parasitol. 22:583–588 [DOI] [PubMed] [Google Scholar]
- 13. Marin-Neto JA, Rassi A, Jr, Morillo CA, Avezum A, Connolly SJ, Sosa-Estani S, Rosas F, Yusuf S, BENEFIT Investigators. 2008. Rationale and design of a randomized placebo-controlled trial assessing the effects of etiologic treatment in Chagas' cardiomyopathy: the BENznidazole Evaluation For Interrupting Trypanosomiasis (BENEFIT). Am. Heart J. 156:37–43 [DOI] [PubMed] [Google Scholar]
- 14. Marinho CRF, Bastos KRB, Sardinha LR, Grisotto MG, Lima MRD, Alvarez JM. 2004. Challenge of Trypanosoma cruzi chronically infected mice with trypomastigotes activates the immune system and reduces subpatent parasitemia levels. J. Parasitol. 90:516–523 [DOI] [PubMed] [Google Scholar]
- 15. Marinho CRF, Bucci DZ, Dagli MLZ, Bastos KRB, Grisotto MG, Sardinha LR, Baptista CRGM, Goncalves CP, Lima MRD, Alvarez JM. 2004. Pathology affects different organs in two mouse strains chronically infected by a Trypanosoma cruzi clone: a model for genetic a studies of Chagas' disease. Infect. Immun. 72:2350–2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Postan M, Dvorak JA, McDaniel JP. 1983. Studies of Trypanosoma cruzi clones in inbred mice. I. A comparison of the course of infection of C3H/HEN- mice with two clones isolated from a common source. Am. J. Trop. Med. Hyg. 32:497–506 [DOI] [PubMed] [Google Scholar]
- 17. Elias RM, Sardinha LR, Bastos KR, Zago CA, da Silva AP, Alvarez JM, Lima MR. 2005. Role of CD28 in polyclonal and specific T and B cell responses required for protection against blood stage malaria. J. Immunol. 174:790–799 [DOI] [PubMed] [Google Scholar]
- 18. Marinho CR, D'Imperio Lima MR, Grisotto MG, Alvarez JM. 1999. Influence of acute-phase parasite load on pathology, parasitism, and activation of the immune system at the late chronic phase of Chagas' disease. Infect. Immun. 67:308–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sardinha LR, Mosca T, Elias RM, do Nascimento RS, Goncalves LA, Bucci DZ, Marinho CR, Penha-Goncalves C, Lima MR, Alvarez JM. 2010. The liver plays a major role in clearance and destruction of blood trypomastigotes in Trypanosoma cruzi chronically infected mice. PLoS Negl. Trop. Dis. 4:e578 doi:10.1371/journal.pntd.0000578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Scott MT, Gosssampson M. 1984. Restricted Igg Isotype Profiles in T-cruzi Infected Mice and Chagas-Disease Patients. Clin. Exp. Immunol. 58:372–379 [PMC free article] [PubMed] [Google Scholar]
- 21. Marinho CRF, Nunez-Apaza LN, Martins-Santos R, Bastos KRB, Bombeiro AL, Bucci DZ, Sardinha LR, Lima MRD, Alvarez JM. 2007. IFN-gamma, but not nitric oxide or specific IgG, is essential for the in vivo control of low-virulence sylvio X10/4 Trypanosoma cruzi parasites. Scand. J. Immunol. 66:297–308 [DOI] [PubMed] [Google Scholar]
- 22. Snapper CM, Paul WE. 1987. Interferon-gamma and B cell stimulatory factor-1 reciprocally regulate Ig isotype production. Science 236:944–947 [DOI] [PubMed] [Google Scholar]
- 23. Palomino SAP, Aiello VD, Higuchi ML. 2000. Systematic mapping of hearts from chronic chagasic patients: the association between the occurrence of histopathological lesions and Trypanosoma cruzi antigens. Ann. Trop. Med. Parasitol. 94:571–579 [DOI] [PubMed] [Google Scholar]
- 24. Tarleton RL. 2001. Parasite persistence in the aetiology of Chagas disease. Int. J. Parasitol. 31:550–554 [DOI] [PubMed] [Google Scholar]
- 25. Vago AR, Macedo AM, Adad SJ, Reis DD, Corrêa-Oliveira R. 1996. PCR detection of Trypanosoma cruzi DNA in oesophageal tissues of patients with chronic digestive Chagas' disease. Lancet 348:891–892 [DOI] [PubMed] [Google Scholar]
- 26. Cazorla SI, Frank FM, Malchiodi EL. 2009. Vaccination approaches against Trypanosoma cruzi infection. Expert Rev. Vaccines 8:921–935 [DOI] [PubMed] [Google Scholar]
- 27. Fontanella GH, De Vusser K, Laroy W, Daurelio L, Nocito AL, Revelli S, Contreras R. 2008. Immunization with an engineered mutant trans-sialidase highly protects mice from experimental Tryponosoma cruzi infection: a vaccine candidate. Vaccine 26:2322–2334 [DOI] [PubMed] [Google Scholar]
- 28. Luhrs KA, Fouts DL, Manning JE. 2003. Immunization with recombinant paraflagellar rod protein induces protective immunity against Trypanosoma cruzi infection. Vaccine 21:3058–3069 [DOI] [PubMed] [Google Scholar]
- 29. Pereira-Chioccola VL, Costa F, Ribeirao M, Soares IS, Arena F, Schenkman S, Rodrigues MM. 1999. Comparison of antibody and protective immune responses against Trypanosoma cruzi infection elicited by immunization with a parasite antigen delivered as naked DNA or recombinant protein. Parasite Immunol. 21:103–110 [DOI] [PubMed] [Google Scholar]
- 30. Duan XF, Yonemitsu Y, Chou B, Yoshida K, Tanaka S, Hasegawa M, Tetsutani K, Ishida H, Himeno K, Hisaeda H. 2009. Efficient protective immunity against Trypanosoma cruzi infection after nasal vaccination with recombinant Sendai virus vector expressing amastigote surface protein-2. Vaccine 27:6154–6159 [DOI] [PubMed] [Google Scholar]
- 31. Limon-Flores AY, Cervera-Cetina R, Tzec-Arjona JL, Ek-Macias L, Sanchez-Burgos G, Ramirez-Sierra MJ, Cruz-Chan JV, VanWynsberghe NR, Dumonteil E. 2010. Effect of a combination DNA vaccine for the prevention and therapy of Trypanosoma cruzi infection in mice Role of CD4(+) and CD8(+) T cells. Vaccine 28:7414–7419 [DOI] [PubMed] [Google Scholar]
- 32. Rigato PO, de Alencar BC, de Vasconcelos JRC, Dominguez MR, Araujo AF, Machado AV, Gazzinelli RT, Bruna-Romero O, Rodrigues MM. 2011. Heterologous plasmid DNA prime-recombinant human adenovirus 5 boost vaccination generates a stable pool of protective long-lived CD8(+) T effector memory cells specific for a human parasite, Trypanosoma cruzi. Infect. Immun. 79:2120–2130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. de Souza EM, Araujo-Jorge TC, Bailly C, Lansiaux A, Batista MM, Oliveira GM, Soeiro MN. 2003. Host and parasite apoptosis following Trypanosoma cruzi infection in in vitro and in vivo models. Cell Tissue Res. 314:223–235 [DOI] [PubMed] [Google Scholar]
- 34. Vitetta ES, Brooks K, Chen YW, Isakson P, Jones S, Layton J, Mishra GC, Pure E, Weiss E, Word C, Yuan D, Tucker P, Uhr JW, Krammer PH. 1984. T-cell-derived lymphokines that induce Igm and Igg secretion in activated murine B-cells. Immunol. Rev. 78:137–157 [DOI] [PubMed] [Google Scholar]
- 35. Cohen IR. 1992. The cognitive principle challenges clonal selection. Immunol. Today 13:441–444 [DOI] [PubMed] [Google Scholar]
- 36. Matzinger P. 2002. An innate sense of danger. Ann. N. Y. Acad. Sci. 961:341–342 [DOI] [PubMed] [Google Scholar]
- 37. de Alencar BC, Persechini PM, Haolla FA, de Oliveira G, Silverio JC, Lannes-Vieira J, Machado AV, Gazzinelli RT, Bruna-Romero O, Rodrigues MM. 2009. Perforin and gamma interferon expression are required for CD4+ and CD8+ T-cell-dependent protective immunity against a human parasite, Trypanosoma cruzi, elicited by heterologous plasmid DNA prime-recombinant adenovirus 5 boost vaccination. Infect. Immun. 77:4383–4395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vasconcelos JR, Bruna-Romero O, Araujo AF, Dominguez MR, Ersching J, de Alencar BC, Machado AV, Gazzinelli RT, Bortoluci KR, Amarante-Mendes GP, Lopes MF, Rodrigues MM. 2012. Pathogen-induced proapoptotic phenotype and high CD95 (Fas) expression accompany a suboptimal CD8+ T-cell response: reversal by adenoviral vaccine. PLoS Pathog. 8:e1002699 doi:10.1371/journal.ppat.1002699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sanchez-Burgos G, Mezquita-Vega RG, Escobedo-Ortegon J, Ramirez-Sierra MJ, Arjona-Torres A, Ouaissi A, Rodrigues MM, Dumonteil E. 2007. Comparative evaluation of therapeutic DNA vaccines against Trypanosoma cruzi in mice. FEMS Immunol. Med. Microbiol. 50:333–341 [DOI] [PubMed] [Google Scholar]
- 40. Zapata-Estrella H, Hummel-Newell C, Sanchez-Burgos G, Escobedo-Ortegon J, Ramirez-Sierra MJ, Arjona-Torres A, Dumonteil E. 2006. Control of Trypanosoma cruzi infection and changes in T-cell populations induced by a therapeutic DNA vaccine in mice. Immunol. Lett. 103:186–191 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.