

Abstract
The prokaryote Chlamydia trachomatis and the protozoan Toxoplasma gondii, two obligate intracellular pathogens of humans, have evolved a similar modus operandi to colonize their host cell and salvage nutrients from organelles. In order to gain fundamental knowledge on the pathogenicity of these microorganisms, we have established a cell culture model whereby single fibroblasts are coinfected by C. trachomatis and T. gondii. We previously reported that the two pathogens compete for the same nutrient pools in coinfected cells and that Toxoplasma holds a significant competitive advantage over Chlamydia. Here we have expanded our coinfection studies by examining the respective abilities of Chlamydia and Toxoplasma to co-opt the host cytoskeleton and recruit organelles. We demonstrate that the two pathogen-containing vacuoles migrate independently to the host perinuclear region and rearrange the host microtubular network around each vacuole. However, Toxoplasma outcompetes Chlamydia to the host microtubule-organizing center to the detriment of the bacterium, which then shifts to a stress-induced persistent state. Solely in cells preinfected with Chlamydia, the centrosomes become associated with the chlamydial inclusion, while the Toxoplasma parasitophorous vacuole displays growth defects. Both pathogens fragment the host Golgi apparatus and recruit Golgi elements to retrieve sphingolipids. This study demonstrates that the productive infection by both Chlamydia and Toxoplasma depends on the capability of each pathogen to successfully adhere to a finely tuned developmental program that aims to remodel the host cell for the pathogen's benefit. In particular, this investigation emphasizes the essentiality of host organelle interception by intravacuolar pathogens to facilitate access to nutrients.
INTRODUCTION
Obligate intracellular pathogens that infect mammals include all viruses, some bacteria such as Chlamydia and Rickettsia spp., and protozoa such as Toxoplasma gondii and Trypanosoma cruzi. To survive, these pathogens have mastered maintaining a delicate balance between exploiting and preserving the cells of their host: they consume host nutrients from the intracellular space that they occupy while keeping host cell integrity in order to sustain intracellular growth. Many obligate intracellular pathogens reside in a vacuolar compartment that represents a relatively safe niche that offers protection from hostile cytosolic innate immune-surveillance pathways and potent inflammatory signaling cascades (1). Intravacuolar pathogens have developed unique processes to invade cells and establish a safe vacuole (2). The origin and composition of the pathogen-containing compartments are largely dictated by the mode of host cell entry and the specificity of the host-pathogen interaction. Vacuolar compartments may be created de novo by the pathogen itself or be derived from host endocytic membranes that are further modified by insertion of microbial factors to become resistant to lysosomal fusion and destruction. Once in a vacuolar compartment, the pathogens need to divert host cell components and co-opt host cell pathways in order to have access to nutrient pools and consequently multiply (3).
Chlamydia trachomatis is an obligate intracellular Gram-negative bacterium that infects a wide range of cell types in humans, with some preference for mucosal epithelial cells. Chlamydial infections are the most common bacterial sexually transmitted infections in humans and are the leading cause of infectious blindness worldwide (4). At the cellular level, C. trachomatis invades cells within 10 min by a specialized form of endocytosis involving chlamydial adhesins and host cell receptors (5, 6), differentiates into a replicate form at ∼30 min postinfection (p.i.), and at ∼3 h p.i. multiplies in its vacuole, termed the inclusion. From 4 h p.i., C. trachomatis-infected mammalian cells undergo dramatic modifications inflicted by the bacterium. These include the remodeling of the host cytoskeleton, e.g., microtubules (7, 8), the recruitment of the host microtubule-organizing center (MTOC) (9, 10), and the attraction of host organelles, e.g., Golgi vesicles and endocytic structures, in order to acquire lipids required for their growth (11–20). As a prototypic obligate intracellular protozoan organism, Toxoplasma gondii is adapted for invasion and multiplication in any nucleated mammalian cell (21). This protozoan parasite (referred to here as “parasite”) causes life-threatening disease in immunocompromised individuals and is responsible for lethal encephalitis in these patients (22). T. gondii actively invades cells within 1 min, creates its own membrane-bound compartment named the parasitophorous vacuole (PV), and immediately undertakes rounds of division every 7 h. Like C. trachomatis, Toxoplasma is also notorious for its ability to extensively modify its host cell and does so in a manner strikingly similar to that reported for C. trachomatis, despite differences in the origin and composition of the inclusion and the PV (22–28). This functional similarity between these two disparate pathogens may be viewed as an example of convergent evolution across phylogenetic domains. Unlike C. trachomatis, Toxoplasma recruits host mitochondria that associate with the PV membrane (PVM) (29, 30). Interestingly, the more invasive Chlamydia psittaci species also recruits host mitochondria around its inclusion (31).
In a previous study, we have established an in vitro cell culture model whereby single fibroblasts were infected by C. trachomatis and Toxoplasma simultaneously (32). In a coinfection system, there is a balance between the success and failure of an infection established by a pathogen that depends on the skills of the pathogen to adhere to its normal developmental program. We showed that a single fibroblast could harbor both chlamydiae and Toxoplasma and that the two pathogens resided in distinct compartments. Toxoplasma held a significant competitive edge over Chlamydia in coinfected cells, as it was able to divert nutrients to the PV with the same efficiency as in monocultures. Consequently, the infectious cycle of the Toxoplasma progressed unimpeded. In contrast, Chlamydia lost the ability to scavenge essential nutrients during coinfection, and the bacterium shifted to a stress-induced persistent mode of growth as a result from being barred from its normal nutrient supplies. Competition between the parasite and the bacterium was further documented by coinfecting with C. trachomatis and slow-growing strains of Toxoplasma or a mutant impaired in nutrient acquisition, whereby chlamydiae developed unhampered. Likewise, in a cell preinfected for 2 days with Chlamydia prior to infection with Toxoplasma, the parasite replicated very slowly. Finally, the tables were turned on Toxoplasma since the parasite's development was arrested in cells coinfected with Toxoplasma and a highly virulent strain of Chlamydia psittaci.
In this study, we expand our understanding of cell biological events underlying the interactions of Chlamydia and Toxoplasma with their mammalian host cell. Specifically, we question whether the co-occurrence of the two pathogens in the same cell does interfere with the innate ability of each to remodel the host cell interior to its own advantage. To provide insight into the cellular events that take place in a dually infected cell, we have examined the distribution of host cell structures relative to the chlamydial inclusion and the PV of Toxoplasma. We show that the host cell undergoes extensive modifications when coinfected by the parasite and the bacterium, as cellular organelles are relocated to the inclusion and the PV. We conclude that Chlamydia and Toxoplasma tend to adhere to their respective intracellular developmental program regardless of the presence of another organism in the cell and that the normal growth of each pathogen (i.e., the production of infectious progeny) is highly dependent on the pathogen's ability to maintain a threshold level of interaction between its vacuole and host cell organelles.
MATERIALS AND METHODS
Reagents and antibodies.
All chemicals were obtained from Sigma Chemical Co. (St. Louis, MO) or Fisher (Waltham, MA) unless indicated otherwise. The C6-ceramide complexed to bovine serum albumin (BSA) was from Molecular Probes (Seattle, WA). The antibodies used for immunofluorescence assays (IFAs) included (i) rabbit or rat polyclonal anti-T. gondii GRA7 (anti-TgGRA7) (26), (ii) mouse monoclonal anti-EF-Tu (33) and rabbit polyclonal anti-IncA (a gift from T. Hackstadt, NIH Rocky Mountain Laboratories, Hamilton, MT), (iii) commercial mouse or rabbit anti-α-tubulin and anti-γ-tubulin, (iv) commercial chicken anti-CERT, (v) commercial rabbit anti-giantin, and (vi) commercial rabbit anti-Tom20 (Santa Cruz Biotech, Santa Cruz, CA). All primary antibodies were used at a dilution of 1:100, except for the anti-TgGRA7 and anti-CERT antibodies, which were used at a dilution of 1:200. The secondary antibodies were goat anti-IgG conjugated to either Alexa 488 or Alexa 594 (Invitrogen) before dilution at 1:2,000.
Propagation of mammalian cells and pathogens.
The human foreskin fibroblasts (HFFs) used in this study were obtained from the American Type Culture Collection (Manassas, VA). HFFs were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin-streptomycin (100 U/ml per 100 μg/ml), and 2 mM l-glutamine. The tachyzoite RH strain (type I) of Toxoplasma gondii was used throughout this study. Red fluorescent protein (RFP)-expressing stable transgenics derived from the RH strain were generously provided by F. Dzierszinski (McGill University, Canada). The parasites were propagated in vitro by serial passage in monolayers of HFFs in DMEM plus 10% FBS (34). The Chlamydia trachomatis lab reference serovar E/UW5-CX was used throughout this study. In one set of experiments, the pathogenic avian type strain Chlamydia psittaci Cal-10 was used. All chlamydiae were propagated in HeLa cells at 37°C with 5% CO2 in DMEM supplemented with 10% FBS, 25 μg/ml gentamicin, and 1.25 μg/ml amphotericin B (Fungizone) as described previously (35).
Infection protocols.
Mammalian cells were seeded onto coverslips in 24-well plates for IFA or in 6-well tissue culture dishes for electron microscopy (EM) and grown at 37°C in a CO2 incubator until 70% confluence. Prior to infection experiments, cells were washed with PBS and incubated for 24 h in antibiotic- and amphotericin B-free medium. To examine the interactions of T. gondii and chlamydiae with their host cells, coinfection and superinfection protocols were used, as previously described (32).
Ceramide uptake assay.
Infected fibroblasts were exposed to 5 μM 6-{[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]hexanoyl}sphingosine (NBD)–C6-ceramide complexed to BSA for 1 h in serum-free medium before examination by live microscopy. In some assays using NBD–C6-ceramide, the cells were infected with the parasites or the bacteria for 24 h and then exposed to 10 μM pyrimethamine and 100 units/ml of penicillin G, respectively, for an additional day before the pulse with the fluorescent lipid.
Fluorescence microscopy.
For IFAs, cells were fixed in a solution consisting of 4% phosphonoformic acid (Polysciences, Inc., Warrington, PA) and 0.02% glutaraldehyde in phosphate-buffered saline (PBS) for 15 min and permeabilized with 0.3% Triton X-100 for 5 min, except for IncA and α- and γ-tubulin immunostaining, for which cells were fixed and permeabilized with 100% methanol for 5 min at −20°C. Samples were then blocked with 3% BSA, dissolved in PBS for 45 min, and probed with primary antibodies diluted in blocking buffer for 1 h to 2 h. Samples were then washed 3 times and probed with secondary antibodies diluted in the blocking buffer for 45 min. Coverslips were mounted onto glass microscope slides using ProLong Gold antifade mounting solution with or without DAPI (4′,6-diamidino-2-phenylindole; Invitrogen). For all fluorescence assays, images were acquired on a Nikon Eclipse E800 microscope equipped with a Spot RT charge-coupled-device camera and processed using Image-Pro-Plus software (Media Cybernetics, Silver Spring, MD) before assembly using Adobe Photoshop software (Adobe Systems, Mountain View, CA). Some images were also viewed with a Nikon Plan Apo ×100 objective using a Nikon 90i microscope, and pictures were taken using a Hammatsu ORCA-ER camera and Volocity software. Images (a z stack per field) were processed using iterative restoration (confidence limit, 98%; iteration limit, 25), and further processing was done using Adobe Photoshop software. For quantification of NBD-ceramide intensity, the total intensity in the green channel was determined for each vacuole and compared with the total fluorescence intensity of the host Golgi apparatus using the following equation: [sum intensity (pathogenic vacuole center)/sum intensity (entire host Golgi apparatus)] ×100. The same quantitative measurements were applied to monitor the fluorescence intensity of microtubules around the inclusions and PVs. Small squares (5 by 5 μm2) were selected from 15 different cells and positioned in the surroundings of the vacuoles. Other squares were positioned within the host cytoplasm at a distance of 20 μm away from the vacuoles for comparison of the fluorescence intensities with microtubules distributed around the vacuoles. The number of Golgi elements or giantin foci present in uninfected and infected cells was measured on three-dimensional (3-D) reconstructed volumes using Volocity software, where individual giantin foci were identified by intensity, and objects less than 0.2 cm3 were excluded from the calculations. The mean volume and standard deviation (SD) were calculated from three independent experiments using Excel software (Microsoft, Redmond, WA).
Electron microscopy.
For thin-section transmission EM (TEM), cells were fixed in 2.5% glutaraldehyde (Electron Microscopy Sciences, Hatfield, PA) in 0.1 M sodium cacodylate buffer (pH 7.4) for 1 h at room temperature and processed as described previously (36) before examination with a Philips CM120 electron microscope (Eindhoven, the Netherlands) under 80 kV.
Statistical analysis.
For the comparison of means, P values were determined by the analysis of variance against the control (ANOVA 2).
RESULTS
Intracellular development of Chlamydia is impaired throughout coinfection with Toxoplasma.
We previously demonstrated that Toxoplasma and Chlamydia trachomatis could simultaneously invade the same cell (either a fibroblast or an epithelial cell) and develop in their respective vacuoles during a 24-h period (32). Toxoplasma exhibited a normal growth rate, whereas the development of C. trachomatis was altered into a stress-induced persistent state. This phenotype was evidenced by (i) the presence of aberrant reticulate bodies (RBs) within the inclusions, (ii) the downregulated expression of late-developmental-stage proteins by the bacteria, and (iii) the reversibility of the persistent state, namely, the restoration of chlamydial growth and late differentiation to infectious elementary bodies (EBs) upon killing of Toxoplasma or supplementation of the culture medium with nutrients. To further explore differences in the growth and development of both the parasite and the bacterium, we examined the PV and inclusion sizes from the onset until late coinfection in dually infected cells. Figure 1A displays PVs labeled for GRA7 (a PVM marker) and inclusions stained for EF-Tu (a cytoplasmic marker for all chlamydial developmental stages) at 3 h, 16 h, and 32 h postcoinfection. The vacuoles of the two pathogens were often found in close proximity to each other in the host perinuclear region. In monocultures, a PV contained an average of 6 and 12 Toxoplasma parasites for the 16-h and 32-h periods of intracellular development, respectively, with each parasite measuring 2 by 7 μm. In comparison, the replicative forms of Chlamydia, the RBs, reached numbers of 50 to 100 and 300 to 600 (per focal section) at 16 h and 32 h p.i., respectively, in monocultures, with each RB homogenously measuring 1 μm in diameter (37). When Toxoplasma was cultivated with Chlamydia, no major differences in the size of the parasites and their number per vacuole were observed at between 3 h and 32 h in coinfected cells (compare Fig. 1A to B). In contrast, the morphological features of the inclusions were dramatically altered in coinfected cells, as smaller inclusions containing enlarged bacteria up to 5 μm in diameter were visible at 16 h postcoinfection (compare Fig. 1A to C). The inclusions expanded in size after 32 h of coculture, but overall, they remained much smaller than inclusions that developed in monoinfected fibroblasts (not shown). At 48 h postcoinfection, the host cell was mainly occupied by very large PVs. At day 3, the parasites egressed from the host cell and were competent to invade new cells, while the bacteria remained enclosed in their inclusions (not shown). We performed quantitative image analysis to compare inclusion size in monoinfected versus coinfected fibroblasts at between 6 h and 42 h p.i. (Fig. 2). Results confirm the much slower growth of inclusions in the presence of PVs, with an inclusion size of 198 ± 33 μm2 after 42 h. In monocultures, the inclusions grew exponentially and were ∼10 times larger than inclusions in coinfected cells, with an inclusion size of 2,166 ± 74 μm2 at 42 h p.i. These observations suggest that, following the cointernalization of Toxoplasma and Chlamydia in the same fibroblast, the bacterium instantly displays a limited capacity to divide, thus generating aberrantly enlarged RBs, while the inclusion development is impaired, thus resulting in the formation of a smaller inclusion. Moreover, in the tested time window, chlamydial growth and development never resumed to normal levels. This strongly suggests that the sharing of an infected cell with Toxoplasma represents a stressful growth condition for Chlamydia. The bacterium then deviates into a stress-induced persistent state (38).
Fig 1.

Morphology of the PV and inclusions of C. trachomatis in cocultures compared to monocultures. (A to C) IFA of T. gondii and C. trachomatis in co- or monocultures. Intravacuolar bacteria and parasites were identified in HFFs using antibodies against EF-Tu (red) and GRA7 (green), respectively, at the indicated times p.i. For direct visual comparison of the vacuolar niches in co- versus monocultures, all images are at the same magnification. Bars, 10 μm.
Fig 2.
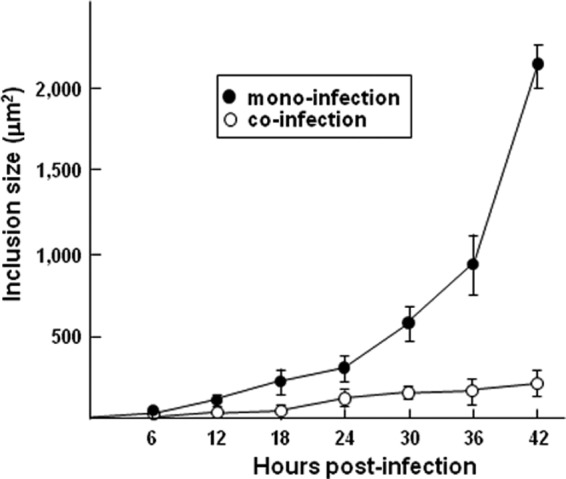
Measurement of inclusion size in mono- and cocultures. To score the size of the chlamydial inclusions, random fields of 75 to 100 EF-Tu-labeled inclusions grown in HFFs at the indicated times in the presence or the absence of Toxoplasma were selected at the indicated times. Data are means ± SDs of 3 independent assays.
The inclusion and the PV do not fuse during coinfection.
The dramatic impact of Toxoplasma on the development of C. trachomatis during coinfection raises the possibility that the Toxoplasma and Chlamydia vacuoles interact with each other to the detriment of the bacterium. A common feature shared by the two pathogens is the formation of membranous fibers that extend away from their respective vacuoles and pervade the cytosol (26, 39–42). These vacuolar membranous extensions form contact with host organelles and other vacuoles located in the cell. The production and distribution of the membranous extensions derived from the inclusion and PV in coinfected cells were analyzed by fluorescence microscopy and compared to those in monoinfected cells. In cells monoinfected for 16 h, we observed a dense network of fibers containing the inclusion membrane protein IncA, up to 10 fibers per inclusion, distributed around the host nucleus (Fig. 3A, panel a) or connecting two inclusions located on opposite sides of the nucleus (Fig. 3A, panel b). In Toxoplasma-infected cells, GRA7-stained extensions from the PVM were visible in the nuclear region (Fig. 3A, panel c) or linking two PVs together (Fig. 3A, panel d). In coinfected cells harboring several PVs or inclusions, we frequently observed chlamydial inclusions that were connected with IncA-labeled tubules (Fig. 3B, panel a) and PVs associated with each other by long GRA7-labeled extensions (Fig. 3B, panel b), as in monoinfections. Under stress conditions, the size and abundance of IncA-laden fibers increase significantly (42, 43). However, we did not observe a significant difference in the number and length of inclusion fibers during coinfection versus monoinfection. In coinfected cells, the extensions emanating from both the PV and inclusion could reach 30 μm in length, suggestive of their remarkable stability. In the vast majority of coinfected cells, the PVM projections (PVMPs) did not interact with the inclusions. However, in ∼5% coinfected cells, we observed chlamydial fibers extending toward the PV and IncA materials accumulated near the PV in coinfected cells, as shown on the 3-D micrographs (Fig. 4). Although IncA is thought to promote fusion events between inclusions, we never observed fusion between a PV and an inclusion or between the respective fibers of these vacuoles.
Fig 3.

Physical interaction between the membranes of the inclusion and the PV in coinfected cells. (A and B) IFA of C. trachomatis (a and b) and Toxoplasma (c and d) in monoinfections (A) or coinfections (B) at 16 h p.i. The inclusion and the PV membranes were immunolabeled for IncA and GRA7, respectively, highlighting membranous extensions wrapping around the host nucleus (panels a and c in panel A) or connecting two vacuoles (panels b and d in panel A). DAPI staining is in blue. Panels a and b in panel B show two inclusions and two PVs connected to each other, respectively, in a manner similar to that in monoinfected cells. Bars, 10 μm.
Fig 4.

Interaction between the PV and the inclusion mediated by chlamydial fibers. IFA of a chlamydial inclusion stained for IncA and Toxoplasma PVM stained for GRA7 in cells coinfected for 16 h. A 3-D reconstruction of a series of optical z stacks of a PV is shown. The top image on the left is an extended-focus view, and images on the right side are the 3-D reconstruction of the z stacks, which are rotated to present different views. Bar, 10 μm.
Both Chlamydia and Toxoplasma recruit host microtubules to their vacuole in coinfected cells.
We next examined the capability of Chlamydia and Toxoplasma to rearrange the host microtubular network around their vacuole and to attract structures connected to microtubules, such as the microtubule-organizing center (MTOC) and the Golgi complex. In monoinfected cells, both C. trachomatis and Toxoplasma usually establish their nest near the host peri-Golgi complex-MTOC region. Each vacuole associates with host centrosomes and is positioned at the center of the microtubular network (24, 26, 44–46). The analogous behavior of the C. trachomatis and Toxoplasma vacuoles prompted us to examine the architecture of the host microtubular network in coinfected cells by immunostaining for α-tubulin (Fig. 5). Microtubules in uninfected cells formed a framework surrounding the nucleus, radiating toward the cell periphery (Fig. 5A). In coinfected cells, inclusions and PVs remained associated with distinct microtubules at 6 h p.i. and were enwrapped by the microtubular network by 24 h p.i., similar to monoinfected cells (Fig. 5B and C). In coinfected cells, the network of microtubules was evenly distributed around each vacuolar compartment. The intensity of fluorescent staining of microtubules around inclusions and PVs in coinfected cells was measured and compared to the fluorescence levels of cortical microtubules dispersed in the host cytoplasm. Figure 5D shows the distribution patterns of the relative fluorescence intensity of microtubules obtained from 150 locations in the host cytoplasm, around the inclusions and around the PVs. The relative fluorescence intensities surrounding the inclusions and the PVs were similar to one another and higher than the fluorescence levels in the host cytoplasm. The mean values of intensities were 97.7 ± 9.3, 134.5 ± 7.2, and 145.1 ± 14.2 for microtubules in the host cytoplasm, around the inclusions, and around the PVs, respectively (P < 0.01). This indicates that in coinfected cells, Chlamydia and Toxoplasma are able to mobilize large numbers of microtubules from the host cytoplasm to their respective vacuoles. For each pathogen, this property appears to be independent of the presence of the other pathogen, as no difference was observed between coinfected and monoinfected cells.
Fig 5.

Association of host microtubules with the chlamydial inclusion and the PV. (A to C) IFA of Toxoplasma- and Chlamydia-infected fibroblasts. Host microtubules were visualized by immunostaining for α-tubulin (green). Parasites and bacteria were labeled for GRA7 and EF-Tu, respectively, or indentified by phase-contrast imaging (single and double arrowheads for T. gondii and C. trachomatis, respectively). DAPI staining is in blue. hn, host nucleus. Bars, 10 μm. (D) Measurements of the fluorescence intensity associated with microtubules around the inclusions and PVs. The relative fluorescence intensities of microtubules were measured in 150 different locations in the host cytoplasm (back bars), around the inclusions (gray bars), and around the PVs (white bars). The small squares shown in red in panel C were selected from 15 different cells and positioned in the surroundings of the vacuoles and within the host cytoplasm. One hundred fifty squares were examined for each group, and the distribution patterns of 150 intensities for each group were plotted.
Toxoplasma wins the battle for the centrosome in coinfected cells.
We next developed morphological assays to assess the subcellular location of host centrosomes in cells dually infected with Chlamydia and Toxoplasma. Fibroblasts infected for 24 h were stained with antibodies against γ-tubulin, a protein marker of the pericentriolar matrix (Fig. 6). In all infected cells, the normal positioning of the centrosomes at the nuclear periphery was disrupted, as these structures were detached from the host nuclear envelope (compare Fig. 6A to B). Host centrosomes were distributed at the PV periphery at a high frequency in mono- and coinfected cells. In the case of coinfected cells harboring multiple PVs, the centrosomes were associated with the largest PV containing the progeny of the first parasite that invaded the cell, as illustrated in panel c of Fig. 6B. Of interest, we did not observe more than two centrosomes per coinfected cell, although supernumerary centrosomes were often seen in cells infected with either Toxoplasma or Chlamydia alone. These observations were validated by quantitative analysis of the number of PVs and inclusions that were less than 1 μm away from the host MTOC (Fig. 6C). In monoinfected cells, the MTOC was seen in close proximity to ∼85% of the PVs and ∼90% of inclusions, while in coinfected cells, ∼95% of the PVs were associated with the MTOC but only ∼3% were associated with the inclusions. The rare instances in which the MTOC was found close to the inclusion were when the two pathogens were in close proximity to one another, and the MTOC appeared to be squeezed between the PV and the inclusion. From these results, it appears that Toxoplasma is more proficient than C. trachomatis in associating with the host MTOC during coinfection.
Fig 6.

Relocation of the host MTOC in coinfected cells. (A) IFA of uninfected fibroblasts showing the subcellular location of the MTOC stained for γ-tubulin (green), which is apposed to the nuclear envelope during interphase (G1) and mitosis (M). (B) IFA of Toxoplasma- and Chlamydia-infected fibroblasts. HFFs were stained for γ-tubulin, and the parasites and bacteria were identified by immunostaining for GRA7 and EF-Tu, respectively. (a) A monoinfected cell and a coinfected cell (delineated by white and yellow dashed lines, respectively), allowing comparison of the distribution of the host MTOC in cells infected with Chlamydia alone and in cells infected with the two pathogens for 24 h; (b and c) the MTOC nearest the largest PV. Single and double arrowheads point to the PV and the inclusion, respectively. Bars, 10 μm. (C) Measurement of the spatial distribution of the MTOC relative to the nucleus in uninfected cells and to the PV and inclusion in infected cells. The bars represent the percentage of MTOCs that are ≤1 μm from either the nucleus in uninfected cells or the PV and the inclusion (incl.) in mono- or coinfected cells. Data are means ± SDs of 3 independent assays, with 80 to 100 vacuoles counted in each experiment (***, P < 0.001).
In cells preinfected with Chlamydia, the host centrosomes are associated with the inclusions, but neither the bacterium nor the parasite develops normally.
Our results indicate that Toxoplasma develops normally in cells coinfected with C. trachomatis, mostly as if nothing was unusual in its environment, clearly establishing a competitive edge of the parasite over Chlamydia. We next aimed to challenge the parasite's ability to multiply in the presence of chlamydial inclusions by infecting fibroblasts first with Chlamydia and allowing the bacteria to grow for 24 h prior to the addition of Toxoplasma to the culture medium. Invasion of C. trachomatis prior to T. gondii would also give the bacteria the opportunity to hijack the host MTOC and perhaps develop normally. To explore this possibility, Chlamydia-preinfected cells were stained for γ-tubulin to localize the MTOC in coinfected cells (Fig. 7). Toxoplasma was able to invade Chlamydia-infected cells by forming a typical PV (Fig. 7A). In the vast majority of coinfected cells (∼97%), the MTOC was close to the inclusions and distant from the PVs, and in very few cells, the MTOC was close to both the inclusion and the PV. Coinfected cells were then stained to examine the size of the vacuoles of each pathogen. In cells previously infected by Chlamydia, Toxoplasma development was impaired, as the majority of the PVs contained only one single parasite at 24 h p.i. (Fig. 7B, panel a). Some parasites could undergo one or two cycles of replication. Quantitative distribution of the number of parasites per PV indicated that ∼50% and ∼25% of the PVs contained one and two parasites, respectively, in monoinfected cells (Fig. 7B, panel b). About 20% of the PVs harbored three or four parasites in cells containing several inclusions. We never observed PVs with eight parasites, as were occasionally found in monoinfections. Examination of the parasite's ultrastructure by TEM revealed the presence of cytoplasmic granules containing stores of amylopectin (Fig. 7C), which are thought to appear when Toxoplasma undergoes encystation upon stress or nutrient starvation (47). The parasites also accumulated abnormal myelinic structures, another sign of distress. The presence of nascent daughter parasites in the mother cell was confirmed by TEM (Fig. 7C, panel b). This suggests that Toxoplasma replication is perturbed in cells preinfected with Chlamydia, perhaps owing to a limitation of available nutrients and/or the exclusion of the host MTOC from the PV.
Fig 7.

Toxoplasma superinfection of cells preinfected with Chlamydia. (A) IFA of Chlamydia in fibroblasts superinfected with Toxoplasma after 24 h. The host MTOC was labeled for γ-tubulin (γ-tub). Parasites (asterisks) were identified by phase image, and bacteria were immunostained with anti-EF-Tu antibodies. DAPI staining is in blue. Bars, 10 μm. (B) (a) IFA illustrating parasite replication (n = parasite number/PV). Bars, 10 μm. (b) Measurement of PV sizes. To assess Toxoplasma development in Chlamydia-preinfected cells (24 h of superinfection with Toxoplasma or 24 h of monoinfection), about 60 PVs were randomly selected and intravacuolar parasites were enumerated. Data on the distribution of Toxoplasma numbers per PV are means ± SDs of 3 independent assays for each condition. (C) (a) EM of Toxoplasma showing amylopectin granules (g), myelinic structures (arrows), and unidentified vacuoles (v). Note that the parasite has recruited host mitochondria (hm) and host ER (hER). n, nucleus. (b) EM of Toxoplasma illustrating two daughter parasites inside the mother cell, as demarcated by the nascent membrane cytoskeleton (inner membrane complex [IMC]; arrowheads). Bars, 500 nm. (D) IFA showing a Chlamydia cell in a monoinfected cell and a coinfected cell with Toxoplasma (delineated by white and yellow dashed lines, respectively) to compare the morphological features of the inclusions. Bar, 10 μm.
In the cells preinfected with Chlamydia for 24 h that were subsequently invaded by the parasite, we also noticed that chlamydial inclusions contained aberrant bodies (Fig. 7A, B, and D). This suggests that, although chlamydiae had formed large inclusions prior to Toxoplasma invasion, the bacteria still underwent stress. A single PV was sufficient to induce a shift of the chlamydial inclusion to a stress-induced persistent state, as the RBs had morphed to aberrant bodies (compare this finding with the morphology of a 48-h inclusion in a monoinfected cell shown in Fig. 7D). This suggests that in C. trachomatis-preinfected cells, the parasite is still able to disrupt the growth of the bacterium even after it has reached its middevelopmental stage.
Host Golgi fragments are equally organized around the PV and the inclusion in coinfected cells.
In most cells, the MTOC and the Golgi apparatus distribute on one side of the nucleus, and they remain connected by components of these two structures. The Golgi apparatus-MTOC region is at the intersection of the endocytic and biosynthetic pathways. Therefore, pathogen positioning in this area of the cell could facilitate the interception of host vesicular traffic through manipulation of the microtubular transport system, which would satisfy pathogen requirements for nutrients. Chlamydia retrieves sphingomyelin and cholesterol from the host Golgi apparatus (11, 13), and the bacterium relies on both endogenous host-derived sphingolipids and exogenously added ceramide in the culture medium to survive (11, 12, 48, 49). This process requires the fragmentation of the Golgi apparatus by degradation of the Golgi matrix protein golgin-84 mediated by Chlamydia (20). Our lipidomic analyses revealed that Toxoplasma contains more than 20 different species of sphingolipids (50). The main source of sphingolipids in the parasite remains undetermined. Some studies reported a biosynthetic pathway for ceramide and glycosphingolipids in Toxoplasma (51, 52). Other studies supported the suggestion that Toxoplasma sphingolipids are derived from the host Golgi apparatus (28).
In this context, we aimed to determine to which extent the Golgi apparatus would be restructured in coinfected cells by examining the morphology of the Golgi apparatus using antibodies against the cis-Golgi protein giantin. In uninfected cells, the Golgi apparatus forms a ribbon-like structure that wraps around the nucleus (Fig. 8A). We confirmed that the Golgi apparatus was largely dispersed in Chlamydia-infected cells. We observed a similar situation in Toxoplasma-infected cells, as the Golgi apparatus was associated with the PV. Cells simultaneously infected with the two pathogens for 14 h displayed dramatic structural alterations of the Golgi apparatus, with displacement of the organelle away from the nucleus, loss of the Golgi apparatus ribbon shape, and wrapping of Golgi elements around both the inclusion and the PV (Fig. 8B). Remarkably, the individual association of either vacuole occurred independently of the number of PVs or inclusions within a cell. On rare occasions where the PV and the inclusion were on opposite sides of the host nucleus, Golgi fragments were seen to be associated with the PV and not with the inclusion (Fig. 8C). Quantitative analyses indicate that the number of giantin-labeled foci increased by ∼10-fold in coinfected cells (containing one PV and one inclusion), whereas the number increased ∼4-fold in monoinfected cells containing one PV and ∼6-fold in monoinfected cells with one inclusion (Fig. 8D). Nearly all of the PVs and inclusions were associated with Golgi elements in single infections, whereas 80% of the inclusions and 98% of the PVs interacted with the Golgi apparatus in coinfected cells.
Fig 8.

Redistribution of the host Golgi complex in coinfected cells. (A to C) IFA of Toxoplasma- and Chlamydia-infected fibroblasts at 24 h p.i. The Golgi complex in HFF during interphase was visualized by staining with anti-giantin antibodies (green), whereas parasites or bacteria were immunostained for GRA7 and EF-Tu, respectively (n = PV number). Single and double arrowheads point to the PV and the inclusion, respectively. Bars, 10 μm. (D) Quantification of Golgi apparatus breakdown in infected cells and association with the PVs and inclusions at 24 h p.i. In randomly selected HFFs labeled for giantin, the number of Golgi elements was enumerated in cells infected for 14 h and compared to that in uninfected cells (blue bars). Data are means ± SDs from 3 infections after normalization, using values collected from uninfected cells. In parallel, the number of PVs and inclusions (n = 50 to 60) closely associated with Golgi elements was measured (gray bars). Interactions were considered positive when the distance between the vacuolar compartment and Golgi apparatus material was ≤200 nm and more than 50% of the surface of the vacuolar compartment was covered by Golgi apparatus material. Tg, T. gondii; Ct, C. trachomatis; uninf., uninfected; inf. infected.
These results reveal that both pathogens are able to fragment the host Golgi apparatus, redirect Golgi apparatus pieces to their vacuoles, and maintain this association as the infection proceeds. With regard to Chlamydia, this process appears to be triggered in the early hours postinfection and to be independent of the recruitment of the host MTOC since this structure is captured by Toxoplasma in coinfected cells.
Chlamydia and Toxoplasma are proficient in retrieving sphingolipids from the host Golgi apparatus during coinfection.
In parallel, we monitored the uptake of host Golgi apparatus ceramide by the two pathogens. To investigate the transport of host ceramide to each pathogen-containing vacuole, we incubated cells with fluorescent ceramide before observation by live fluorescence microscopy (Fig. 9A). After 1 h of incubation, intense staining of the host Golgi apparatus was apparent in uninfected cells. In monoinfected cells, the fluorescent lipids were observed on Golgi elements distributed around the inclusions or the PVs and on the bacteria and parasites themselves. The plasma membranes of the two pathogens were stained, as was the limiting membrane of the inclusion and the PV. Transport of ceramide to the inclusion or the PV was ∼90% inhibited by penicillin and pyrimethamine, respectively, indicating that normal exponential growth of the two pathogens is required for lipid uptake (Fig. 9B). The inclusions and the PVs displayed similar amounts of accumulated ceramide (or derived lipids; Fig. 9B). These results reveal that both pathogens can successfully scavenge exogenous ceramide from the host Golgi apparatus. The ability to retrieve ceramide as efficiently during coinfection as during monoinfection suggests that T. gondii and C. trachomatis may have evolved different, i.e., noncompeting, mechanisms to incorporate Golgi apparatus-derived ceramide.
Fig 9.

Uptake of exogenous ceramide by Toxoplasma and Chlamydia during coinfection and distribution of the host CERT protein in infected cells. (A) Live fluorescence microscopy of uninfected or infected cells incubated with NBD-ceramide for 1 h. For infections, HFFs were incubated with C. trachomatis or T. gondii for 24 h before adding NBD-ceramide to the medium. In parallel experiments, cells were infected with the bacteria or the parasites and then treated with penicillin (+pen) and pyrimethamine (+pyr), respectively, prior to exposure to ceramide. Bars, 10 μm. (B) Quantitative fluorescence levels of NBD-ceramide incorporated into the PVs and inclusions of co- or monoinfected cells, in the absence or the presence of penicillin (+pen) and pyrimethamine (+pyr). Error bars indicate SEMs (n = 3 per condition) (**, P < 0.005). At least 60 images per condition were collected by confocal microscopy using sequential scanning and imported into Volocity software. Fluorescence intensity of staining in the PV or in the inclusion was expressed as a percentage of the total fluorescence intensity of staining found within the host Golgi apparatus. (C) Immunolocalization of the host CERT protein in uninfected cells with anti-CERT antibodies and cells infected with C. trachomatis and/or T. gondii for 24 h (n = PV number). Bars, 10 μm.
Recent studies identified the endoplasmic reticulum (ER)-to-Golgi ceramide transfer protein CERT (53) to be a host factor specifically recruited to the chlamydial inclusion and involved in sphingolipid delivery to the C. trachomatis inclusion (14, 19). To examine the localization of the CERT protein in our experimental system, we labeled infected cells with antibodies against the CERT protein. In uninfected cells, the CERT protein was found on small vesicles clustered in the perinuclear area (Fig. 9C). In Chlamydia-infected cells, we confirmed the redistribution of CERT protein from these cytoplasmic vesicles to the inclusion surface. In Toxoplasma-infected cells, the CERT-containing vesicles were intact and not associated with the PV. In coinfected cells, CERT staining was visible on the membrane of each inclusion regardless of the number of PVs in the cell and of the viability of the parasite. These results indicate that the presence of Toxoplasma in the cell does not disrupt in any way the recruitment of host CERT by Chlamydia. Toxoplasma does not compete with the bacterium to obtain ceramide and acquires this lipid likely via a CERT-independent pathway.
Both Toxoplasma and Chlamydia psittaci share host mitochondria.
It has been reported that unlike C. trachomatis, the inclusion of Chlamydia psittaci forms a tight association with many host mitochondria from 10 h p.i. (spacing of 5 nm between the inclusion membrane and host mitochondrion [31]), possibly through host kinesin phosphorylation (54). When kinesin activity is blocked and mitochondria are immobilized, C. psittaci growth is delayed, most probably through loss or restriction of energy availability. The T. gondii PVM also exhibits a remarkable association with host mitochondria (29, 30) mediated by parasite proteins (55), and this recruitment already occurs at 1 min p.i. Owing to the similar affinity of the C. psittaci and Toxoplasma vacuoles for host mitochondria, we examined the interaction of the two pathogens with the network of mitochondria in coinfected cells and compared the interaction with that in monoinfected cells. Mitochondria were immunostained for the outer mitochondrial membrane receptor Tom20. In uninfected cells, the network of mitochondria distributed around the nucleus and extended in the cytoplasm into long branches (Fig. 10A). In cells infected with C. psittaci for 24 h, the pattern of host mitochondria dramatically changed, as previously observed (31), and these organelles closely surrounded the entire circumference of the inclusion, leaving few mitochondria around the host nucleus and dispersed in the cytoplasm (Fig. 10B). When Toxoplasma was infecting cells for 24 h, each PV was equally decorated by host mitochondrial profiles, all around the periphery (Fig. 10C). We previously demonstrated that simultaneous coinfection of C. psittaci and Toxoplasma was detrimental for the parasite, likely owing to the faster replication of the bacterium than the parasite (32). Our results show that despite its growth being severely altered in coinfected cells, Toxoplasma was able to recruit host mitochondria as efficiently in coinfected as in monoinfected cells (Fig. 10D). This was observed in all coinfected cells observed regardless of the number of PVs per cell (panels a to c in Fig. 10D). The C. psittaci inclusions were also systematically covered by host mitochondria in coinfected cells. In very rare cases (∼4%) where several PVs were present in the cell, the inclusions developed poorly, never fused with each other, and remained small with a mean size of 37 ± 8 μm2 (normal inclusion size at 24 h p.i., 558 ± 46 μm2; Fig. 10D, panel c). Interestingly, in these coinfected cells, the atrophic inclusions were unable to associate with host mitochondria, unlike Toxoplasma.
Fig 10.

Redistribution of host mitochondria in cells coinfected with Toxoplasma and Chlamydia psittaci. (A to D) IFA of Toxoplasma- and C. psittaci-infected fibroblasts at 24 h p.i. Host mitochondria were immunostained with anti-Tom20 antibodies. C. psittaci was identified by DAPI staining and arrows on phase-contrast pictures. In panel c of panel D, 3 small inclusions are encircled. RFP-expressing Toxoplasma was used, and arrowheads on phase-contrast pictures point to the PVs. Bars, 10 μm.
These data illustrate that when in the same cell, Toxoplasma and C. psittaci are able to recruit host mitochondria and share this organelle, suggesting that this property is a factor contributing to successful productive infection. It is tempting to propose that the poor development of C. psittaci in cells containing multiple PVs in some instances may be caused by the absence of host mitochondria associated with the inclusions.
DISCUSSION
The intracellular pathogens C. trachomatis and Toxoplasma target and exploit host cell pathways and organelles throughout their intracellular infection cycle as part of a strategy to establish an environment beneficial for replication (56–58). Both pathogens usurp host microtubules, associate with the MTOC, cause fragmentation of the Golgi apparatus, intercept Golgi vesicles, co-opt endocytic organelles, and scavenge sphingolipids and sterols from these organelles. The abilities shared by Chlamydia and Toxoplasma to usurp the same host organelles implies that, if placed inside the same cell, the two pathogens should be in direct rivalry and that only the winner of that competition would be able to grow and develop normally (i.e., to yield infectious progeny) within the infected cell. In this study, we have examined whether the presence of Toxoplasma and C. trachomatis in the same mammalian cell alters the normal recruitment of host cell structures mediated by each pathogen. In particular, we have explored the capabilities of the bacterium and parasite to recruit host microtubules, the MTOC, and Golgi elements during coinfection. Our results reveal that the relative positioning and morphology of host microtubules, the MTOC, and the Golgi apparatus are more dramatically modified in cells infected by both pathogens than monoinfected cells, indicating that the damage inflicted by the parasite and bacterium on the host cell is additive. The inclusions and PVs are abundantly coated by host microtubules. The two pathogens distribute the fragments of the host Golgi apparatus equally around their respective vacuoles. The host MTOC is detached from the nucleus and entirely monopolized by Toxoplasma. These features suggest that following internalization, Chlamydia and Toxoplasma trigger an intracellular program whereby each pathogen takes advantage of the host cell in order to replicate in a favorable pathogen-specific environment, in spite of the presence of the other pathogen.
We propose the following sequence of events when C. trachomatis and Toxoplasma infect a cultured fibroblast concurrently. C. trachomatis invades a cell by an endocytic-like process and subsequently transforms the vacuolar compartment into a typical inclusion that fuses with other inclusions. These steps occur before the migration of the chlamydial inclusion to the host perinuclear area and take comparatively longer than the process of invasion and PV formation mediated by Toxoplasma (∼1 min). The newly formed PVs do not fuse with each other but travel directly to the host nucleus within ∼30 min. Thus, the parasite arrives in the perinuclear area before the bacterium, conferring on it a significant advantage for associating with the host MTOC. Nonetheless, even if chlamydial inclusions are kept at a distance from the host MTOC, they are still able to recruit host microtubules and to associate with the host Golgi apparatus. This suggests that the control of microtubule distribution exerted by the MTOC is not required for the bacterium to attract host organelles. The dramatic fragmentation of the Golgi apparatus and reorganization around the inclusions further suggest that the process of host golgin-84 cleavage may still be effective in coinfected cells (20). In coinfected cells, the bacteria can efficiently retrieve host ceramide from the Golgi apparatus, but they are no longer able to scavenge cholesterol–low-density lipoprotein (32). Moreover, replicating bacteria become abnormally enlarged and do not progress to infectious EBs, suggesting that bacterial cell division is progressively slowing down while late differentiation is blocked. In marked contrast, Toxoplasma parasites present in coinfected cells still appear to be highly proficient at exploiting host cell structures, almost as if C. trachomatis was not there, and grow similarly as in monocultures. If, however, C. trachomatis is allowed to grow intracellularly and monopolize host cell structures by itself for 24 h prior to coinfection, the replication of the parasite is then slower and the parasite accumulates sugars, similar to the cyst form of T. gondii. Interestingly, in cells preinfected with Chlamydia for 24 h, Toxoplasma causes the immediate arrest of chlamydial division, suggesting that even at a middle/early late stage of C. trachomatis development, chlamydiae are still susceptible to stress imposed by the incoming parasite, as reported for other stressors like antibiotics (59).
We previously showed that a possible cause for the observed stress response of Chlamydia when coinfecting with Toxoplasma is the deprivation of essential metabolites that are scavenged by Toxoplasma at the expense of the bacterium (32). Another factor that may contribute to the stress-induced persistent state of C. trachomatis during coinfection may be the failure to associate with the host MTOC by the inclusion. Many studies report that C. trachomatis modifies the host cell cycle by selectively targeting different cellular pathways (10, 60, 61). Although a potential selective advantage for Chlamydia in recruiting host centrosomes has not been demonstrated, it is possible to speculate that by stalling host cell division before cytokinesis, the bacterium ensures for itself a stable and spacious environment, sufficient to sustain its developmental cycle. For Toxoplasma, the host MTOC is likely a significant target, as this structure is systematically found to be associated with the PVs and remains at the PV surface throughout infection. A previous study reported that T. gondii infection induces normally quiescent cells to enter the S phase (62), subsequently preventing cell cycle progression (63). These perturbations in the host cell cycle machinery upon T. gondii infection may also influence how the parasite modulates its own replication scheme while intracellular.
The C. trachomatis inclusion promotes its own fusion with numerous host organelles, including Golgi vesicles and multivesicular bodies (1). Toxoplasma's behavior is diametrically opposed with regard to fusion with host organelles (64). Coinfection of T. gondii with Trypanosoma cruzi (65), Mycobacterium tuberculosis (66), or Coxiella burnetii (67) and chlamydiae (this study) supports the nonfusogenic nature of the T. gondii vacuole, likely as a consequence of not having a matching recognition signal. Chlamydial inclusions are connected with IncA-labeled fibers and PVs associated with each other by long GRA7-labeled extensions during coinfections. We observed apparent PV-inclusion connections via Chlamydia-derived fibers, whose significance is unclear. It remains possible that the chlamydial fibers are not targeting the PV per se but are targeting the host Golgi apparatus distributed around the PV in order to capture material from the Golgi apparatus.
Another example of competition between two different intracellular pathogens that covet the same organelles is illustrated by Toxoplasma and Chlamydia psittaci, which both recruit host mitochondria. In cells coinfected with Toxoplasma and C. psittaci, the host mitochondrial network undergoes considerable reorganization as the two pathogens adeptly compete to hijack host mitochondria, with both succeeding in redistributing mitochondria around their respective vacuoles. The high frequency and efficacy for the recruitment of host mitochondria by Toxoplasma and the maintenance of this interaction throughout infection, even in the presence of the virulent C. psittaci, reveal the importance of the PV-mitochondrion association for Toxoplasma growth. This association would facilitate the access of nutrients, e.g., lipids, ATP, or lipoate present in mitochondria, to the parasite. The fast growth of C. psittaci depends on the host cell for energy-rich metabolites such as ATP (68). One possible source of ATP would be the host mitochondria closely apposed to the inclusion membrane. C. psittaci has an ATP-ADP exchange mechanism that functionally acts as a reverse mitochondrion. The bacterium takes up ATP while expelling ADP, a feature that might cause mitochondria to approach and/or remain in close contact with the inclusion. We observed that the underdeveloped inclusions of C. psittaci lack host mitochondria around the inclusions. It is therefore possible that ATP is required for promoting fusion between early inclusions. Alternatively, the growth of the C. psittaci inclusion may be impaired at the onset of coinfection by an unidentified mechanism that makes the bacterium unable to later recruit host mitochondria and salvage ATP to pursue its normal development.
In this study, we have investigated the mutual influence exerted by Toxoplasma and chlamydiae on each other in single coinfected cells. Our results have provided new insights into the complex mode of intracellular development of both pathogens and have led to the identification of points of vulnerability in their respective developmental cycles. The interior of a mammalian cell provides a stable and nutrient-rich milieu to intravacuolar pathogens. However, the presence of a vacuolar membrane between chlamydiae or Toxoplasma and the host cytosol restricts direct access to cytosolic nutrients. This investigation has emphasized the critical importance for both intravacuolar pathogens to be competent to interact with and modify host endomembranes. Only when these pathogens are able to maintain these attributes are they proficient to retrieve the nutrients and energy-rich metabolites from host organelles that are necessary to sustain their intracellular survival and growth.
ACKNOWLEDGMENTS
We are grateful to the members of the I. Coppens and P. M. Bavoil laboratories for their helpful discussions during the course of this work. We are grateful to T. Hackstadt for the anti-IncA antibodies. We also have a special gratitude for the work of H. Zhang in coinfecting cells and performing the IFA. We thank the competent technical staff from the Microscopy Facility at Johns Hopkins University.
This work was supported by National Institutes of Health grants RO1 AI06767 to I.C. and U19 AI084044 to P.M.B.
Footnotes
Published ahead of print 14 December 2012
REFERENCES
- 1. Kumar Y, Valdivia RH. 2009. Leading a sheltered life: intracellular pathogens and maintenance of vacuolar compartments. Cell Host Microbe 5:593–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Casadevall A. 2008. Evolution of intracellular pathogens. Annu. Rev. Microbiol. 62:19–33 [DOI] [PubMed] [Google Scholar]
- 3. Fraser-Liggett CM. 2005. Insights on biology and evolution from microbial genome sequencing. Genome Res. 15:1603–1610 [DOI] [PubMed] [Google Scholar]
- 4. Schachter J. 1999. Infection and disease epidemiology, p 139–169 In Stephens RS. (ed), Chlamydia: intracellular biology, pathogenesis, and immunity. ASM Press, Washington, DC [Google Scholar]
- 5. Byrne GI, Moulder JW. 1978. Parasite-specified phagocytosis of Chlamydia psittaci and Chlamydia trachomatis by L and HeLa cells. Infect. Immun. 19:598–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dautry-Varsat A, Subtil A, Hackstadt T. 2005. Recent insights into the mechanisms of Chlamydia entry. Cell. Microbiol. 7:1714–1722 [DOI] [PubMed] [Google Scholar]
- 7. Clausen JD, Christiansen G, Holst HU, Birkelund S. 1997. Chlamydia trachomatis utilizes the host cell microtubule network during early events of infection. Mol. Microbiol. 25:441–449 [DOI] [PubMed] [Google Scholar]
- 8. Schramm N, Wyrick PB. 1995. Cytoskeletal requirements in Chlamydia trachomatis infection of host cells. Infect. Immun. 63:324–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grieshaber SS, Grieshaber NA, Hackstadt T. 2003. Chlamydia trachomatis uses host cell dynein to traffic to the microtubule-organizing center in a p50 dynamitin-independent process. J. Cell Sci. 116:3793–3802 [DOI] [PubMed] [Google Scholar]
- 10. Johnson KA, Tan M, Sütterlin C. 2009. Centrosome abnormalities during a Chlamydia trachomatis infection are caused by dysregulation of the normal duplication pathway. Cell. Microbiol. 11:1064–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hackstadt T, Rockey DD, Heinzen RA, Scidmore MA. 1996. Chlamydia trachomatis interrupts an exocytic pathway to acquire endogenously synthesized sphingomyelin in transit from the Golgi apparatus to the plasma membrane. EMBO J. 15:964–977 [PMC free article] [PubMed] [Google Scholar]
- 12. Hackstadt T, Scidmore M, Rockey D. 1995. Lipid metabolism in Chlamydia trachomatis-infected cells: directed trafficking of Golgi-derived sphingolipids to the chlamydial inclusion. Proc. Natl. Acad. Sci. U. S. A. 92:4877–4881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carabeo RA, Mead DJ, Hackstadt T. 2003. Golgi-dependent transport of cholesterol to the Chlamydia trachomatis inclusion. Proc. Natl. Acad. Sci. U. S. A. 100:6771–6776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Derré I, Swiss R, Agaisse H. 2011. The lipid transfer protein CERT interacts with the Chlamydia inclusion protein IncD and participates to ER-Chlamydia inclusion membrane contact sites. PLoS Pathog. 7:e1002092 doi:10.1371/journal.ppat.1002092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Capmany A, Damiani MT. 2010. Chlamydia trachomatis intercepts Golgi-derived sphingolipids through a Rab14-mediated transport required for bacterial development and replication. PLoS One 5:e14084 doi:10.1371/journal.pone.0014084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Elwell CA, Jiang S, Kim JH, Lee A, Wittmann T, Hanada K, Melancon P, Engel JN. 2011. Chlamydia trachomatis co-opts GBF1 and CERT to acquire host sphingomyelin for distinct roles during intracellular development. PLoS Pathog. 7:e1002198 doi:10.1371/journal.ppat.1002198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beatty WL. 2006. Trafficking from CD63-positive late endocytic multivesicular bodies is essential for intracellular development of Chlamydia trachomatis. J. Cell Sci. 119:350–359 [DOI] [PubMed] [Google Scholar]
- 18. Beatty WL. 2008. Late endocytic multivesicular bodies intersect the chlamydial inclusion in the absence of CD63. Infect. Immun. 76:2872–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Elwell CA, Engel JN. 2012. Lipid acquisition by intracellular Chlamydiae. Cell. Microbiol. 14:1010–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Heuer D, Rejman Lipinski A, Machuy N, Karlas A, Wehrens A, Siedler F, Brinkmann V, Meyer TF. 2009. Chlamydia causes fragmentation of the Golgi compartment to ensure reproduction. Nature 457:731–735 [DOI] [PubMed] [Google Scholar]
- 21. Sibley LD. 2003. Toxoplasma gondii: perfecting an intracellular life style. Traffic 4:581–586 [DOI] [PubMed] [Google Scholar]
- 22. Luft BJ, Remington JS. 1992. Toxoplasmic encephalitis in AIDS. Clin. Infect. Dis. 15:211–222 [DOI] [PubMed] [Google Scholar]
- 23. Sweeney KR, Morrissette NS, LaChapelle S, Blader IJ. 2010. Host cell invasion by Toxoplasma gondii is temporally regulated by the host microtubule cytoskeleton. Eukaryot. Cell 9:1680–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Walker ME, Hjort EE, Smith SS, Tripathi A, Hornick JE, Hinchcliffe EH, Archer W, Hager KM. 2008. Toxoplasma gondii actively remodels the microtubule network in host cells. Microbes Infect. 10:1440–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Coppens I, Sinai AP, Joiner KA. 2000. Toxoplasma gondii exploits host low-density lipoprotein receptor-mediated endocytosis for cholesterol acquisition. J. Cell Biol. 149:167–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Coppens I, Dunn JD, Romano JD, Pypaert M, Zhang H, Boothroyd JC, Joiner KA. 2006. Toxoplasma gondii sequesters lysosomes from mammalian hosts in the vacuolar space. Cell 125:261–274 [DOI] [PubMed] [Google Scholar]
- 27. Romano JD, Bano N, Coppens I. 2008. New host nuclear functions are not required for the modifications of the parasitophorous vacuole of Toxoplasma. Cell. Microbiol. 10:465–476 [DOI] [PubMed] [Google Scholar]
- 28. de Melo EJ, de Souza W. 1996. Pathway of C6-NBD-ceramide on the host cell infected with Toxoplasma gondii. Cell. Struct. Funct. 21:47–52 [DOI] [PubMed] [Google Scholar]
- 29. de Melo EJ, de Carvalho TU, de Souza W. 1992. Penetration of Toxoplasma gondii into host cells induces changes in the distribution of the mitochondria and the endoplasmic reticulum. Cell Struct. Funct. 17:311–317 [DOI] [PubMed] [Google Scholar]
- 30. Sinai AP, Webster P, Joiner KA. 1997. Association of host cell endoplasmic reticulum and mitochondria with the Toxoplasma gondii parasitophorous vacuole membrane: a high affinity interaction. J. Cell Sci. 110:2117–2128 [DOI] [PubMed] [Google Scholar]
- 31. Matsumoto A, Bessho H, Uehira K, Suda T. 1991. Morphological studies of the association of mitochondria with chlamydial inclusions and the fusion of chlamydial inclusions. J. Electron Microsc. 40:356–363 [PubMed] [Google Scholar]
- 32. Romano JD, de Beaumont C, Carrasco JA, Ehrenman K, Bavoil PM, Coppens I. A novel co-infection model with Toxoplasma and Chlamydia trachomatis highlights the importance of host cell manipulation for nutrient scavenging. Cell. Microbiol., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang YX, Shi Y, Zhou M, Petsko GA. 1994. Cloning, sequencing, and expression in Escherichia coli of the gene encoding a 45-kilodalton protein, elongation factor Tu, from Chlamydia trachomatis serovar F. J. Bacteriol. 176:1184–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roos DS, Donald RG, Morrissette NS, Moulton AL. 1994. Molecular tools for genetic dissection of the protozoan parasite Toxoplasma gondii. Methods Cell. Biol. 45:27–63 [DOI] [PubMed] [Google Scholar]
- 35. Tan C, Hsia RC, Shou H, Carrasco JA, Rank RG, Bavoil PM. 2010. Variable expression of surface-exposed polymorphic membrane proteins in in vitro-grown Chlamydia trachomatis. Cell. Microbiol. 12:174–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Coppens I, Joiner KA. 2003. Host but not parasite cholesterol controls Toxoplasma cell entry by modulating organelle discharge. Mol. Biol. Cell 14:3804–3820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wilson DP, Mathews S, Wan C, Pettitt AN, McElwain DL. 2004. Use of a quantitative gene expression assay based on micro-array techniques and a mathematical model for the investigation of chlamydial generation time. Bull. Math. Biol. 66:523–537 [DOI] [PubMed] [Google Scholar]
- 38. Beatty WL, Morrison RP, Byrne GI. 1994. Persistent chlamydiae: from cell culture to a paradigm for chlamydial pathogenesis. Microbiol. Rev. 58:686–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brown WJ, Skeiky YA, Probst P, Rockey DD. 2002. Chlamydial antigens colocalize within IncA-laden fibers extending from the inclusion membrane into the host cytosol. Infect. Immun. 70:5860–5864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rome ME, Beck JR, Turetzky JM, Webster P, Bradley PJ. 2008. Intervacuolar transport and unique topology of GRA14, a novel dense granule protein in Toxoplasma gondii. Infect. Immun. 76:4865–4875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Delevoye C, Nilges M, Dehoux P, Paumet F, Perrinet S, Dautry-Varsat A, Subtil A. 2008. SNARE protein mimicry by an intracellular bacterium. PLoS Pathog. 4:e1000022 doi:10.1371/journal.ppat.1000022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Paumet F, Wesolowski J, Garcia-Diaz A, Delevoye C, Aulner N, Shuman HA, Subtil A, Rothman JE. 2009. Intracellular bacteria encode inhibitory SNARE-like proteins. PLoS One 4:e7375 doi:10.1371/journal.pone.0007375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Suchland RJ, Rockey DD, Weeks SK, Alzhanov DT, Stamm WE. 2005. Development of secondary inclusions in cells infected by Chlamydia trachomatis. Infect. Immun. 73:3954–3962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Campbell S, Richmond SJ, Yates P. 1989a. The development of Chlamydia trachomatis inclusions within the host eukaryotic cell during interphase and mitosis. J. Gen. Microbiol. 135:1153–1165 [DOI] [PubMed] [Google Scholar]
- 45. Campbell S, Richmond SJ, Yates PS. 1989b. The effect of Chlamydia trachomatis infection on the host cell cytoskeleton and membrane compartments. J. Gen. Microbiol. 135:2379–2386 [DOI] [PubMed] [Google Scholar]
- 46. Majeed M, Kihlstrom E. 1991. Mobilization of F-actin and clathrin during redistribution of Chlamydia trachomatis to an intracellular site in eukaryotic cells. Infect. Immun. 59:4465–4472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Coppin A, Dzierszinski F, Legrand S, Mortuaire M, Ferguson D, Tomavo S. 2003. Developmentally regulated biosynthesis of carbohydrate and storage polysaccharide during differentiation and tissue cyst formation in Toxoplasma gondii. Biochimie 85:353–361 [DOI] [PubMed] [Google Scholar]
- 48. Scidmore MA, Fischer ER, Hackstadt T. 1996. Sphingolipids and glycoproteins are differentially trafficked to the Chlamydia trachomatis inclusion. J. Cell Biol. 134:363–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. van Ooij C, Kalman L, van Ijzendoorn S, Nishijima M, Hanada K, Mostov K, Engel JN. 2000. Host cell-derived sphingolipids are required for the intracellular growth of Chlamydia trachomatis. Cell. Microbiol. 2:627–637 [DOI] [PubMed] [Google Scholar]
- 50. Lige B, Romano JD, Bandaru VV, Ehrenman K, Levitskaya J, Sampels V, Haughey NJ, Coppens I. 2011. Deficiency of a Niemann-Pick, type C1-related protein in Toxoplasma is associated with multiple lipidoses and increased pathogenicity. PLoS Pathog. 7:e1002410 doi:10.1371/journal.ppat.1002410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sonda S, Sala G, Ghidoni R, Hemphill A, Pieters J. 2005. Inhibitory effect of aureobasidin A on Toxoplasma gondii. Antimicrob. Agents Chemother. 49:1794–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Azzouz N, Rauscher B, Gerold P, Cesbron-Delauw MF, Dubremetz JF, Schwarz RT. 2002. Evidence for de novo sphingolipid biosynthesis in Toxoplasma gondii. Int. J. Parasitol. 32:677–684 [DOI] [PubMed] [Google Scholar]
- 53. Yamaji T, Kumagai K, Tomishige N, Hanada K. 2008. Two sphingolipid transfer proteins, CERT and FAPP2: their roles in sphingolipid metabolism. Life 60:511–518 [DOI] [PubMed] [Google Scholar]
- 54. Escalante-Ochoa C, Ducatelle R, Charlier G, De Vos K, Haesebrouck F. 1999. Significance of host cell kinesin in the development of Chlamydia psittaci. Infect. Immun. 67:5441–5446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sinai AP, Joiner KA. 2001. The Toxoplasma gondii protein ROP2 mediates host organelle association with the parasitophorous vacuole membrane. J. Cell Biol. 154:95–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Saka HA, Valdivia RH. 2010. Acquisition of nutrients by Chlamydiae: unique challenges of living in an intracellular compartment. Curr. Opin. Microbiol. 13:4–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cocchiaro JL, Valdivia RH. 2009. New insights into Chlamydia intracellular survival mechanisms. Cell. Microbiol. 11:1571–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Laliberté J, Carruthers VB. 2008. Host cell manipulation by the human pathogen Toxoplasma gondii. Cell. Mol. Life Sci. 65:1900–1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Skilton RJ, Cutcliffen LT, Barlow D, Wang Y, Salim O, Lambden PR, Clarke IN. 2009. Penicillin induced persistence in Chlamydia trachomatis: high quality time lapse video analysis of the developmental cycle. PLoS One 4:e7723 doi:10.1371/journal.pone.0007723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Knowlton AE, Brown HM, Richards TS, Andreolas LA, Patel RK, Grieshaber SS. 2011. Chlamydia trachomatis infection causes mitotic spindle pole defects independently from its effects on centrosome amplification. Traffic 12:854–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Brown HM, Knowlton AE, Grieshaber SS. 2012. Chlamydial infection induces host cytokinesis failure at abscission. Cell. Microbiol. 14:1554–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lavine MD, Arrizabalaga G. 2009. Induction of mitotic S-phase of host and neighboring cells by Toxoplasma gondii enhances parasite invasion. Mol. Biochem. Parasitol. 164:95–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Molestina RE, El-Guendy N, Sinai AP. 2008. Infection with Toxoplasma gondii results in dysregulation of the host cell cycle. Cell. Microbiol. 10:1153–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mordue DG, Håkansson S, Niesman I, Sibley LD. 1999. Toxoplasma gondii resides in a vacuole that avoids fusion with host cell endocytic and exocytic vesicular trafficking pathways. Exp. Parasitol. 92:87–99 [DOI] [PubMed] [Google Scholar]
- 65. Meirelles MN, De Souza W. 1983. Interaction of lysosomes with endocytic vacuoles in macrophages simultaneously infected with Trypanosoma cruzi and Toxoplasma gondii. J. Submicrosc. Cytol. 15:889–896 [PubMed] [Google Scholar]
- 66. Black CM, Bermudez LE, Young LS, Remington JS. 1990. Co-infection of macrophages modulates interferon gamma and tumor necrosis factor-induced activation against intracellular pathogens. J. Exp. Med. 172:977–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sinai AP, Paul S, Rabinovitch M, Kaplan G, Joiner KA. 2000. Coinfection of fibroblasts with Coxiella burnetii and Toxoplasma gondii: to each their own. Microbes Infect. 2:727–736 [DOI] [PubMed] [Google Scholar]
- 68. Hatch TP, Al-Hossaing E, Silverman JA. 1982. Adenosine nucleotide and lysine transport in Chlamydia psittaci. J. Bacteriol. 150:662–670 [DOI] [PMC free article] [PubMed] [Google Scholar]