

Abstract
Plague caused by Yersinia pestis manifests itself in bubonic, septicemic, and pneumonic forms. Although the U.S. Food and Drug Administration recently approved levofloxacin, there is no approved human vaccine against plague. The capsular antigen F1 and the low-calcium-response V antigen (LcrV) of Y. pestis represent excellent vaccine candidates; however, the inability of the immune responses to F1 and LcrV to provide protection against Y. pestis F1− strains or those which harbor variants of LcrV is a significant concern. Here, we show that the passive transfer of hyperimmune sera from rats infected with the plague bacterium and rescued by levofloxacin protected naive animals against pneumonic plague. Furthermore, 10 to 12 protein bands from wild-type (WT) Y. pestis CO92 reacted with the aforementioned hyperimmune sera upon Western blot analysis. Based on mass spectrometric analysis, four of these proteins were identified as attachment invasion locus (Ail/OmpX), plasminogen-activating protease (Pla), outer membrane protein A (OmpA), and F1. The genes encoding these proteins were cloned, and the recombinant proteins purified from Escherichia coli for immunization purposes before challenging mice and rats with either the F1− mutant or WT CO92 in bubonic and pneumonic plague models. Although antibodies to Ail and OmpA protected mice against bubonic plague when challenged with the F1− CO92 strain, Pla antibodies were protective against pneumonic plague. In the rat model, antibodies to Ail provided protection only against pneumonic plague after WT CO92 challenge. Together, the addition of Y. pestis outer membrane proteins to a new-generation recombinant vaccine could provide protection against a wide variety of Y. pestis strains.
INTRODUCTION
Yersinia pestis, the causative agent of bubonic, septicemic, and pneumonic plague, is a Gram-negative zoonotic pathogen, and human infections are often acquired through the infected-flea vector carried by domestic and/or wild animals (1). The microorganism possesses multiple virulence determinants that are both chromosomally and extrachromosomally encoded on its three plasmids: pCD1, pMT1/pFra, and pPCP1/pPla (2). Although bubonic plague, inflicted by flea bites, in humans represents the most common form worldwide (http://www.gov.mb.ca/health/publichealth/cdc/protocol/plague.pdf), it can lead to secondary septicemic and pneumonic plague, with a nearly 100% mortality rate. Primary pneumonic plague results by inhaling aerosolized droplets charged with Y. pestis, and the organism can then rapidly spread from person to person, possibly leading to mass causalities (http://www.cdc.gov/ncidod/dvbid/plague/info.htm).
Presently, there is no licensed plague vaccine in the United States (3), which is alarming in the light of epidemics and potential bioterrorism threats associated with this lethal pathogen. The current subunit vaccines under investigation include the capsular antigen F1 and the low-calcium-response V antigen (LcrV). LcrV is part of the type 3 secretion system (T3SS) and is encoded by the pCD1 plasmid (3). F1 and LcrV vaccines are tested as either purified individual antigens or as a fusion of these proteins (F1-V or V-F1) (4–6; see also reference 3 and references therein). Although efficacious in rodents and some nonhuman primate models (4–6; see also reference 3 and references therein), these recombinant vaccines would not be as efficacious against F1− strains of Y. pestis that exist in nature and are as virulent as the wild-type (WT) Y. pestis (7, 8). However, since LcrV is highly immunogenic and represents a protective antigen of Y. pestis, anti-LcrV antibodies alone would provide significant protection to the host against challenge with the F1− strains even if antibodies to F1 were inconsequential. Indeed, antibodies to recombinant LcrV alone were shown to provide protection in mice against both bubonic and pneumonic plague caused by encapsulated and F1− Y. pestis strains (9), whereas antibodies to F1 protected animals from the F1+ strain (10). Likewise, it was shown that an F1-V fusion protein, as well as F1 or LcrV immunogens, individually protected mice against pneumonic plague induced by WT or its F1− virulent isogenic Y. pestis strain (11). In the same vein, Heath et al. (12) reported the protection of mice against bubonic and pneumonic plague by antibodies to an F1-V fusion protein against challenge with both F1+and F1− strains of Y. pestis. However, they noted that immunization of animals with the F1-V fusion protein led to better protection compared to mice that were vaccinated with either F1 or LcrV antigen alone against the F1+ strain (12).
More importantly, the sequence of LcrV has diverged in various Y. pestis strains, and thus antibody responses to one variant may not provide optimal protection against Y. pestis strains that have LcrVs with varied amino acid sequences (7, 8). In one study, an LcrV variant (LcrV5214), mutated for 5 amino acid residues (thus unable to interact with the Toll-like receptor 2), was expressed in a live-attenuated Salmonella enterica Typhimurium strain; when this recombinant Salmonella strain was tested as a potential oral vaccine in mice, it did not protect animals against developing bubonic plague (13). Later, Sun et al. (14) also indicated that when the native LcrV-encoding gene on the pCD1 plasmid was replaced with its variant LcrV2345 (with similar 5-amino-acid substitutions, as described above) in Y. pestis KIM6 strain (deleted of the pigmentation [pgm] locus) and both were used to induce bubonic and pneumonic plague in mice, no difference in bacterial virulence was observed between the two strains. Thus, immune responses only to F1 and LcrV to provide protection against Y. pestis F1− strains or against strains that harbor variants of LcrV are a real concern.
Considering the limitations associated with LcrV and F1 antigens mentioned above, it seems necessary that additional antigens be incorporated into novel recombinant plague vaccine preparations. In that vein, we used immunoblotting with rat hyperimmune sera, obtained from WT Y. pestis CO92-infected and levofloxacin-rescued animals, to identify novel plague antigens that could be incorporated into a new-generation subunit plague vaccine. Using mass spectrometric analysis, we identified four outer membrane protein (OMP) antigens, namely, F1, attachment-invasion locus (Ail/OmpX), plasminogen-activating protease (Pla), and outer membrane protein A (OmpA), to which the rat antisera reacted. Since anti-F1 antibodies are protective against pneumonic plague in a mouse model (see reference 3 and references therein), we primarily focused our studies on the other three OMPs (Ail/OmpX, OmpA, and Pla), of which two (Ail/OmpX and Pla) are bona fide virulence factors of Y. pestis (15, 16).
It is important to mention here that the genetic background of the mouse strains does contribute to their susceptibility or resistance to developing plague. For example, both F1 and fimbrial protein PsaA were needed for the full virulence of Y. pestis in terms of inducing bubonic and pneumonic plague in C57BL/6J mice (17). However, the Δcaf1 mutant exhibited a more attenuated phenotype in developing bubonic plague in C57BL/6J mice compared to BALB/cJ animals (17). Because of the diversity in the genetic makeup of humans, we evaluated whether the active immunization of outbred Swiss-Webster mice and Brown Norway rats with purified, recombinant Ail/OmpX, OmpA, and Pla could generate protective antibodies against bubonic and pneumonic plague induced by both F1− and WT CO92 strains.
Pla is a 312-amino-acid aspartate multifunctional protease encoded by the pPCP1 plasmid and has been shown to enhance bacterial adherence to the extracellular matrix (18, 19). In addition, Pla activates circulating human plasminogen into plasmin that degrades components of the extracellular matrix (20, 21), thus facilitating bacterial dissemination to peripheral organs. Pla also modulates bacterial virulence by degrading Yersinia OMPs and their secretion (22, 23) and by interacting with lipopolysaccharide, a major component of the outer membrane in Gram-negative bacteria (24).
Attachment invasion locus (Ail/OmpX) protein belongs to the Ail/Lom family of OMPs and provides protection to Y. pestis from complement-dependent killing (25). Furthermore, Ail is a dominant adhesion molecule that plays an important role in the delivery of T3SS effectors into targeted host cells during infection (26). In both rat and mouse bubonic plague models of infection, a Δail mutant exhibited reduced systemic spread as well as greatly attenuated virulence (greater in rats than in mice); however, a productive adaptive immune response was generated after a high-dose challenge, as evidenced by significant serum antibody levels against the Y. pestis F1 antigen (16). Likewise, Kolodziejek et al. reported that the deletion of the ail gene from Y. pestis CO92 led to a highly attenuated mutant strain in a rat model of pneumonic plague (27). In a Y. pestis KIM5 background strain (deleted of the pgm locus), Ail was shown to be more important for host cell binding, in inducing host cell cytotoxicity, and for virulence in a mouse model of infection when the bacteria were given by the intravenous route than Pla and that Pla mediation of Yop secretion was independent of its protease activity (28). Ail specifically binds fibronectin, an extracellular matrix protein, and possibly acts as a bridge receptor between Ail and the host cells (29). Interestingly, anti-fibronectin antibodies inhibited the KIM5-mediated cytotoxicity of the host cells in an Ail-dependent fashion, demonstrating the necessity of this interaction for Yop delivery into the host (29).
OmpA plays a significant role in anchoring the outer membrane to the cell wall peptidoglycan and has pore-forming activity, producing nonspecific diffusion channels (30, 31). Indeed, OmpA in several pathogens has been shown to mediate resistance to complement and antibacterial peptides, as well as play a role in invasion and intracellular survival (32–34). Recently, OmpA was identified as a prosurvival factor for macrophage-engulfed Y. pestis, and a ΔompA mutant strain was outcompeted by the WT strain in a mouse coinfection assay, suggesting a role it may play during dissemination within the host (35).
The present study was undertaken to specifically demonstrate whether antibodies to Ail, Pla, and OmpA provide protection to animals against bubonic and pneumonic plague caused by WT Y. pestis CO92 and its F1− mutant strain.
MATERIALS AND METHODS
Strains, plasmids, and media.
The bacterial strains and plasmids used in the present study are listed in Table 1. Y. pestis strains were grown at 28°C in heart infusion broth (HIB; Bacto, Sparks, MD) or on sheep blood agar plates in the Centers for Disease Control and Prevention-approved select agent laboratory. Escherichia coli cultures were grown at 37°C in Luria-Bertani (LB) broth and on LB agar plates. The antibiotic kanamycin was used at a working concentration 50 μg/ml.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristicsa | Source or reference |
|---|---|---|
| Strains | ||
| Y. pestis | ||
| CO92 BEI | Fully virulent WT strain | BEIb |
| Δcaf1 | Deletion mutant of the caf1 gene (F1) | 39 |
| E. coli BL21(DE3) | F− ompT gal dcm lon hsdSB(rB− mB−) λ(DE3 [lacI lacUV5-T7 gene 1 ind1 sam7 nin5]) | Novagen |
| Plasmids | ||
| pET-30a(+) | T7 promoter-based prokaryotic expression vector; Kmr | Novagen |
| pET-20b(+) | T7 promoter-based prokaryotic expression vector; Apr | Novagen |
| pET-30/ail | ail gene of Y. pestis cloned at BglII/SalI sites of pET-30a(+) vector for purification of Ail protein; Kmr | This study |
| pET-30/ompA | ompA gene of Y. pestis cloned at NdeI/XhoI sites of pET-30a(+) vector for purification of OmpA protein; Kmr | This study |
| pET-20/caf1 | caf1 gene of Y. pestis cloned at NdeI/XhoI sites of pET-20b(+) vector for purification of F1 protein; Apr | This study |
| pET-20/pla | pla gene of Y. pestis cloned at NdeI/XhoI sites of pET-20b(+) vector for purification of Pla protein; Apr | 38 |
Apr, ampicillin resistance; Kmr, kanamycin resistance.
BEI, The Biodefense and Emerging Infections Research Resources Repository, Manassas, VA.
Reagents and commercial antigens.
Restriction enzymes, Taq DNA polymerase, and T4 DNA ligase were purchased from New England BioLabs (Beverly, MA). A His SpinTrap kit and columns charged with Ni2+-Sepharose were purchased from GE Healthcare Biosciences Corp. (Piscataway, NJ). Imject Alum used as an adjuvant was purchased from Thermo Scientific Pierce Biotechnology (Rockford, IL). The F1-LcrV (F1-V) fusion protein was obtained from the Biodefense and Emerging Infections Research Resources Repository, Manassas, VA.
DNA isolation and PCR assays.
Y. pestis CO92 genomic DNA (gDNA) was isolated by utilizing a DNeasy tissue kit from Qiagen (Valencia, CA). PCR assays were performed under standard conditions as follows: an initial denaturation for 5 min at 94°C, followed by 30 cycles of denaturation for 30 s at 94°C, annealing for 30 s at 65°C, and extension for 30 s at 72°C, with a final extension for 7 min at 72°C. Amplified products were purified using a PCR purification kit (Qiagen). Plasmid DNA was isolated using a QIAprep spin miniprep kit (Qiagen). All primers (Table 2) were synthesized by Integrated DNA Technologies, Inc. (Coralville, IA).
Table 2.
Primers used in this study
| Primerb | Sequence (5′–3′)a | Purpose |
|---|---|---|
| ail/BglIIF | TCATAGATCTAATGAATAAGACATTACTGGTCTCT | Cloning of the ail gene into the pET-30a(+) vector |
| ail/SalIR | GGTCGTCGACTTAGAACCGGTAACCCGCGCCAAGC | Cloning of the ail gene into the pET-30a(+) vector |
| ompA/NdeIF | AGCCATATGAAAAAGACAGCTATCGCATTAGC | Cloning of the ompA gene into the pET-30a(+) vector |
| ompA/XhoIR | AACCTCGAGAGCCTGTGGCTGAGTCACAAC | Cloning of the ompA gene into the pET-30a(+) vector |
| caf1/NdeIF | CACATATGAAAAAAATCAGTTCCGTTATCG | Cloning of the caf1 gene into the pET-20b(+) vector |
| caf1/XhoIR | CACTCGAGTTGGTTAGATACGGTTACGGTTACAG | Cloning of the caf1 gene into the pET-20b(+) vector |
Underlining indicates restriction endonuclease site.
F, forward; R, reverse.
Transformation of E. coli BL21(DE3) strain.
Preparation of competent cells and plasmid DNA transformations of E. coli BL21(DE3) strain were performed by using the Z-Competent transformation kit from Zymo Research (Orange, CA), as described previously (36).
Hyperimmune sera from rats infected with WT Y. pestis CO92 and rescued with levofloxacin.
Brown Norway rats (n = 9/group) were challenged with WT CO92 (10 50% lethal doses [LD50] of 5,000 CFU administered intranasally [i.n.]) and then given levofloxacin (5 mg/kg/day given intraperitoneally [i.p.]), the minimal protective dose, at 24, 36, 42, and 48 h postinfection (p.i.) for 6 days. At 34 days p.i., the surviving animals were rechallenged with 10 LD50 of WT CO92. The animals were bled after 64 days, and the immune sera from the survivors were used for performing Western blot analysis against total cell lysates of WT CO92. Sera from uninfected but levofloxacin-treated animals served as a negative control.
Western blot analysis.
Whole-cell lysates of WT CO92 were subjected to sodium dodecyl sulfate (SDS)–10 to 16% gradient polyacrylamide gel electrophoresis (PAGE), followed by immunoblotting with rat hyperimmune or preimmune sera (see above) at a dilution of 1:1,000. After treatment of the membranes with goat anti-rat secondary antibodies conjugated with horseradish peroxidase (HRP), enhanced chemiluminescence (ECL) substrate was used to visualize the protein bands by using an ECL Western blotting detection reagent kit (GE Healthcare).
Passive transfer of hyperimmune rat sera to naive animals.
The hyperimmune rat sera obtained from Y. pestis-infected and levofloxacin-rescued rats that were given the antibiotic regimen 24 h p.i. for 6 days (see above) were pooled and used for the present study. Naive rats were administered 100 μl of the hyperimmune sera by the i.p. route. After 4 h, the passively immunized rats were i.n. challenged with 20 LD50 of WT Y. pestis CO92, and the mortality in animals was monitored. As controls, naive rats received either preimmune rat sera or phosphate-buffered saline (PBS).
Mouse passive antibody transfer.
Swiss-Webster mice (n = 15) were i.n. infected with 1 LD50 of WT Y. pestis CO92 (500 CFU). After 30 days, we terminally bled the survivors (n = 8), and their sera were used for the passive immunization of naive mice. Briefly, 100 μl of pooled immune or preimmune sera was injected i.p. into naive mice (n = 10/group). After 4 h, passively immunized mice were i.n. challenged with 10 LD50 of WT CO92 (5,000 CFU) and observed for survival.
Differences in the immune responses of mice and rats.
WT Y. pestis CO92 was i.n. administered to animals (mice, n = 10/group; rats, n = 9/group) at the target challenge dose of 10,000 CFU, which correlated with 20 LD50 for both the rodent species. At 24 h p.i., levofloxacin was administered once daily (10 mg/kg) for 6 days via the i.p. route. The control animal groups did not receive the antibiotic but were infected. All of the animal studies were performed with either Swiss-Webster mice or Brown Norway rats.
Animal survival was monitored and recorded for 29 days p.i. On day 30 of the study, all survivors were rechallenged with WT CO92 via the i.n. route (20 LD50) and monitored for an additional 34 days. At the same time point, animals in the rechallenge control groups were i.n. infected for the first time with 20 LD50 of WT CO92.
Cloning of ail, ompA, pla, and caf1 genes in E. coli and purification of the recombinant proteins.
Using the pET-30a(+) vector, plasmid constructs containing the PCR-amplified ail and ompA genes (using the primers listed in Table 2) were generated. The amino-terminal (NH2) histidine (His)-tagged Ail and OmpA antigens from E. coli BL21(DE3) were purified by using the His SpinTrap kit (GE Healthcare) according to the recommended protocol under denaturing conditions with 8 and 4 M urea, respectively. Briefly, recombinant E. coli cultures were grown to log phase in LB broth at 37°C with agitation. Cells were then induced with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 4 h. After sonication and centrifugation, membrane proteins were solubilized in the binding buffer containing 4 to 8 M urea and loaded onto the His column. After a thorough washing of the column (3×), the bound proteins were eluted with 500 mM imidazole and urea. The purity of recombinant proteins was examined by Coomassie blue and SYPRO-Ruby staining (Bio-Rad, Hercules, CA), and the concentration of antigens was determined by spectrophotometric readings of the optical density at 280 nm (OD280) and a BCA protein assay kit (Thermo Scientific), which is based on Lowry's method (37).
Likewise, the NH2-terminal His-tagged Pla was purified in a single-step affinity chromatography using Ni2+-Sepharose chromatography under denaturing conditions, as described previously (38). This antigen was insoluble and did not activate plasminogen. On the contrary, the carboxyl-terminal His-tagged F1 was Ni2+-Sepharose affinity chromatography purified in a soluble form under native conditions. The purity of these antigens was also examined by Coomassie blue and SYPRO-Ruby staining of SDS–12% polyacrylamide gels.
Continuous-elution electrophoresis and mass spectrometry.
A saturated 10-ml culture of Y. pestis CO92 was grown in HIB medium at 28°C with agitation (180 rpm) from a frozen −80°C stock. Subcultures in 90 ml of fresh HIB medium were incubated for 2 h under the same conditions. The temperature of the culture was then shifted to 37°C for the next 4 h of incubation. The bacterial cells were pelleted and then resuspended in 10 ml of SDS-PAGE sample buffer containing β2-mercaptoethanol. The sample was heated at 100°C for 10 min in a dry bath and plated to confirm the sterility of the sample. An aliquot of the sample (1 ml) was loaded onto a 10% polyacrylamide gel and electrophoresed under conditions specified by the manufacturer by using a Model 491 Prep Cell (Bio-Rad), which is designed to purify proteins from complex mixtures by continuous-elution electrophoresis. Once the blue dye of the sample migrated through 3/4 of the gel, the proteins were eluted from the column using elution buffer (50 mM ammonium formate, 0.01% SDS [pH 8.0]), and 4 ml of each fraction (for a total of 150 to 200 ml) was collected. The samples were concentrated to 1/4 of the volume in a Savant SpeedVac (Biodirect, Inc., Taunton, MA) and then dialyzed against PBS. An aliquot of the samples was subjected to SDS-PAGE and SYPRO-Ruby staining. In a parallel gel, Western blot analysis was performed with hyperimmune rat sera to identify immunoreactive bands. The matching protein bands were manually removed from the stained gel, trypsin digested, and subjected to matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) at the Protein Chemistry Core Laboratory at the University of Texas Medical Branch (UTMB).
Immunization of animals.
Five- to six-week-old female Swiss-Webster mice (n = 10/group; Taconic Farms, Germantown, NY) and 5-week-old female Brown Norway rats (n = 6/group; Harlan Laboratories, Houston, TX) were immunized on day 0, followed by two boosters (given 2 weeks apart) with the purified antigens at concentrations of 20 μg per animal after being mixed with the Imject Alum (1:1) by the intramuscular (i.m.) route. The control animals received adjuvant only. Blood was drawn prior to immunization and 1 week after the second booster. All of the animal immunization protocols were performed in an Animal Biosafety Level 2 (ABSL-2) facility within the Galveston National Laboratory (GNL), UTMB.
Animal challenges.
One week after the second antigen booster, the immunized animals were moved to an ABSL-3 facility within the GNL and infected by the i.n. and s.c. routes with WT Y. pestis CO92 or an isogenic Δcaf1 mutant of Y. pestis CO92 (39). All of the animal challenge experiments were performed in the ABSL-3 facility. Briefly, mice and rats were anesthetized with a mixture of xylazine-ketamine or isoflurane. The animals were observed daily for signs of morbidity and mortality during the course of the experiment. For mice immunized with Ail, OmpA, and Pla, we challenged them with 500 LD50 (2.5 × 104 CFU) of the Δcaf1 mutant of Y. pestis CO92 by the s.c. route to induce bubonic plague (39). Likewise, we infected immunized mice with 15 LD50 (i.n., 7,500 CFU) of the Δcaf1 mutant to induce pneumonic plague.
For rats immunized with F1-V antigen, we used an i.n. challenge dose of 5 × 106 CFU for infection, representing 10,000 LD50, of WT CO92. For rats immunized with Ail, OmpA, and Pla antigens, we used an i.n. challenge dose of 4 × 103 to 5 × 103 CFU for infection, which represented 8 to 10 LD50, and an s.c. dose of 7 × 102 to 8 × 102 CFU, which corresponded to 7 to 8 LD50 of WT CO92. All animal experiments were performed under a protocol approved by the Institutional Animal Care and Use Committee at UTMB.
Analysis of antibody production by ELISA.
For enzyme-linked immunosorbent assays (ELISAs), 96-well microtiter plates (Evergreen Scientific, Los Angeles, CA) were coated with 10 ng of Ail, OmpA, Pla, F1, or F1-V antigen/well at 4°C overnight. After blocking and washing (40), sera from naive and immunized mice and rats were serially diluted and incubated with the affixed antigens in the microtiter plates for 1 h at room temperature. After several washes, HRP-conjugated goat anti-mouse/rat secondary antibodies were added to the wells at a dilution of 1:10,000 for 1 h at room temperature, followed by several washes and the addition of the substrate 3,3′,5,5′-tetramethylbenzidine (TMB). After a 20-min colorimetric development period, the reaction was quenched by the addition of 2 N H2SO4, and the plate/well absorbance was read at OD450 using a spectrophotometer.
For Western blot analysis, purified antigens (OmpA, Ail, Pla, and F1-V; 100 ng) were subjected to SDS–12% PAGE and immunoblotting. The blots were probed with sera to specific antigens.
Antibodies to whole bacteria in the hyperimmune rat sera.
Y. pestis CO92 cells were grown to log phase and, after centrifugation, were resuspended in PBS at a concentration of 5 × 109 CFU/ml. Subsequently, the low-binding 96-well microtiter plates were coated with poly-l-lysine (10 μg/ml in PBS, 50 μl/well) for 30 min at room temperature, followed by the addition of 50 μl of the bacterial suspension/well, after washing off the residual lysine. The plates were centrifuged at 1,000 rpm for 5 min at 4°C, followed by the addition of 50 μl of a 0.5% glutaraldehyde solution/well and incubation for 15 min. The glutaraldehyde solution was gently removed, and the wells were washed several times with PBS in the biological safety hood in the select agent laboratory. The wells were then treated with 200 μl/well of a solution of 0.1 M glycine–0.1% bovine serum albumin (BSA) in water, followed by incubation for 30 min at room temperature. The wells were washed with PBS, followed by blocking of the wells of the plates with 200 μl of PBS–1% BSA/well for 1 h at room temperature. After the blocking solution was removed, 100 μl of the primary antibodies (naive versus hyperimmune sera, 10-fold diluted) in PBS–0.1% BSA were added for 1 h. After five washes with PBS–0.05% Tween 20 solution, 100 μl of HRP-conjugated secondary antibodies (diluted 1:10,000 in PBS–0.1% BSA) was added for 1 h. After thorough washing, 100 μl of the substrate (TMB) was added as described above. The reaction was quenched by adding 50 μl of 2 N H2SO4, and the color intensity was read at OD450.
Statistics.
Wherever appropriate, data were analyzed using unpaired Student t test. The animal data were analyzed by using either the Fisher exact test, Kaplan-Meier's survival estimates, or an unpaired Student t test with P values of ≤0.05 considered significant.
RESULTS
Brown Norway rats infected with WT Y. pestis CO92 and rescued by levofloxacin were protected against developing subsequent pneumonic plague.
With the goal of producing hyperimmune sera raised against plague outer membrane antigens, we challenged rats with WT CO92 (10 LD50 by the i.n. route) and then administered levofloxacin (5 mg/kg/day, the MIC) at 24, 36, 42, and 48 h p.i. for 6 days. As noted (Fig. 1A), 100% of the animals survived initial challenge when the levofloxacin treatment was delayed up to 42 h. On the contrary, >50% of the rats died when the antibiotic treatment was delayed to 48 h after infection. All of the levofloxacin-treated groups of rats (24 to 42 h, P = 0.0011; 48 h, P = 0.0032) were significantly protected based on the Kaplan-Meier's survival estimates compared to the control group of animals that were infected but not treated with levofloxacin. As expected, all of the control rats died by day 6 after initial infection (Fig. 1A).
Fig 1.
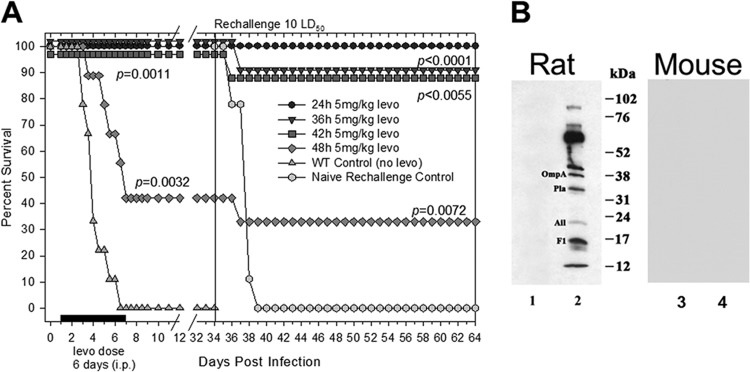
(A) Delayed levofloxacin (levo) treatment of rats infected intranasally with 10 LD50 of WT Y. pestis CO92 was protective against developing pneumonic plague. Antibiotic treatment (5 mg/kg/day) was initiated by 24, 36, 42, and 48 h p.i. and continued for 6 days thereafter. The solid black bar on the x axis represents 6 days of antibiotic treatment starting 24 h p.i. Animals were rechallenged with 10 LD50 of WT CO92 on day 34 (indicated by a vertical line) and monitored for another 30 days. The data were analyzed statistically using Kaplan-Meier survival estimates, and the P values are indicated. (B) Detection by immunoblot analysis and identification of OmpA, Pla, Ail, and F1 target antigens by MALDI-TOF (lane 1, preimmune; lane 2, immune) is shown from sera of rats infected by the i.n. route with 10 LD50 of WT Y. pestis CO92 and rescued by levofloxacin (designated as rat). No protein bands were detected when pooled sera from Y. pestis-infected and levofloxacin-rescued mice were used (designated as mouse) under conditions identical to those for rat sera. Lane 3, preimmune mouse sera; lane 4, immune mouse sera.
Upon rechallenge of the survivors on day 34, 90 to 100% of the animals were still protected in those groups that received levofloxacin 24 to 42 h p.i. (P < 0.0055 to 0.0001), whereas this protection dropped to 35% in the group of rats in which the levofloxacin treatment was delayed to 48 h. However, this protection was still statistically significant compared to the control animals (P = 0.0072). It should be noted that no levofloxacin was given to the animals after rechallenge. The age/sex-matched control infected rats died of plague by day 5 (Fig. 1A). These data demonstrated that levofloxacin-treated animals developed a protective immunity to Y. pestis. Similar results were observed when animals were administered levofloxacin at 10 mg/kg/day at the same time points (data not shown).
To then determine whether the hyperimmune sera from these rats would react with WT CO92 proteins, we performed immunoblot analysis. We used pooled sera from rats that were infected with Y. pestis CO92 and given levofloxacin 24 h p.i. (Fig. 1A). In doing so, we detected 10 to 12 discrete bands in the size range of 12 to 102 kDa that reacted with rat immune (Fig. 1B, lane 2) but not preimmune sera (lane 1). Clearly, genes encoding these target antigens were of interest to us.
Passive transfer of hyperimmune rat sera to naive rats was protective against inducing pneumonic plague.
To demonstrate whether passively transferred hyperimmune rat sera, used for Western blot analysis, also provided protection to naive rats against developing pneumonic plague, animals were administered either immune or preimmune sera via the i.p. route prior to i.n. challenge with the WT Y. pestis CO92. Prior to performing this passive immunization experiment, we determined the antibody titer in the hyperimmune rat sera against the whole Y. pestis cells by performing whole-cell ELISA (Fig. 2A). In these passive immunization studies, the animals were never treated with levofloxacin. As shown in Fig. 2A, significant antibody titers (1:108) were observed in Y. pestis-infected and levofloxacin-rescued rats compared to the preimmune control sera.
Fig 2.
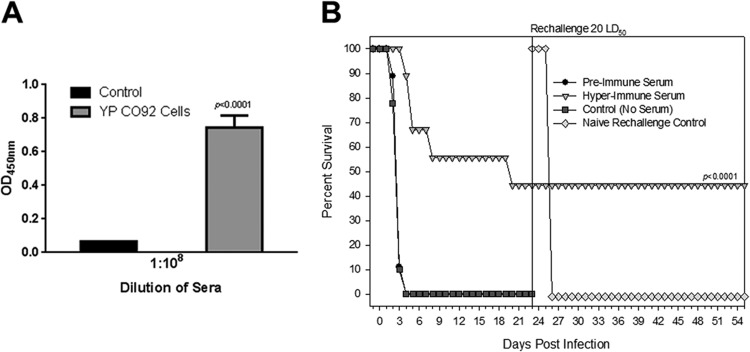
(A) Evaluation of antibody titers against the whole WT CO92 bacterial cells by ELISA. The hyperimmune rat sera obtained after Y. pestis infection and rescuing of the animals with levofloxacin given 24 h p.i. were used for ELISA. The sera were pooled from all of the surviving animals, and the experiment was performed in triplicate. The arithmetic means ± the standard deviations are plotted. The pooled preimmune sera were used as a control. The sera were 10-fold diluted to determine endpoint antibody titers. (B) Passive transfer of pooled hyperimmune rat sera to naive rats provided protection to the latter when challenged with 20 LD50 of WT Y. pestis CO92. On day 23, the surviving rats were rechallenged with the same LD50 of WT CO92 (indicated by a vertical line). The data were statistically analyzed by using Kaplan-Meier survival estimates, and the P value is provided. Animals that were injected with preimmune sera served as a negative control. For rechallenge, we used same age- and sex-matched naive rats as a control to demonstrate that the given dose of Y. pestis led to 100% mortality due to pneumonic plague.
At 4 h after administration of the hyperimmune or preimmune sera, rats were i.n. challenged with 20 LD50 of WT CO92, and all animals that either did not receive any sera or were injected with the preimmune sera died by day 4 (Fig. 2B). However, rats that received hyperimmune sera demonstrated 55% protection until day 19, which then dropped to 44%, still a significant difference relative to the above-mentioned two control groups (P < 0.0001). On day 23, we rechallenged the surviving animals that received hyperimmune sera with the same LD50 of CO92, and no further deaths occurred (Fig. 2B). The rationale of this study was 2-fold and included (i) determination of whether passively administered Y. pestis-specific hyperimmune rat sera could protect naive animals against pneumonic plague and (ii) determination of whether surviving animals could mount an independently active immune response against the plague bacterium that could protect rats during a rechallenge. Animals from the same batch and age that received preimmune sera served as a negative control for the rechallenge study, and all of them died (as expected) (Fig. 2B). These data demonstrated that immunoreactive antigens detected by Western blotting (Fig. 1B) were capable of generating protective antibodies which prevented rats from developing pneumonic plague. Interestingly, parallel mouse studies indicated that passive transfer of sera from Y. pestis-infected mice (given 1 LD50 by the i.n. route [see Materials and Methods]) to naive mice did not provide any protection (data not shown).
Differences in the adaptive immune response of mice and rats during pneumonic plague.
Mice and rats were i.n. challenged with 20 LD50 of WT CO92 in parallel. After 24 h p.i., animals received levofloxacin at 5 to 10 mg/kg/day via the i.p. route for 6 days. As expected, both the mouse and the rat untreated challenge control animals (without levofloxacin) died by day 4 p.i. In sharp contrast, 100% of the mice and rats survived when given levofloxacin 24 h p.i., confirming that this concentration of the antibiotic was indeed protective for both animal models (Fig. 3). This level of survival was statistically significant compared to the challenge control group beginning on day 3, as determined by the Kaplan-Meier and Fisher exact tests (P < 0.0001).
Fig 3.
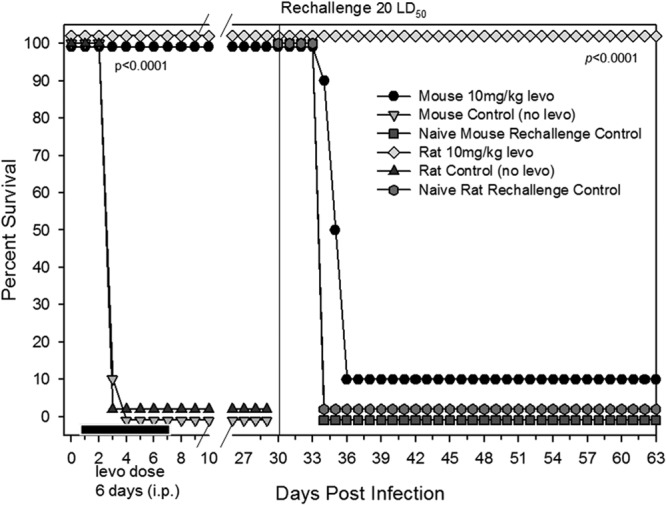
Mice and rats that were infected with 20 LD50 of WT CO92 and then treated with levofloxacin at 10 mg/kg/day starting at 24 h p.i. for 6 consecutive days were 100% protected from developing pneumonic plague. The solid black bar along the x axis represents levofloxacin treatment of animals 24 h after infection, and the antibiotic regimen was given over a period of 6 days. On day 30 (indicated by a vertical line), the survivors were rechallenged with the same LD50 of WT CO92. Age- and sex-matched animals were used as rechallenge controls. The data were statistically analyzed by using Kaplan-Meier survival estimates, and the P values are reported.
On day 30 p.i., survivors (both mice and rats) were rechallenged to demonstrate the development of protective immunity against reinfection with 20 LD50 of WT CO92. The age-matched naive control animals were similarly infected, and all succumbed to plague as expected. Importantly, levofloxacin-treated rats demonstrated 100% survival after rechallenge (P < 0.0001 relative to the control group), whereas only 10% of the levofloxacin-treated mice survived rechallenge (Fig. 3).
Overall, these data tend to suggest key differences in the immune responses of mice versus rats to pneumonic plague. We speculate that mice do not develop as robust an adaptive immune response as do rats for protection against subsequent plague infections. Investigations of differences between infected B- and T-cell responses of the two species have not yet been published, representing a gap in our understanding of the rodent models of pneumonic plague. Our speculation was based on the fact that sera from Y. pestis-infected and levofloxacin-rescued mice (Fig. 3) did not react with any WT CO92 protein bands upon Western blot analysis at a dilution of 1:1,000 (Fig. 1B). This was in sharp contrast to the hyperimmune sera obtained on day 34 and 64 (Fig. 1A) from rats which reacted with 10 to 12 discrete bands. Consequently, we used both rodent species in our studies.
Identification of immunoreactive antigens of WT CO92 that reacted with rat hyperimmune sera.
Based on continuous-elution electrophoresis, Western blot analysis of isolated proteins, and mass spectrometry, we identified four Y. pestis CO92 proteins (<40 kDa) that reacted strongly with our rat hyperimmune sera (Fig. 1B). One of them was F1 (16 kDa), and the remaining three were Ail/OmpX (20 kDa), Pla (34 kDa), and Salmonella-like OmpA (38 kDa) (Fig. 4). It was of interest to note that F1 migrated more slowly than expected, representing the aggregative nature of this protein (41). The bands which migrated close to 17 or 14 kDa on the Western blot (Fig. 4) also represented Ail/OmpX, based on MALDI-TOF analysis. Finally, the bands which migrated between 36 and 52 kDa were identified as DNA-directed RNA polymerase alpha subunit (36 kDa), OmpC (41 kDa), and elongation factor Tu (∼50 kDa).
Fig 4.
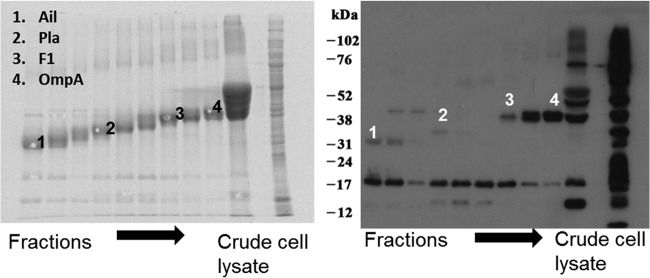
Continuous elution SDS–10% PAGE using a Model 491 Prep Cell. Whole-cell lysates of WT CO92 were run on the gel, and the fractions were collected. Every four fractions were pooled, concentrated, dialyzed, subjected to SDS–12% PAGE, and stained with SYPRO-Ruby (left panel). An aliquot of the starting crude material was also subjected to electrophoresis. A parallel gel was subjected to Western blot analysis using hyperimmune rat sera (Fig. 1B) (right panel). The Western blot bands that corresponded to the SYPRO-Ruby-stained protein bands were picked and subjected to MALDI-TOF for identification. The identity of spots is indicated. The presence of multiple bands on the Western blot represented either aggregated or degraded version of the same proteins, as confirmed by mass spectrometry of several bands. The migration of molecular mass markers is also shown.
Plasmid constructions and purification of Y. pestis antigens.
The genes for ail, ompA, pla, and caf1 were cloned into pET expression vectors with appropriate restriction sites and sequenced (Tables 1 and 2). Ail, OmpA, and Pla consist of 179, 363, and 312 amino acid residues, respectively, and were purified by affinity chromatography under denaturing conditions with urea because of their insoluble nature. F1 consists of 178 amino acid residues and was purified under native conditions.
Protective effects of antibodies to F1, LcrV, Pla, OmpA, and Ail/OmpX against Y. pestis infection in a mouse model.
Before evaluating protection conferred by Pla, OmpA, or Ail/OmpX recombinant proteins, we first tested the efficacy of F1 and LcrV subunit vaccines in providing protection to mice against pneumonic plague to establish a comparative benchmark. Mice were immunized via the i.m. route (20 μg of purified F1-V or F1 in alum) on days 0, 15 and 30 and then challenged after 45 days. We observed 50% protection in mice immunized with F1 after i.n. challenge with 100 LD50 of WT Y. pestis CO92, which corresponded to high F1 antibody titers in the animal sera (Fig. 5), whereas this protection was 100% when the animals were immunized with F1-V.
Fig 5.
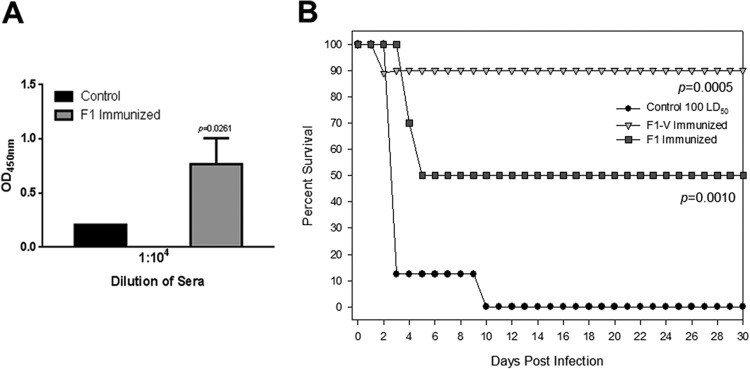
Active immunization of mice with F1 and LcrV provided protection to animals against pneumonic plague when challenged by the i.n. route with 100 LD50 of WT CO92. Antibody titers to F1 antigen in the mouse sera compared to that of preimmune sera (control) are shown in panel A, while animal protection data are shown in panel B. The data were statistically analyzed using Kaplan-Meier survival estimates, and the P values are presented.
Following i.m. immunizations with OmpA, Ail/OmpX, and Pla, mice mounted antibody responses against all three antigens, although the titers varied. The titers of anti-Pla and anti-OmpA antibodies were the lowest, while the anti-Ail/OmpX antibody titers developed were similar to that of F1-V antigen (Fig. 6A). These antibodies specifically reacted with their respective purified antigens on Western blots (see insets). Mice immunized with the aforementioned antigens were subsequently infected via the s.c. route with 500 LD50 (which corresponds to approximately 75 to 100 LD50 of the WT Y. pestis CO92) of our Δcaf1 mutant strain of Y. pestis CO92 (39). The Δcaf1 mutant was chosen for the infection studies since, in this mutant, the Ail/OmpX, OmpA, and Pla surface antigens would be more fully exposed to neutralizing antibodies. All of the unimmunized, naive control animals died by day 20, although 90% of the animals were dead by day 10. Similarly, antibodies to Pla did not provide any protection to mice (data not shown). In sharp contrast, antibodies to both OmpA and Ail/OmpX provided protection to mice resulting in 40 to 50% animal survival, respectively. In fact, Ail/OmpX- and OmpA-immunized mouse survival curves were significantly different than unimmunized mouse survival curves (P = 0.001 to 0.0066) (Fig. 6B).
Fig 6.

(A) Evaluation of antibody titers in the sera of mice after active immunization with OmpA, Ail, Pla, and F1-V proteins. The individual animal serum was titrated in duplicate after serial 5- or 10-fold dilution. Preimmune sera were used as controls. The insets show the detection of the purified antigen using specific serum to a particular antigen (lane 1, by using preimmune serum; lane 2, by using postimmunization serum). (B) Active immunization of mice with F1-V, OmpA, and Ail provided protection to mice against developing bubonic plague. Animals were challenged with the Δcaf1 mutant at the indicated dose. (C) Pla-immunized mice were 60% protected against i.n. challenge of Δcaf1 mutant at a 15 LD50. The data were statistically analyzed by using Kaplan-Meier survival estimates, and the P values are presented. *, P when the data were analyzed using an unpaired Student t test. This value was 0.08 with the Kaplan-Meier survival estimates.
F1-V immunization clearly provided the greatest degree of protection whereby 100% of immunized mice survived s.c. challenge (P < 0.0001) (Fig. 6B). In one group, we immunized mice with all three antigens Ail/OmpX, OmpA, and Pla; there was no additive or synergistic effect on protection against bubonic plague. In fact, the protective effects of antibodies to three antigens in cocktail were similar to that noted for Ail/OmpX alone (data not shown).
Interestingly, in a pneumonic plague mouse model, only antibodies to Pla provided protection to animals when challenged with 15 LD50 (7,500 CFU) of Δcaf1 mutant of CO92 (Fig. 6C) despite Pla immunization not protecting against a bubonic plague challenge (data not shown). At this dose, while 90% of the unimmunized mice died by day 9, 60% of the Pla-immunized animals were protected from developing pneumonic plague (P = 0.01) (Fig. 6C). Neither Ail/OmpX nor OmpA immunizations protected mice from developing pneumonic plague (data not shown) despite immunizations with both of the aforementioned antigens providing protection against bubonic plague in mice (Fig. 6B). We have shown earlier (39) that the LD50 of WT CO92 and its caf1 mutant were essentially similar in a mouse model of pneumonic plague.
LD50 determination of WT Y. pestis CO92 in a rat model of bubonic and pneumonic plague.
In addition to testing the efficacy of Ail/OmpX, OmpA, and Pla putative vaccine candidates in mice (which we routinely do) (42), we also sought to employ a rat model of infection, as we have done in the past (43), for comparative purposes. However, prior to undergoing immunization (with our three candidate antigens) and challenge studies, we first determined the LD50 of the WT Y. pestis CO92 for both bubonic and pneumonic plague, so we conducted a dose-response experiment in a rat model (n = 5/group). The LD50s in rats by the i.n. and s.c. routes were calculated to be 500 and 100 CFU, respectively, for WT CO92. Therefore, for rat challenge studies, we used 8 to 10 LD50 (4.0 × 103 to 5.0 × 103 CFU) and 8 LD50 (8.0 × 102 CFU) for evoking pneumonic and bubonic plague, respectively, which typically cause 100% mortality in animals between 3 and 8 days depending upon the route of infection. As shown in Fig. 7, both of the aforementioned routes of infection resulted in 0% rat survival by day 8 p.i. when infected with 1 × 103 CFU (i.n.) and 4 × 103 CFU (s.c.).
Fig 7.
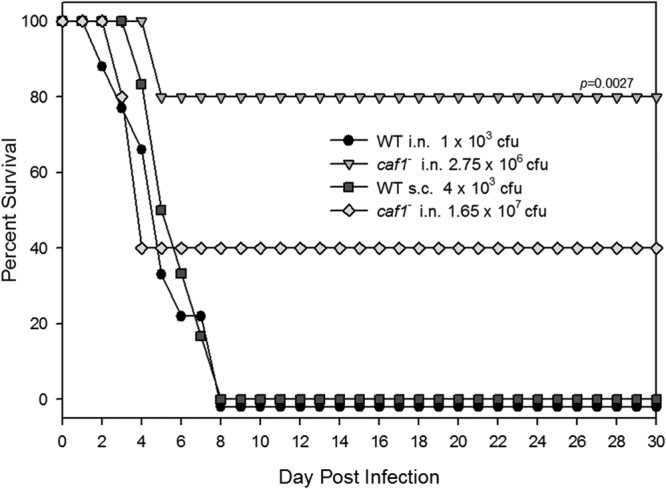
Evaluation of the Δcaf1 mutant and its corresponding WT Y. pestis CO92 in the rat model of pneumonic and bubonic plague. The animals were infected either via the i.n. or s.c. route at the indicated doses. Rat survival was recorded over a period of 30 days. The data were statistically analyzed by using Kaplan-Meier survival estimates, and the P values are presented.
For evaluating the Δcaf1 mutant in rats, we used two i.n. challenge doses of 2.75 × 106 CFU and 1.65 × 107 CFU, which corresponded to WT equivalent of 5,500 and 33,000 LD50s., respectively. At the lower challenge dose (2.75 × 106 CFU), 80% of infected mice survived out to 30 days p.i. (P = 0.0027 relative to the WT i.n. challenge rat survival), while the higher dose (1.65 × 107 CFU) resulted in as high as 40% rat survival (Fig. 7). Unlike in mice (Fig. 6C), the Δcaf1 mutant was highly attenuated in the rat i.n. challenge model of infection, and, as a result, we used the WT CO92 Y. pestis strain in our rat studies when testing the efficacy of Ail/OmpX, OmpA, and Pla as protective antigens.
Protective effects of antibodies to Ail, OmpA, and Pla during bubonic and pneumonic plague in a rat model.
As we had previously reported for our mouse studies (Fig. 6A), prior to carrying out animal survival experiments on Pla-, OmpA-, or Ail/OmpX-immunized rats, we first sought to demonstrate whether humoral responses against the aforementioned antigens were being generated in rats. Indeed, we were able to demonstrate somewhat comparable antibody titers in rats to all three studied antigens in addition to F1-V (Fig. 8A). Using the Y. pestis CO92 WT strain (as opposed to the Δcaf1 mutant) in our rat challenges, for reasons described above, we determined that whereas antibodies to Ail/OmpX were protective against developing pneumonic plague (P = 0.0268) at a LD50 of 8.6 (Fig. 8B), anti-OmpA and anti-Pla antibodies failed to provide any protection (data not shown). Our F1-V subunit vaccine resulted in 100% rat protection out to day 30 p.i. (P < 0.0001) (Fig. 8B). In a bubonic plague model, none of the antibodies (to Ail/OmpX, Pla, or OmpA) provided any protection to rats (at 7 LD50).
Fig 8.
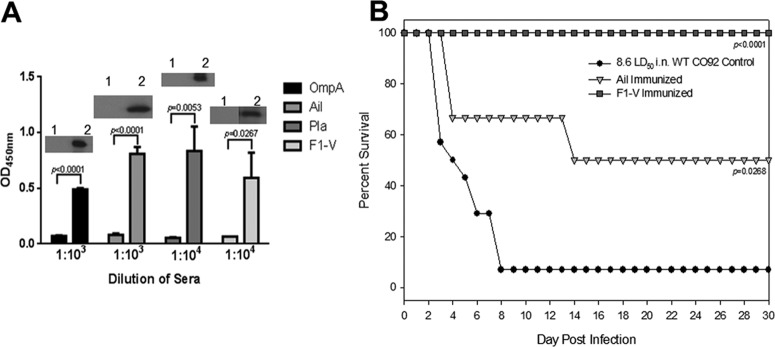
(A) Evaluation of antibody titers in the sera of rats after active immunization with OmpA, Ail, Pla, and F1-V proteins. Individual animal sera were titrated in duplicate after serial 10-fold dilution. Preimmune sera were used as controls. The insets show the detection of the purified antigen using specific serum to a particular antigen (lane 1, by using preimmune serum; lane 2, by using postimmunization serum). (B) Active immunization of rats with F1-V and Ail provided protection to animals against developing pneumonic plague. The animals were challenged with the WT CO92 at a LD50 of 8.6. The data were statistically analyzed by using Kaplan-Meier survival estimates, and the P values are presented.
DISCUSSION
Much of the emphasis on plague vaccine development has centered on F1 and LcrV antigens, particularly in subunit vaccine research (see reference 3 and references therein). However, we argue that focusing exclusively on F1 and LcrV as the sole protective antigens suitable for exploitation as vaccine candidates is shortsighted. This is so on account of natural variation in the lcrV open reading frames of various Y. pestis isolates (7, 8, 44), as well as on account of the existence of highly virulent Y. pestis isolates that lack the F1 antigen altogether (7, 8). Taken together, the search for additional protective antigens should continue and was the basis of the present study.
To directly address this, we sought to identify novel protective Y. pestis antigens and determined their effectiveness as putative vaccine candidates against both pneumonic and bubonic mouse and rat models of infection. Both routes of infection were studied since disease outbreaks could occur via flea bite or inhalational, either naturally or intentionally, and we wanted to develop a more comprehensive and holistic assessment of the protective nature of our antigens in question. In this study, for example, Pla did not protect mice against bubonic plague but did confer protection against the pneumonic form of the disease, while the converse was true for the Ail/OmpX and OmpA antigens.
In that same vein, we used both mouse and rat models of infection since we were one of the first groups to use rats to study pneumonic plague pathogenesis (43) and have since learned that each model can reveal data that the other model does not (42, 43). In support of this statement, we presented (in this report) that mice and rats did not generate the same protective immune responses against Y. pestis infection in levofloxacin-rescued animals. Furthermore, unlike in mice (39), the Δcaf1 mutant was highly attenuated in the rat i.n. model of infection as shown in this study. Indeed, we, and other investigators, have shown that F1 does not contribute to Y. pestis pathogenesis during pneumonic plague but does participate in contributing to virulence during bubonic plague in mice (38, 45). However, vaccines composed of either F1 or F1 and V antigens, as well as the V-F1 fusion protein contribute to developing protection against pneumonic plague in nonhuman primates (6, 46, 47). In earlier studies, protection was shown in a mouse model against both bubonic and pneumonic plague by passive immunization with monoclonal antibodies to F1 (48) and the recombinant F1-V antigen fusion vaccine (12). These data indicated that immunoprotective antigens need not be virulence factors for the bacterium. As a result, we had to use the WT CO92 Y. pestis strain in our rat studies when testing the efficacy of our putative Ail/OmpX, OmpA, and Pla subunit vaccines.
Only Ail/OmpX protected rats against pneumonic plague, while antibodies to other antigens were not protective against bubonic and pneumonic plague in rats. These findings serve to underscore the importance of using multiple infection model systems when determining the efficacy of novel vaccine candidates. Whether these differences were related to different subclasses of antibodies produced or activation of different subsets of T cells needs to be further explored as a future study. Based on the data presented in Fig. 1B, it is apparent that sera from Y. pestis-infected and levofloxacin-rescued mice did not have similar Y. pestis antigen-specific antibodies that were observed in similarly infected and treated rats.
It has been reported that the genetic makeup of the host dictates susceptibility or resistance to Y. pestis infection. For example, C57BL/6J mice were shown to be more susceptible to developing bubonic plague than BALB/cJ mice by the Δcaf1 mutant strain of Y. pestis (17). Likewise, C57BL/6J and BALB/c substrains, such as BALB/cByJ and BALB/cAnNHsd, were highly susceptible to infection with the Y. pestis KIM5 strain compared to the BALB/cJ mouse strain exhibiting 250-fold-higher LD50s for KIM5 (49). This resistance of BALB/cJ mice to Y. pestis infection correlated well with the presence of a plague resistance locus (prl1) that maps to the major histocompatibility region on chromosome 17, which carries innate and adaptive immunity-associated genes (49). Therefore, subtle differences in the immune responses of mice and rats to Y. pestis infection and the much higher LD50 of the Δcaf1 mutant in a rat model of pneumonic plague compared to the WT Y. pestis CO92 could be genetically linked to specific rodent species and requires further evaluation.
Finally, since antigens were purified under denaturing conditions, this could have impacted the level of protection noted in animals. Since antibodies to conformational epitopes to these antigens could be important, we will explore this possibility by expressing these antigen-encoding genes in different expression vector systems, which could provide us with soluble antigens.
Interestingly, all four of the protein targets identified by our hyperimmune Western blot screen, have been previously designated as either bona fide virulence factors or as virulence-associated factors. When considering the collective literature demonstrating the importance of Pla, OmpA, and Ail/OmpX in Y. pestis's pathogenesis, the fact that our hyperimmune Western blot screen identified these three outer membrane proteins (in addition to F1) becomes less surprising. However, what remained unknown was whether any of these outer membrane antigens could serve as useful subunit vaccine candidates.
In the present study, we made considerable progress in identifying novel vaccine candidate antigens for a potential Y. pestis subunit vaccine. Such a vaccine candidate could use one, two, or even all three of our candidates in cocktail either alone or in addition to F1-V. This is particularly important since active immunization of cynomolgus macaques with F1-V provided significant protection against pneumonic plague, while African green monkeys were not well protected against developing pneumonic plague (7). Therefore, a cocktail of vaccine antigens may provide optimal protection against different forms of plague in multiple models.
Overall, based on our data, antibodies to Ail/OmpX and OmpA provided protection to mice against bubonic but not pneumonic plague and anti-Pla antibodies were protective against pneumonic but not bubonic plague. In a rat model, anti-Ail/OmpX antibodies were protective against developing pneumonic plague. The immunological basis for this variation is unknown at this time, but this reinforces the need to test new immunogens in multiple animal model systems to fully elucidate their potential as important vaccine components. Indeed, it has been reported that while F/LcrV-based vaccines are protective in cynomolgus macaques but the protection level was highly variable (between 0 and 75%) against pneumonic plague in African green monkeys (50). Whether such differences could be pivotal in providing protection against bubonic versus pneumonic plague in different animal models represent one scenario which needs to be further investigated.
Finally, although antibodies to F1-V provided 100% protection to animals against plague when infected by the WT Y. pestis CO92, this protection is expected to be much lower when infection occurs with the F1− strains of Y. pestis or those Y. pestis strains which have different variants of LcrV. In such scenarios, addition of other antigens, such as Ail/OmpX, Pla, and/or OmpA could be beneficial as antibodies against them would certainly help to negate severe plague infections in vaccinated people. This would allow widening the window for other treatment options, such as antibiotics to save plague-infected individuals.
Although our initial study indicated that immunization of mice with equal amounts of Ail/OmpX, OmpA, and Pla did not provide any added benefit (in terms of animal protection), different ratios of these antigens must be tested, and any competition among these antigens in producing protective antibodies should be explored. Indeed, F1 and LcrV antigens must be used in a specific ratio during immunization to demonstrate optimal protection in animals (5, 6, 47). We were able to demonstrate that OmpA, Pla, and Ail/OmpX were all protective in either bubonic or pneumonic Y. pestis challenges in either mouse or rat models of infection. It will be important in the future to evaluate whether the addition of the above-mentioned antigens to an F1-V subunit vaccine could provide enhanced protection against Y. pestis strains that are devoid of the capsule or carry different variants of LcrV. Such a multivalent vaccine could generate both a robust and broad adaptive immune response targeting multiple surface-exposed Y. pestis antigens. Taking such an approach could yield the most efficacious Y. pestis vaccine to date.
ACKNOWLEDGMENTS
Studies conducted for this manuscript were supported by the NIH/NIAID grants AI064389 and N01 AI30065 (A.K.C.). J.A.R. was supported by the National Aeronautics and Space Administration cooperative agreement NNX08B4A47A, as well as AI064389. C.J.V.L. was supported by the NIH/NIAID T32 predoctoral training grant on biodefense (AI060549). We also acknowledge a UC7 grant, which facilitated our research in the Galveston National Laboratory.
Footnotes
Published ahead of print 12 December 2012
REFERENCES
- 1. Ligon BL. 2006. Plague: a review of its history and potential as a biological weapon. Semin. Pediatr. Infect. Dis. 17:161–170 [DOI] [PubMed] [Google Scholar]
- 2. Chain PS, Carniel E, Larimer FW, Lamerdin J, Stoutland PO, Regala WM, Georgescu AM, Vergez LM, Land ML, Motin VL, Brubaker RR, Fowler J, Hinnebusch J, Marceau M, Medigue C, Simonet M, Chenal-Francisque V, Souza B, Dacheux D, Elliott JM, Derbise A, Hauser LJ, Garcia E. 2004. Insights into the evolution of Yersinia pestis through whole genome comparison with Yersinia pseudotuberculosis. Proc. Natl. Acad. Sci. U. S. A. 101:13826–13831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rosenzweig JA, Jejelowo O, Sha J, Erova TE, Brackman SM, Kirtley ML, van Lier CJ, Chopra AK. 2011. Progress on plague vaccine development. Appl. Microbiol. Biotechnol. 91:265–286 [DOI] [PubMed] [Google Scholar]
- 4. Quenee LE, Berube BJ, Segal J, Elli D, Ciletti NA, Anderson D, Schneewind O. 2010. Amino acid residues 196–225 of LcrV represent a plague protective epitope. Vaccine 28:1870–1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Qiu Y, Liu Y, Qi Z, Wang W, Kou Z, Zhang Q, Liu G, Liu T, Yang Y, Yang X, Xin Y, Li C, Cui B, Huang S, Liu H, Zeng L, Wang Z, Yang R, Wang H, Wang X. 2010. Comparison of immunological responses of plague vaccines F1 + rV270 and EV76 in Chinese-origin rhesus macaque, Macaca mulatta. Scand. J. Immunol. 72:425–433 [DOI] [PubMed] [Google Scholar]
- 6. Chichester JA, Musiychuk K, Farrance CE, Mett V, Lyons J, Mett V, Yusibov Y. 2009. A single component two-valent LcrV-F1 vaccine protects non-human primates against pneumonic plague. Vaccine 27:3471–3474 [DOI] [PubMed] [Google Scholar]
- 7. Smiley ST. 2008. Current challenges in the development of vaccines for pneumonic plague. Expert Rev. Vaccines 7:209–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Titball RW, Williamson ED. 2004. Yersinia pestis (plague) vaccines. Expert. Opin. Biol. Ther. 4:965–973 [DOI] [PubMed] [Google Scholar]
- 9. Anderson GW, Jr, Leary SE, Williamson ED, Titball RW, Welkos SL, Worsham PL, Friedlander AM. 1996. Recombinant V antigen protects mice against pneumonic and bubonic plague caused by F1-capsule-positive and -negative strains of Yersinia pestis Infect. Immun. 64:4580–4585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Friedlander AM, Welkos SL, Worsham PL, Andrews GP, Heath DG, Anderson GW, Jr, Pitt ML, Estep J, Davis K. 1995. Relationship between virulence and immunity as revealed in recent studies of the F1 capsule of Yersinia pestis. Clin. Infect. Dis. 21:S178–S181 [DOI] [PubMed] [Google Scholar]
- 11. Anderson GW, Jr, Heath DG, Bolt CR, Welkos SL, Friedlander AM. 1998. Short- and long-term efficacy of single-dose subunit vaccines against Yersinia pestis in mice. Am. J. Trop. Med. Hyg. 58:793–799 [DOI] [PubMed] [Google Scholar]
- 12. Heath DG, Anderson GW, Jr, Mauro JM, Welkos SL, Andrews GP, Adamovicz J, Friedlander AM. 1998. Protection against experimental bubonic and pneumonic plague by a recombinant capsular F1-V antigen fusion protein vaccine. Vaccine 16:1131–1137 [DOI] [PubMed] [Google Scholar]
- 13. Branger CG, Sun W, Torres-Escobar A, Perry R, Roland KL, Fetherston J, Curtiss R., III 2010. Evaluation of Psn, HmuR, and a modified LcrV protein delivered to mice by live attenuated Salmonella as a vaccine against bubonic and pneumonic Yersinia pestis challenge. Vaccine 29:274–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sun W, Curtiss R, III 2012. Amino acid substitutions in LcrV at putative sites of interaction with Toll-like receptor 2 do not affect the virulence of Yersinia pestis. Microb. Pathog. 53:198–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hinnebusch BJ, Fischer ER, Schwan TG. 1998. Evaluation of the role of the Yersinia pestis plasminogen activator and other plasmid-encoded factors in temperature-dependent blockage of the flea. J. Infect. Dis. 178:1406–1415 [DOI] [PubMed] [Google Scholar]
- 16. Hinnebusch BJ, Jarrett CO, Callison JA, Gardner D, Buchanan SK, Plano GV. 2011. Role of the Yersinia pestis Ail protein in preventing a protective polymorphonuclear leukocyte response during bubonic plague. Infect. Immun. 79:4984–4989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weening EH, Cathelyn JS, Kaufman G, Lawrenz MB, Price P, Goldman WE, Miller VL. 2011. The dependence of the Yersinia pestis capsule on pathogenesis is influenced by the mouse background. Infect. Immun. 79:644–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kukkonen M, Korhonen TK. 2004. The omptin family of enterobacterial surface proteases/adhesins: from housekeeping in Escherichia coli to systemic spread of Yersinia pestis. Int. J. Med. Microbiol. 294:7–14 [DOI] [PubMed] [Google Scholar]
- 19. Lobo LA. 2006. Adhesive properties of the purified plasminogen activator Pla of Yersinia pestis. FEMS Microbiol. Lett. 262:158–162 [DOI] [PubMed] [Google Scholar]
- 20. Saksela O. 1985. Plasminogen activation and regulation of pericellular proteolysis. Biochim. Biophys. Acta 823:35–65 [DOI] [PubMed] [Google Scholar]
- 21. Kukkonen M, Lahteenmaki K, Suomalainen M, Kalkkinen N, Emody L, Lang H, Korhonen TK. 2001. Protein regions important for plasminogen activation and inactivation of α2-antiplasmin in the surface protease Pla of Yersinia pestis. Mol. Microbiol. 40:1097–1111 [DOI] [PubMed] [Google Scholar]
- 22. Sodeinde OA, Subrahmanyam YV, Stark K, Quan T, Bao Y, Goguen JD. 1992. A surface protease and the invasive character of plague. Science 258:1004–1007 [DOI] [PubMed] [Google Scholar]
- 23. Sodeinde OA, Sample AK, Brubaker RR, Goguen JD. 1988. Plasminogen activator/coagulase gene of Yersinia pestis is responsible for degradation of plasmid-encoded outer membrane proteins. Infect. Immun. 56:2749–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kukkonen M, Suomalainen M, Kyllonen P, Lahteenmaki K, Lang H, Virkola R, Helander IM, Holst O, Korhonen TK. 2004. Lack of O-antigen is essential for plasminogen activation by Yersinia pestis and Salmonella enterica. Mol. Microbiol. 51:215–225 [DOI] [PubMed] [Google Scholar]
- 25. Bartra SS, Styer KL, O'Bryant DM, Nilles ML, Hinnebusch BJ, Aballay A, Plano GV. 2008. Resistance of Yersinia pestis to complement-dependent killing is mediated by the Ail outer membrane protein. Infect. Immun. 76:612–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Felek S, Krukonis ES. 2009. The Yersinia pestis Ail protein mediates binding and Yop delivery to host cells required for plague virulence. Infect. Immun. 77:825–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kolodziejek AM, Schnider DR, Rohde HN, Wojtowicz AJ, Bohach GA, Minnich SA, Hovde CJ. 2010. Outer membrane protein X (Ail) contributes to Yersinia pestis virulence in pneumonic plague and its activity is dependent on the lipopolysaccharide core length. Infect. Immun. 78:5233–5243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Felek S, Tsang TM, Krukonis ES. 2010. Three Yersinia pestis adhesins facilitate Yop delivery to eukaryotic cells and contribute to plague virulence. Infect. Immun. 78:4134–4150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tsang TM, Felek S, Krukonis ES. 2010. Ail binding to fibronectin facilitates Yersinia pestis binding to host cells and Yop delivery. Infect. Immun. 78:3358–3368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Park JS, Lee WC, Yeo KJ, Ryu KS, Kumarasiri M, Hesek D, Lee M, Mobashery S, Song JH, Kim SI, Lee JC, Cheong C, Jeon YH, Kim HY. 2012. Mechanism of anchoring of OmpA protein to the cell wall peptidoglycan of the gram-negative bacterial outer membrane. FASEB J. 26:219–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sugawara E, Nikaido H. 1992. Pore-forming activity of OmpA protein of Escherichia coli. J. Biol. Chem. 267:2507–2511 [PubMed] [Google Scholar]
- 32. Fu H, Belaaouaj AA, Dahlgren C, Bylund J. 2003. Outer membrane protein A-deficient Escherichia coli activates neutrophils to produce superoxide and shows increased susceptibility to antibacterial peptides. Microbes Infect. 5:781–788 [DOI] [PubMed] [Google Scholar]
- 33. Llobet E, March C, Giménez P, Bengoechea JA. 2009. Klebsiella pneumoniae OmpA confers resistance to antimicrobial peptides. Antimicrob. Agents Chemother. 53:298–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Prasadarao NV, Wass CA, Weiser JN, Stins MF, Huang SH, Kim KS. 1996. Outer membrane protein A of Escherichia coli contributes to invasion of brain microvascular endothelial cells. Infect. Immun. 64:146–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bartra SS, Gong X, Lorica CD, Jain C, Nair MK, Schifferli D, Qian L, Li Z, Plano GV, Schesser K. 2011. The outer membrane protein A (OmpA) of Yersinia pestis promotes intracellular survival and virulence in mice. Microb. Pathog. 52:41–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Erova TE, Pillai L, Fadl AA, Sha J, Wang S, Galindo AK, Chopra CL. 2006. DNA adenine methyltransferase influences the virulence of Aeromonas hydrophila. Infect. Immun. 74:410–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265–275 [PubMed] [Google Scholar]
- 38. Agarkov A, Chauhan S, Lory PJ, Gilbertson SR, Motin VL. 2008. Substrate specificity and screening of the integral membrane protease Pla. Bioorg. Med. Chem. Lett. 18:427–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sha J, Endsley JJ, Kirtley ML, Foltz SM, Huante MB, Erova TE, Kozlova EV, Popov VL, Yeager LA, Zudina IV, Motin VL, Peterson JW, DeBord KL, Chopra AK. 2011. Characterization of an F1 deletion mutant of Yersinia pestis CO92, pathogenic role of F1 antigen in bubonic and pneumonic plague, and evaluation of sensitivity and specificity of F1 antigen capture-based dipsticks. J. Clin. Microbiol. 49:1708–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sha J, Pillai L, Fadl AA, Galindo CL, Erova TE, Chopra AK. 2005. The type III secretion system and cytotoxic enterotoxin alter the virulence of Aeromonas hydrophila. Infect. Immun. 73:6446–6457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zavialov AV, Knight SD. 2007. A novel self-capping mechanism controls aggregation of periplasmic chaperone Caf1M. Mol. Microbiol. 64:153–164 [DOI] [PubMed] [Google Scholar]
- 42. Agar SL, Sha J, Foltz SM, Erova TE, Walberg KG, Parham TE, Baze WB, Suarez G, Peterson JW, Chopra AK. 2008. Characterization of a mouse model of plague after aerosolization of Yersinia pestis CO92. Microbiology 154:1939–1948 [DOI] [PubMed] [Google Scholar]
- 43. Agar SL, Sha J, Foltz SM, Erova TE, Walberg KG, Parham TE, Baze WB, Suarez G, Peterson JW, Chopra AK. 2009. Characterization of the rat pneumonic plague model: infection kinetics following aerosolization of Yersinia pestis CO92. Microbes Infect. 11:205–214 [DOI] [PubMed] [Google Scholar]
- 44. Anisimov AP, Dentovskaya SV, Panfertsev EA, Svetoch TE, Kopylov PK, Segelke BW, Zemla A, Telepnev MV, Motin VL. 2010. Amino acid and structural variability of Yersinia pestis LcrV protein. Infect. Genet. Evol. 10:137–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Friedlander AM, Welkos SL, Worsham PL, Andrews GP, Heath DG, Anderson GW, Jr, Pitt ML, Estep J, Davis K. 1995. Relationship between virulence and immunity as revealed in recent studies of the F1 capsule of Yersinia pestis. Clin. Infect. Dis. 21:178–181 [DOI] [PubMed] [Google Scholar]
- 46. Davis KJ, Fritz DL, Pitt ML, Welkos SL, Worsham PL, Friedlander AM. 1996. Pathology of experimental pneumonic plague produced by fraction 1-positive and fraction 1-negative Yersinia pestis in African green monkeys (Cercopithecus aethiops). Arch. Pathol. Lab. Med. 120:156–163 [PubMed] [Google Scholar]
- 47. Williamson ED, Packer PJ, Waters EL, Simpson AJ, Dyer D, Hartings J, Twenhafel N, Pitt MLM. 2011. Recombinant (F1+V) vaccine protects cynomolgus macaques against pneumonic plague. Vaccine 29:4771–4777 [DOI] [PubMed] [Google Scholar]
- 48. Anderson GW, Jr, Worsham PL, Bolt CR, Andrews GP, Welkos SL, Friedlander AM, Burans JP. 1997. Protection of mice from fatal bubonic and pneumonic plague by passive immunization with monoclonal antibodies against the F1 protein of Yersinia pestis. Am. J. Trop. Med. Hyg. 56:471–473 [DOI] [PubMed] [Google Scholar]
- 49. Turner JK, McAllister MM, Xu JL, Tapping RI. 2008. The resistance of BALB/cJ mice to Yersinia pestis maps to the major histocompatibility complex of chromosome 17. Infect. Immun. 76:4092–4099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Smiley ST. 2008. Immune defense against pneumonic plague. Immunol. Rev. 225:256–271 [DOI] [PMC free article] [PubMed] [Google Scholar]