

Abstract
We have previously reported that the trypanosome-specific proteins P34 and P37 form a unique preribosomal complex with ribosomal protein L5 and 5S rRNA in the nucleoplasm. We hypothesize that this novel trimolecular complex is necessary for stabilizing 5S rRNA in Trypanosoma brucei and is essential for the survival of the parasite. In vitro quantitative analysis of the association between the proteins L5 and P34 is fundamental to our understanding of this novel complex and thus our ability to exploit its unique characteristics. Here we used in vitro fluorescence resonance energy transfer (FRET) to analyze the association between L5 and P34. First, we demonstrated that FRET can be used to confirm the association between L5 and P34. We then determined that the binding constant for L5 and P34 is 0.60 ± 0.03 μM, which is in the range of protein-protein binding constants for RNA binding proteins. In addition, we used FRET to identify the critical regions of L5 and P34 involved in the protein-protein association. We found that the N-terminal APK-rich domain and RNA recognition motif (RRM) of P34 and the L18 domain of L5 are important for the association of the two proteins with each other. These results provide us with the framework for the discovery of ways to disrupt this essential complex.
INTRODUCTION
African trypanosomes are responsible for trypanosomiasis in humans and nagana in domestic animals. Trypanosoma brucei has a complicated life cycle that requires adaptation to life within a mammalian host and an insect vector, the tsetse fly (1). Throughout this life cycle, RNA binding proteins (RBPs) play the central roles in many adaptive processes, including regulation of gene expression and protein synthesis (2, 3), making them particularly important in this organism.
RBPs play essential roles in the many aspects of ribosomal biogenesis. Ribosomal biogenesis in eukaryotes is an essential and conserved process that requires the processing and assembly of four rRNAs (18, 28, 5.8, and 5S rRNAs) with over 80 ribosomal proteins. Much of this process occurs in the nucleolus (4–6). However, unlike the other three rRNAs, 5S rRNA is independently transcribed by RNA polymerase III within the nucleoplasm and must be transported into the nucleolus by the L5 ribosomal protein (7, 8). L5 is the only known eukaryotic ribosomal protein that forms a preribosomal L5-5S rRNA ribonucleoprotein (RNP) complex to stabilize and facilitate the trafficking of 5S rRNA to the nucleolus (7, 9, 10). Mutations in critical residues within L5 lead to loss of 5S rRNA and ribosomal biogenesis defects, indicating that it is essential to cell viability. T. brucei L5 is comprised of a predicted RanBP1_WASP domain in its N-terminal region, an L18 domain in the central region, and a C-terminal domain that possesses residues critical for 5S rRNA binding.
Our laboratory has previously identified two trypanosome-specific RBPs, P34 and P37, which are essential for the viability of T. brucei (11). P34 and P37 are highly similar, the major difference between them being the presence of an additional 18 amino acids in the P37 N terminus. Both proteins are comprised of an APK-rich N-terminal domain, a central domain containing an RNA recognition motif (RRM), and a C-terminal region containing KKDX repeats. P34 is dominantly expressed in the procyclic stage, while P37 is mainly expressed in the bloodstream stage. Previous studies in the laboratory have indicated that P34 and P37 specifically bind 5S rRNA and are essential for ribosome biogenesis and survival of the parasite. We have also shown that they are required for several steps in the ribosomal biogenesis pathway in T. brucei, including association of the 60S subunit with XpoI and Nmd3 and subsequent export from the nucleus and possibly in the joining of the large and small ribosomal subunits (11–14).
Recent studies have also demonstrated that P34 and P37 associate with both ribosomal protein L5 and 5S rRNA and form a preribosomal complex in the nucleoplasm that is unique to trypanosomes (12, 13). Loss of P34 and P37 using RNA interference causes a decrease in 5S rRNA level and 80S ribosome formation, with an overall decrease in protein synthesis (11). This phenotype is similar to that of L5 knockouts in yeast (Saccharomyces cerevisiae), as indicated above (14). Moreover, examination of the sequence of T. brucei L5 showed that it contains a substitution of the highly conserved arginine (yeast position 285) by a noncharged alanine in the conserved C terminus previously implicated in binding to 5S rRNA (15). Experimental evidence confirmed that this L5 binds to 5S rRNA with a lower binding constant than that of other eukaryotic L5 proteins (12). These findings indicate that P34 and P37 associate with L5 and 5S rRNA to form a unique preribosomal complex and suggest that the trypanosome-specific proteins function to complement L5 for stabilizing and trafficking 5S rRNA in the early steps of ribosome biogenesis.
In vitro quantitative analysis of the protein-protein association between L5 and P34 within this unique complex is fundamental to our understanding of the preribosomal particle and the potential for using this complex as a therapeutic target. Here we used in vitro fluorescence resonance energy transfer (FRET) to analyze the association between the T. brucei P34 and L5. First, we demonstrated that FRET can be used to examine the association between P34 and L5. Then we determined the binding constant for L5 and P34 and used FRET to study the regions of L5 and P34 involved in protein-protein association. In each case, we have confirmed the results using a more traditional, though less quantitative, immune capture approach.
MATERIALS AND METHODS
Recombinant proteins.
P34 (GenBank accession no. AF020695), L5 (GenBank accession no. XM_822569), enhanced yellow fluorescent protein (eYFP; Clontech), and cerulean fluorescent protein (cerulean FP; generous gift from Mark Carrington, Cambridge) (16) were amplified by PCR using the forward (F) and reverse (R) primers P34P34 F and R, L5 F and R, and green fluorescent protein (GFP) F and R separately (Table 1). Primers for the P34 N terminus and C terminus and RRM truncates and primers for the L5 N terminus and C terminus and L18 domain (Table 1) were used to amplify each domain separately by PCR. Cerulean-L5 and eYFP-P34 were generated by overlap extension PCR with no linker between the fluorescent tag and eYFP-P34. The first set of primers for cerulean-L5 was GFP F and cerulean-L5 R and L5 F and R, and the second set was GFP F and L5 R. The first set of primers for eYFP-P34 was GFP F and eYFP-P34 R and P34 F and R, and the second set was GFP F and P34 R.
Table 1.
Primers used to amplify the full-length and truncated versions of L5 and P34, cerulean-L5, and eYFP-P34
| Primera | Sequence |
|---|---|
| P34 F | 5′-CAGCTGATGGCCCCAAAGTCTGC-3′ |
| P34 R | 5′-TCACTGCTTCCTCTTGGCATC-3′ |
| L5 F | 5′-ATGACATTCGTTAAGATTG-3′ |
| L5 R | 5′-TTACTTCGCACGGTCACGG-3′ |
| P34 N-terminus F | 5′-ATGGCCCCAAAGTCTGCTACAAAGTCTGCT-3′ |
| P34 N-terminus R | 5′-TCAGTGGGTGCCGGCAGCGGGGCGTGCCTT-3′ |
| P34 C-terminus F | 5′-TCAACTCGCTCGCTTCAGAAGGAAAAGGAC-3′ |
| P34 C-terminus R | 5′-TCACTGCTTCCTCTTGGCATCCTTCTTTTCATTCT-3′ |
| P34 RRM F | 5′-AACGGTGTGTATGTGAAGAACTGGGGTCAG-3′ |
| P34 RRM R | 5′-TCACGAGCGAGTTGAAAGAGCCACACGTAG-3′ |
| L5 N-terminus F | 5′-ATGACATTCGTTAAGGTTGTGAAGAACAAGGCG-3′ |
| L5 N-terminus R | 5′-TCACTCGCGGCGGCGGCGGTACTTCACCTGGAA-3′ |
| L5 C-terminus F | 5′-CCTCACCGCCCCAATCGGTTCCCCGGTTACAAC-3′ |
| L5 C-terminus R | 5′-TTACTTCGCACGGTCACGGATGCGCTCAATCAC-3′ |
| L5 L18 F | 5′-GGGAAGACGGATTACCACGCACGCCGCCGCATG-3′ |
| L5 L18 R | 5′-TACGGCGAGGCCACCATCCACAGCACCCTTCAA-3′ |
| GFP F | 5′-ATGATGAGCAAGGGCGAGG-3′ |
| GFP R | 5′-AGAACCACCTCCCCCCTTG-3′ |
| Cerulean-L5 R | 5′-ACAACCTTAACGAATGTCATAGAACCACCTCCCCCCTTG-3′ |
| eYFP-P34 R | 5′-CAGACTTTGGGGCCATAGAACCACCTCCCCC-3′ |
F, forward; R, reverse.
Cerulean FP, eYFP, cerulean-L5, eYFP-P34, P34, and L5 truncates were cloned into pTrc-His1 TOPO (Invitrogen) for expression as a polyhistidine-fusion protein in TOP10 One Shot cells (Invitrogen). P34 was cloned into plasmid pQE-1 (Qiagen) and expressed as a polyhistidine-fusion protein in Escherichia coli strain M15, as previously described (12, 17). All recombinant protein expression plasmids were confirmed by DNA sequence analysis. The expression and purification of recombinant proteins were performed as previously described (12, 17). Ten microliters of each elution fraction was analyzed by SDS-PAGE with Coomassie blue staining, and 100 ng of each fraction was analyzed by Western blot analysis using anti-P34/P37 (18), anti-L5 (12), antipolyhistidine (H1029; Sigma), or anti-GFP (A10260; Life Technologies). Fractions containing recombinant protein were pooled and desalted using a PD-10 column (GE) with storage buffer (10 mM Tris [pH 7.6], 150 mM KCl, 0.1 mM EDTA, 0.1 mM dithiothreitol [DTT], 0.1% NP-40, 0.1 mM phenylmethylsulfonyl fluoride [PMSF]) and flash-frozen at −80°C in 400-μl aliquots.
The polyhistidine tag of purified recombinant P34 was removed by treatment with TAGZyme (Qiagen), and the polyhistidine tag of purified recombinant L5 was removed by treatment with EKMax enterokinase (Life Technologies).
All fluorescent protein concentrations were determined using the peak absorption of a set of dilutions and the extinction coefficient for cerulean of 43,000 M−1 cm−1 at 433 nm and that for eYFP of 84,000 M−1 cm−1 at 514 nm (16, 19). Concentrations of other recombinant proteins were determined by Bio-Rad protein assay (9).
Absorption and fluorescence measurements.
Samples were measured in 96-well clear bottom plates using the Synergy H1 hybrid reader (Biotek). Absorption spectra were recorded from 375 nm to 600 nm with 1-nm intervals. Emission spectra were collected from 450 nm to 600 nm (excitation, 400 nm; 1-nm slit width; sensitivity, 100). Excitation at 400 nm improves the dynamic range as the excitation of eYFP is at a minimum level (20). Excitation at 400 nm leads to emission of cerulean at 475 nm. When FRET occurs, excitation at 400 nm leads to emission of eYFP at 530 nm (16, 21). Proteins and RNA were incubated in binding buffer (10 mM Tris [pH 7.6], 150 mM KCl, 0.1 mM EDTA, 0.1 mM DTT, 0.1% NP-40) for 15 min at room temperature. All measurements were repeated at least three times with at least two preparations of proteins.
Immune capture experiments.
Protein A-labeled magnetic beads (Dynabeads, Life Technologies) were washed in phosphate-buffered saline (PBS) and blocked in PBS-bovine serum albumin (BSA) (1 mg/ml). Antibody was added to the beads and incubated at 4°C for 1 h. Dimethyl pimelimidate (Thermo Scientific, Pierce) dissolved in 0.2 M triethanolamine (Sigma-Aldrich) in PBS was used to cross-link the antibody to the beads (three room temperature incubations of 30 min each separated by brief washes in 0.2 M triethanolamine). After cross-linking, the reaction was quenched with 50 mM ethanolamine (Sigma-Aldrich) in PBS (two incubations of 10 min each). Excess antibody was eluted in two washes with 1 M glycine (pH 3). The antibody-coated beads were washed with PBS-Tween (PBS-T) and then incubated with antigen at 4°C for 1 h. Supernatants were collected, beads were washed with PBS-T three times, and the captured antigen was eluted in SDS-PAGE loading buffer at 70°C for 10 min. Immune capture fractions were analyzed by Western blot analysis using the antipolyhistidine monoclonal antibody H1029 (Sigma).
RESULTS
Cerulean-L5 and eYFP-P34 associate, as demonstrated by FRET.
Previous results from our laboratory have demonstrated that P34, L5, and 5S rRNA form a trimolecular complex, and P34 directly associates with L5 using immune capture methods (12). Electrophoretic mobility shift assays have also shown that P34 and L5 directly associate with 5S rRNA individually (12, 17). To further study the protein-protein interaction between them more quantitatively and to examine the interaction in the presence of interfering molecules, we explored the use of FRET as a direct measure of binding between partners. We first constructed plasmids that express the protein with appropriate tags. Recombinant cerulean-L5 and eYFP-P34 were expressed in E. coli as polyhistidine-tagged proteins and purified with Ni-nitrilotriacetic acid (NTA) agarose columns. The eluted proteins were analyzed by SDS-PAGE and Western blot analysis. SDS-PAGE analysis (Fig. 1A, upper panel) suggests that purities are above 95%, and the proteins are expressed at the correct molecular masses (63 kDa for cerulean-L5 and 62 kDa for eYFP-P34). Western blot analysis (Fig. 1A, lower panel) using anti-L5 and anti-P34/P37 antibodies shows that reactive epitopes of L5 and P34 are not altered due to the presence of the fluorescent tags. To confirm the fluorescence properties of the expressed fluorescent protein fusions, the absorption and emission spectra (at an excitation wavelength of 400 nm) of the purified recombinant cerulean-L5 and eYFP-P34 were recorded. Normalized absorption and emission spectra of fluorescence-tagged proteins are shown in Fig. 1B. Cerulean-L5 showed two absorption (blue) peaks at 424 nm and 433 nm and an emission (green) peak at 476 nm with a shoulder at 502 nm, while eYFP-P34 showed two absorption (red) peaks at 483 nm and 519 nm and a single emission (purple) peak at 530 nm. These results are consistent with the previously reported fluorescence properties of cerulean and eYFP (16, 20). As a comparison, L5 and P34 are nonfluorescent in this range (data not shown). This indicates that properties of the fluorescent tags are not affected by the addition of L5 or P34.
Fig 1.

Expression, purification, and spectral analysis of recombinant proteins used in FRET analysis. (A) Each eluted fraction (E1 to E4) from the 50% Ni-NTA agarose column was analyzed by 12% SDS-PAGE followed by Coomassie blue staining (upper panel) and Western blot analysis using anti-L5 (lower panel left) and anti-P34 (lower panel, right) antibodies. (B) Normalized (Norm.) absorption spectra and emission spectra (excitation at 400 nm) of cerulean-L5 and eYFP-P34. These assays were repeated three times with three different preparations of recombinant proteins.
Since the FRET signal has an R−6 dependence on the distance between the fused fluorescent tags, a separation of the tags beyond the Forster radius (10 nm) decreases the FRET signal to zero (29). Therefore, many factors, including the size of the molecules and the location of the binding surfaces, alter the efficiency of energy transfer. To observe whether FRET occurs due to the specific binding of L5 and P34, emission spectra of solutions of 1 μM cerulean-L5 (blue), 1 μM eYFP-P34 (purple), 1 μM cerulean-L5 plus 1 μM eYFP (green), and 1 μM cerulean-L5 plus 1 μM eYFP-P34 (red) (excitation at 400 nm) were recorded and are shown in Fig. 2. The solution containing 1 μM cerulean-L5 plus 1 μM eYFP-P34 (red) shows decreased cerulean emission around 475 nm as well as a sharply increased eYFP emission around 530 nm. This increase is significantly larger than can be accounted for by the direct excitation of 1 μM cerulean-L5 (blue), 1 μM eYFP-P34 (purple), or 1 μM cerulean-L5 plus 1 μM eYFP (green). Other incubation times were assayed with similar results (data not shown). Therefore, these results indicate that L5 and P34 have associated and their fluorescent tags are within the range required for energy transfer to occur.
Fig 2.

Cerulean-L5 and eYFP-P34 associate, as demonstrated by FRET. Shown are fluorescence emission spectra (excitation, 400 nm) demonstrating FRET between 1 μM cerulean-L5 and 1 μM eYFP-P34 fusion proteins (red), emission of the individual components at this concentration (blue and purple), and the emission of 1 μM cerulean-L5 plus 1 μM eYFP (green). This assay was repeated three times with three different preparations of recombinant proteins.
Equilibrium binding constant of cerulean-L5 and eYFP-P34 as determined by FRET.
In order to determine the equilibrium binding constant KD from FRET data, we performed a titration in which the acceptor-tagged protein eYFP-P34 was added to its donor-tagged partner cerulean-L5, while the emission spectra were monitored using the Synergy H1 hybrid reader (Biotek). The emission spectra of the titration of eYFP-P34 (from 0 μM to 8 μM) into 1 μM cerulean-L5 are shown in Fig. 3A. During the titration of eYFP-P34, emission at 530 nm increased in proportion to the number of bound pairs (20, 22).
Fig 3.
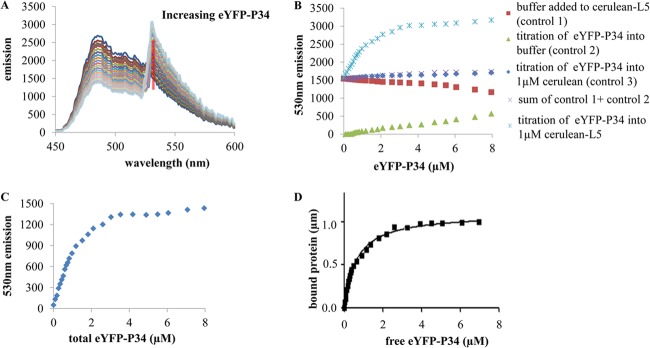
Equilibrium binding constant of cerulean-L5 and eYFP-P34 as determined by FRET. FRET assays were performed with a titration of eYFP-P34 into cerulean-L5. (A) Steady-state FRET binding assay. The emission spectra were recorded during the titration of eYFP-P34 into 1 μM cerulean-L5. eYFP-P34 was added in increments from 0 μM to 8 μM. (B) Fluorescence emission at 530 nm for the FRET binding assay. The assay results shown include measurement of dilution, with buffer added to cerulean-L5 (control 1), measurement of direct excitation, with eYFP-P34 added to buffer (control 2), the control for nonspecific interactions, with eYFP-P34 added to 1.0 μM cerulean (control 3), the sum of controls 1 and 2, and eYFP-P34 added to cerulean-L5. (C) Fluorescence emission at 530 nm as a function of total eYFP-P34 concentration. Controls accounting for dilution (control 1) and direct excitation (control 2) were subtracted. (D) Bound protein was determined from the FRET data as a function of the free eYFP-P34 concentration, fitted by binding hyperbola for one binding site. These assays were repeated three times with two different preparations of proteins.
The 530-nm peak emission intensities are plotted in Fig. 3B. To control for dilution, dilution of the 1 μM cerulean-L5 by an identical titration of buffer leads to a linear decrease (Fig. 3B, control 1, red rectangles). The same titration of eYFP-P34 into buffer gives a measure of direct excitation of eYFP (Fig. 3B, control 2, green triangles) and is also linear. To control for nonspecific interactions, eYFP-P34 was titrated into 1 μM unfused cerulean, which does not associate with P34 (Fig. 3B, control 3, blue diamonds). This leads to a linear increase in 530-nm emission. The sum of the dilution and direct excitation controls (Fig. 3B, control 1 and control 2, purple X's) is almost identical to that obtained for the titration of eYFP-P34 into unfused cerulean (Fig. 3B, control 3, blue diamonds), indicating that nonspecific interaction is negligible. Most significantly, the titration of eYFP-P34 into 1 μM cerulean-L5 showed a curved plot of cerulean-L5 plus eYFP-P34 (Fig. 3B, blue asterisks), which is characteristic of saturable, specific, and biomolecular binding.
A plot of the binding data with the dilution (control 1 and control 2) and direct excitation (control 3) controls subtracted is presented in Fig. 3C. The saturation level, determined by an exponential fit, corresponds to 1 μM cerulean-L5 bound. Using this relationship between the FRET signal and bound species and a binding stoichiometry of 1:1 (20, 23), data were converted into the relative amount of bound cerulean-L5 and eYFP-P34. The free eYFP-P34 is calculated in a subtraction for each point and with the axis rescaled to give the bound protein versus free eYFP-P34 binding curve shown in Fig. 3D, fitted with the binding hyperbola for one binding site (24) using Graphpad Prism 5.0.1 software. The equilibrium binding constant is calculated from the fitting curve, giving Kd = 0.60 ± 0.03 μM, which is in the range of protein-protein binding constants of RNA binding proteins (25).
The N terminus and RRM of P34 are required for L5 association.
FRET is ideal for the study of binding partners in the presence of interacting molecules, and we utilized this approach to examine the regions of each protein participating in the P34-L5 interaction (20, 22, 26). For this application, potential interacting proteins or domains are added to solutions of tagged binding partners. The decrease in the emission peak indicates the dissociation of the initial bound complex and the association between the newly added untagged proteins with one of the tagged proteins. In order to determine the regions of P34 responsible for binding to L5, full-length L5 and P34, as well as N-terminal, C-terminal, and RRM truncates of P34, were expressed in E. coli and purified. The diagram for full-length P34 and P34 truncates is shown in Fig. 4A. To validate the use of FRET in our system, we first added full-length untagged P34 into the solution containing 1 μM cerulean-L5 plus 1 μM eYFP-P34 (red). Figure 4B shows that addition of untagged P34 caused a significant decrease in the emission at 530 nm, and the decrease in the emission at 530 nm is proportional to the amount of added untagged P34. These data indicate that untagged full-length P34 competes with eYFP-P34 for binding to cerulean-L5, decreasing the FRET signal.
Fig 4.

The N-terminus and RRM domains of P34 are required for the association with L5. FRET analysis was used to determine the regions of P34 involved in L5 association. (A) Schematic diagrams of P34 truncates. FL, full length; N, N-terminal domain; RRM, RRM central domain; C, C-terminal domain. (B) Emission spectra of 1 μM cerulean-L5 plus 1 μM eYFP-P34 with P34 titration. (C) Emission spectra of 1 μM cerulean-L5 plus 1 μM eYFP-P34 with 1 μM full-length P34 or addition of 1 μM P34 truncates. (D) Immune capture assay. Full-length P34 and P34 truncates were incubated with L5 in the binding buffer. Anti-L5 antibodies were used to capture the associating proteins. The immunoprecipitate (P) and the supernatant (S) fractions were analyzed by Western blot (WB) analysis using anti-His antibodies. “beads alone,” no antibody; “input,” 10% of total proteins used for immune capture assay; M, molecular mass (kDa) markers. These assays were repeated three times with three different preparations of recombinant proteins.
Individual untagged P34 truncates were then added to the cerulean-L5 and eYFP-P34 solution. Compared with the addition of untagged full-length P34 (blue), Fig. 4C shows that the addition of 1 μM untagged C-terminal truncate of P34 into cerulean-L5 and eYFP-P34 solution (green) only slightly decreases the emission at 530 nm. The addition of 1 μM untagged N-terminal (black) or RRM (purple) of P34 truncate results in a greater decrease in the 530 nm emission peak than 1 μM untagged C-terminal P34 (green), but the decrease is not as large as that caused by 1 μM untagged full-length P34 (blue). This indicates that both the N terminus and RRM are important for binding of P34 to L5.
We also performed immune capture assays to confirm the binding regions between L5 and P34. In these experiments, we utilized purified full-length L5 without a polyhistidine tag and full-length P34 and P34 truncates with polyhistidine tags. Immune capture fractions (Fig. 4D) using a peptide antibody against L5 were subsequently analyzed using the antipolyhistidine monoclonal antibody. The proteins were detected in the input fraction (positive control, lanes 1, 6, 11, and 16), and the beads alone do not interact nonspecifically with full-length P34 and P34 truncates (negative control, lanes 5, 10, 15, and 20). The absence of signal in the bead control corresponds to weak, nonspecific interactions, which are disrupted in subsequent washes (data not shown). The C-terminal truncate of P34 (10 kDa) can only be detected in the supernatant fraction (lane 12), indicating that the C terminus of P34 alone is not capable of association with L5. Importantly, full-length P34 (34 kDa, lane 3), RRM (26 kDa, lane 13), and N-terminal truncates (11 kDa, lane 8) of P34 can be detected in the precipitate. This indicates that the P34 RRM and N-terminal domains are involved in association with L5, which is consistent with the FRET data. RRM and N-terminal truncates of P34 are also found in the supernatant (S), indicating that, at stoichiometric amounts, not all of the RRM and N-terminal truncates of P34 are interacting with L5 under these conditions.
The L18 domain of L5 is required for P34 association.
To study the regions of L5 involved in binding with P34, the full-length L5 and the N-terminal, C-terminal, and L18 domains of L5 were expressed in E. coli and purified. The diagram for the full-length L5 and L5 truncates is shown in Fig. 5A. Untagged full-length L5 and L5 truncates were added to the cerulean-L5 and eYFP-P34 solution. Figure 5B shows that compared with the emission of solution containing 1 μM cerulean-L5 plus 1 μM eYFP-P34 (red), the addition of full-length L5 (blue) largely decreases the emission at 530 nm. The addition of 1 μM N-terminal (black) and C-terminal (green) domains of L5 does not significantly affect the FRET signal of the cerulean-L5–eYFP-P34 interaction. However, the addition of 1 μM L18 domain of L5 (purple) decreases the FRET signal of the cerulean-L5–eYFP-P34 interaction nearly as well as the full-length L5 protein (blue), indicating that the L18 domain is the major domain of L5 involved in the association with P34.
Fig 5.
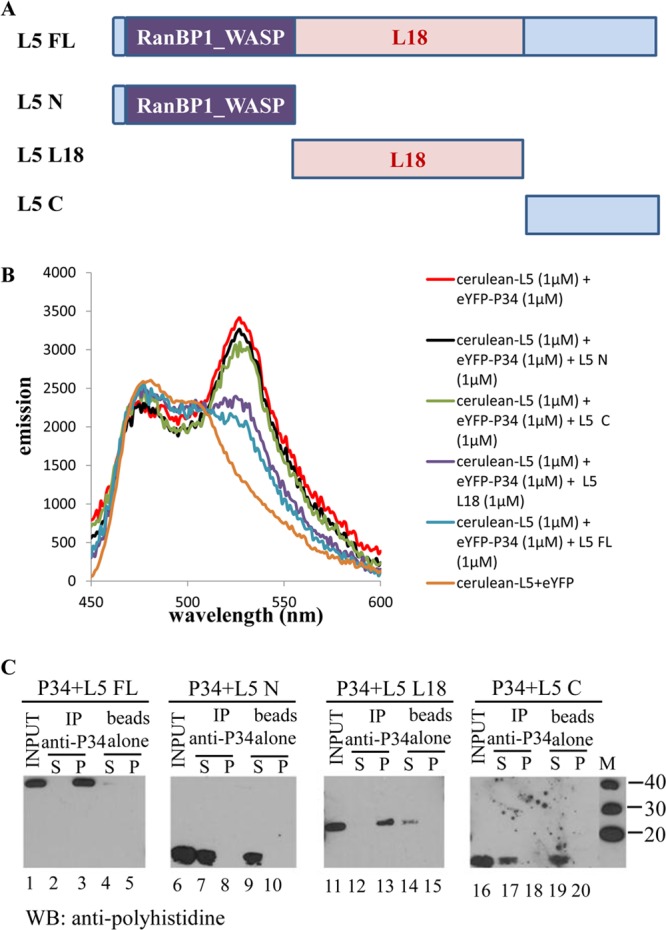
The L18 domain of L5 is required for association with P34. FRET analysis was used to determine the regions of L5 involved in P34 association. (A) Schematic diagrams of L5 truncates. FL, full length; N, N-terminal domain; L18, L18 internal domain; C, C-terminal domain. (B) Emission spectra of 1 μM cerulean-L5 plus 1 μM eYFP-P34 with 1 μM full-length L5 or addition of 1 μM L5 truncates. (C) Immune capture assay. Full-length L5 and L5 truncates were incubated with P34 in the binding buffer. Anti-P34 antibodies were used to capture down the associating proteins. The immunoprecipitate (P) and the supernatant (S) fractions were analyzed by Western blot (WB) analysis using anti-His antibodies. “beads alone,” no antibody; “input,” 10% of total proteins used for immune capture assay; M, molecular mass (kDa) markers; IP, immunoprecipitation. These assays were repeated three times with three different preparations of recombinant proteins.
Immune capture assays were also performed to confirm the regions of L5 involved in binding with P34. We utilized purified full-length P34 without the polyhistidine tag and full-length and truncated L5 with the polyhistidine tag. Immune capture fractions (Fig. 5C) using a peptide antibody against P34 were subsequently analyzed using the antipolyhistidine monoclonal antibody. For the negative control, the beads alone do not interact nonspecifically with full-length L5 (lane 5) or the N terminus (lane 10), L18 domain (lane 15), and C terminus (lane 20) of L5. N-terminal (13 kDa, lane 7) and C-terminal (10 kDa, lane 17) truncates of L5 can only be detected in the supernatant fraction of the immune capture, indicating that N terminus and C terminus of L5 are not important for the association with P34. Consistent with the findings from FRET experiments, full-length L5 (40 kDa, lane 3) and the L18 domain of L5 (24 kDa, lane 13) can be detected in the precipitate fraction, confirming that the L18 domain of L5 is involved in association with L5.
DISCUSSION
In T. brucei, we have reported that trypanosome-specific proteins P34 and P37 form a complex with L5 and 5S rRNA in the nucleoplasm (12). This novel preribosomal complex, which is not found in other eukaryotic cells, is hypothesized to compensate for the decreased binding affinity of T. brucei L5 and to augment its role in binding, stabilizing, and trafficking 5S rRNA. To study the association of this complex, we have utilized FRET to quantify the protein-protein interaction in vitro. FRET is a powerful approach to determine protein binding affinities, analyze multiprotein association, and directly screen for inhibitors of protein-protein interaction (20, 26).
In order to study the preribosomal complex in T. brucei, L5 and P34 were N-terminally tagged with cerulean and eYFP, respectively, which are a spectrally well-suited FRET pair and possess enhanced quantum yields. Absorption and emission spectra were examined to confirm that the fluorescence properties of the tags are not affected by the addition of the proteins (Fig. 1B). Moreover, immune capture assays showed that tagged protein eYFP-P34 was captured with untagged L5 and cerulean-L5 was captured with untagged P34, indicating that tagged proteins possess the same activities as the untagged P34 and L5, which bind to each other. We next observed FRET due to the specific binding of L5 and P34, in contrast to the nonspecific binding controls (Fig. 2).
FRET has been confirmed to be able to determine protein-protein binding affinities as accurately as isothermal titration calorimetry (20). To measure the binding constant of P34 and L5, we utilized the increase in acceptor emission at 530 nm as a direct measure of the quantity of bound protein, yielding data for binding curves (Fig. 3), fitted with the binding hyperbola with one binding site. The binding constant determined for the interaction, Kd = 0.60 ± 0.03 μM, is in the range of protein-protein binding constants of RNA binding proteins (25). The binding affinity between L5 and P34 is lower than that between L5 and 5S rRNA or P34 and 5S rRNA; however, we anticipate that it will be enhanced in the presence of 5S rRNA (12). A detailed knowledge of the binding parameters is important when attempting to disrupt an essential protein-protein interaction (27).
To determine the regions of L5 and P34 involved in protein-protein association, untagged P34 and L5 truncates were added to cerulean-L5 and eYFP-P34 solutions. The decrease in emission at 530 nm indicates that the binding of fluorescence-tagged protein to tagged partner has decreased due to binding with untagged protein. Immune capture assays were also performed to confirm the FRET results.
Both P34 and P37 possess highly basic APK-rich N termini, an RRM in the internal region, and KKDX repeats in the C terminus (28). The RRM comprises four-stranded β-sheets packed against two α-helices (29). Two conserved motifs located in the central β-sheets are thought to be involved in RNA interaction (29, 30). It is known that RRMs are involved not only in RNA-protein binding but also in protein-protein interaction (29, 30). In previous experiments, we found that the RRMs of P34 and P37 serve to mediate a protein-RNA interaction with 5S rRNA (M. Ciganda, unpublished data), as well as protein-protein interaction with an acidic region in NOPP44/46 (28), which is a trypanosome-specific family of tyrosine-phosphorylated RNA binding proteins primarily localized to the nucleolus (31, 32). Figure 4C shows that the addition of the N terminus and RRM of P34 to the FRET interacting pair led to a decrease in the emission at 530 nm. Figure 4D shows that N terminus and RRM of P34 were immune captured together with L5. These results indicate that the N terminus and RRM of P34 are important for the binding to L5. It is possible that the C terminus also plays a role but does not fold correctly as a fragment. It has been reported that the protein interaction mediated through β-sheets of RRM prevents RNA binding. In contrast, protein interaction through α-helices, leaving β-sheets solvent exposed, allows RNA binding (30). Since P34 is able to form a complex with 5S rRNA and L5, this suggests that the α-helices of P34 RRMs, but not the β-sheets, are involved in the protein-protein interaction.
L5 consists of a predicted RanBP1_WASP domain in its N terminus and an L18 domain in the internal region. RanBP1_WASP is a domain that is known to bind proline-rich sequences for protein-protein interaction (33). The L18 domain of L5 is homologous to the bacterial ribosomal protein L18, which is responsible for 5S rRNA-protein binding (34, 35). Figure 5B shows that the addition of the L18 domain of L5 decreased the emission of the FRET interacting pair at 530 nm, suggesting that the L18 domain of L5 is involved in association with P34. The L18 domain contains three α-helices and four-stranded β-sheets (35). Most of the residues involved in 5S rRNA binding are located on the face of the β-sheets (35). It has been reported that the RRM can associate with other protein domains through their α-helices, and the interaction is stabilized by salt bridges or hydrophobic interactions (30, 36, 37). This suggests that the RRMs of P34 may associate with the L18 domain of L5 via their α-helices, leaving their β-sheet exposed to interact with 5S rRNA.
Previous work has shown that the RRM of P34 is involved in both protein-protein interaction (28) and protein-5S rRNA interaction (Ciganda, unpublished). It is known that the L18 domain of L5 is important for binding to 5S rRNA (35). Thus, domains of P34 and L5 for protein-protein and protein-5S rRNA interaction may be close to one another or overlap. Previous data demonstrated that the association of P34 and L5 does not require 5S rRNA. However, the data did suggest that 5S rRNA enhanced the interaction of the two proteins (12). These data suggest that the protein-protein binding affinities of P34 and L5 binding may be enhanced by 5S rRNA binding, which may change the conformation of L5 or P34 and thereby enhance the protein-protein association to form a 5S rRNA-L5-P34 trimolecular complex. Future experiments will focus on the role of 5S rRNA in this novel preribosomal particle.
Previous data showed that P34, P37, and L5 bind to different sites of 5S rRNA, although footprint analysis suggested some overlap of region protected by P34 and P37 with the L5 binding domain (17). Further experiments in our laboratory are under way to study the association of 5S rRNA and proteins and to determine which specific residues within the domains we have identified are critical for the protein-protein and protein-RNA interactions in the complex. Fluorescence-tagged proteins will also be expressed in T. brucei. In vivo FRET experiments will clearly demonstrate how these proteins associate with each other and with the developing ribosome through the ribosome biogenesis pathway in T. brucei.
Although the ribosomes are, in general, highly conserved, they possess subtle sequence and/or conformational differences, which enable drug selectivity and facilitate therapeutic usage. There are number of antibiotics which target ribosomes at distinct locations within functionally relevant sites (38). For example, the antibiotics chloramphenicol, clindamycin, and streptogramin target the peptidyl-transferase center (PTC) of the ribosome to treat bacterial infections (39, 40). The structure-specific interactions between internal ribosomal entry site (IRES) of viral RNA and host 40S ribosome subunit have been proven to be a novel target for therapeutic intervention. Several small molecules have been identified to inhibit IRES activity either in vitro or in cell culture by high-throughput screens (41). A small RNA, SLRef, was found to interact specifically with human La protein and the ribosomal protein S5 and to inhibit ribosome assembly and selectively inhibit hepatitis C virus (HCV) RNA translation (42). Since this trypanosome-specific preribosomal complex is essential for 5S rRNA stability, ribosome assembly and cell viability in these parasites, it may prove to be a target for drug targeting. Previous data from cryo-electron microscopy (cryo-EM) studies on T. cruzi ribosomes suggest that there are expansion segments unique to trypanosome rRNAs that are available for protein interaction (43). We postulate that P34 and P37 as well as other trypanosome-specific proteins interact with these expansion segments and perform critical functions that could be targeted. The FRET assay has been described as an ideal method for the discovery and validation of drugs targeting protein-protein and protein-RNA interactions (44–46). The assay we present here provides a method for high-throughput screening for inhibitors of this protein-protein interaction.
ACKNOWLEDGMENT
This work was supported by NIH grant GM092719.
Footnotes
Published ahead of print 21 December 2012
REFERENCES
- 1. Vickerman K. 1985. Developmental cycles and biology of pathogenic trypanosomes. Br. Med. Bull. 41:105–114 [DOI] [PubMed] [Google Scholar]
- 2. Gunzl A. 2010. The pre-mRNA splicing machinery of trypanosomes: complex or simplified? Eukaryot. Cell 9:1159–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rudenko G. 2010. Epigenetics and transcriptional control in African trypanosomes. Essays Biochem. 48:201–219 [DOI] [PubMed] [Google Scholar]
- 4. Takahashi N, Yanagida M, Fujiyama S, Hayano T, Isobe T. 2003. Proteomic snapshot analyses of preribosomal ribonucleoprotein complexes formed at various stages of ribosome biogenesis in yeast and mammalian cells. Mass Spectrom. Rev. 22:287–317 [DOI] [PubMed] [Google Scholar]
- 5. Hernandez-Verdun D, Roussel P, Thiry M, Sirri V, Lafontaine DL. 2010. The nucleolus: structure/function relationship in RNA metabolism. Wiley Interdiscip. Rev. RNA 1:415–431 [DOI] [PubMed] [Google Scholar]
- 6. Fatica A, Tollervey D. 2002. Making ribosomes. Curr. Opin. Cell Biol. 14:313–318 [DOI] [PubMed] [Google Scholar]
- 7. Steitz JA, Berg C, Hendrick JP, La Branche-Chabot H, Metspalu A, Rinke J, Yario T. 1988. A 5S rRNA/L5 complex is a precursor to ribosome assembly in mammalian cells. J. Cell Biol. 106:545–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Allison LA, Romaniuk PJ, Bakken AH. 1991. RNA-protein interactions of stored 5S RNA with TFIIIA and ribosomal protein L5 during Xenopus oogenesis. Dev. Biol. 144:129–144 [DOI] [PubMed] [Google Scholar]
- 9. Ciganda M, Williams N. 2011. Eukaryotic 5S rRNA biogenesis. Wiley Interdiscip. Rev. RNA 2:523–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nazar RN. 2004. Ribosomal RNA processing and ribosome biogenesis in eukaryotes. IUBMB Life 56:457–465 [DOI] [PubMed] [Google Scholar]
- 11. Hellman KM, Ciganda M, Brown SV, Li J, Ruyechan W, Williams N. 2007. Two trypanosome-specific proteins are essential factors for 5S rRNA abundance and ribosomal assembly in Trypanosoma brucei. Eukaryot. Cell 6:1766–1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ciganda M, Prohaska K, Hellman K, Williams N. 2012. A novel association between two trypanosome-specific factors and the conserved L5-5S rRNA complex. PLoS One 7:e41398 doi:10.1371/journal.pone.0041398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pitula J, Ruyechan WT, Williams N. 2002. Two novel RNA binding proteins from Trypanosoma brucei are associated with 5S rRNA. Biochem. Biophys. Res. Commun. 290:569–576 [DOI] [PubMed] [Google Scholar]
- 14. Deshmukh M, Tsay YF, Paulovich AG, Woolford JL., Jr 1993. Yeast ribosomal protein L1 is required for the stability of newly synthesized 5S rRNA and the assembly of 60S ribosomal subunits. Mol. Cell. Biol. 13:2835–2845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yeh LC, Lee JC. 1995. Contributions of multiple basic amino acids in the C-terminal region of yeast ribosomal protein L1 to 5 S rRNA binding and 60 S ribosome stability. J. Mol. Biol. 246:295–307 [DOI] [PubMed] [Google Scholar]
- 16. Rizzo MA, Springer GH, Granada B, Piston DW. 2004. An improved cyan fluorescent protein variant useful for FRET. Nat. Biotechnol. 22:445–449 [DOI] [PubMed] [Google Scholar]
- 17. Ciganda M, Williams N. 2012. Characterization of a novel association between two trypanosome-specific proteins and 5S rRNA. PLoS One 7:e30029 doi:10.1371/journal.pone.0030029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang J, Ruyechan W, Williams N. 1998. Developmental regulation of two nuclear RNA binding proteins, p34 and p37, from Trypanosoma brucei. Mol. Biochem. Parasitol. 92:79–88 [DOI] [PubMed] [Google Scholar]
- 19. Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. 2002. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 20:87–90 [DOI] [PubMed] [Google Scholar]
- 20. Martin SF, Tatham MH, Hay RT, Samuel ID. 2008. Quantitative analysis of multi-protein interactions using FRET: application to the SUMO pathway. Protein Sci. 17:777–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Markwardt ML, Kremers GJ, Kraft CA, Ray K, Cranfill PJ, Wilson KA, Day RN, Wachter RM, Davidson MW, Rizzo MA. 2011. An improved cerulean fluorescent protein with enhanced brightness and reduced reversible photoswitching. PLoS One 6:e17896 doi:10.1371/journal.pone.0017896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Song Y, Madahar V, Liao J. 2011. Development of FRET assay into quantitative and high-throughput screening technology platforms for protein-protein interactions. Ann. Biomed. Eng. 39:1224–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Motulsky H, Christopoulos A. 2003. Fitting models to biological data using linear and nonlinear regression: a practical guide to curve fitting. GraphPad Software, Inc., San Diego, CA [Google Scholar]
- 24. Greenhouse JB, Kass RE, Tsay RS. 1987. Fitting nonlinear models with ARMA errors to biological rhythm data. Stat. Med. 6:167–183 [DOI] [PubMed] [Google Scholar]
- 25. Trowitzsch S, Weber G, Luhrmann R, Wahl MC. 2008. An unusual RNA recognition motif acts as a scaffold for multiple proteins in the pre-mRNA retention and splicing complex. J. Biol. Chem. 283:32317–32327 [DOI] [PubMed] [Google Scholar]
- 26. Stankovic-Valentin N, Kozaczkiewicz L, Curth K, Melchior F. 2009. An in vitro FRET-based assay for the analysis of SUMO conjugation and isopeptidase cleavage. Methods Mol. Biol. 497:241–251 [DOI] [PubMed] [Google Scholar]
- 27. Cochran AG. 2000. Antagonists of protein-protein interactions. Chem. Biol. 7:R85–R94 [DOI] [PubMed] [Google Scholar]
- 28. Pitula JS, Park J, Parsons M, Ruyechan WT, Williams N. 2002. Two families of RNA binding proteins from Trypanosoma brucei associate in a direct protein-protein interaction. Mol. Biochem. Parasitol. 122:81–89 [DOI] [PubMed] [Google Scholar]
- 29. Clery A, Blatter M, Allain FH. 2008. RNA recognition motifs: boring? Not quite. Curr. Opin. Struct. Biol. 18:290–298 [DOI] [PubMed] [Google Scholar]
- 30. Maris C, Dominguez C, Allain FH. 2005. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 272:2118–2131 [DOI] [PubMed] [Google Scholar]
- 31. Das A, Peterson GC, Kanner SB, Frevert U, Parsons M. 1996. A major tyrosine-phosphorylated protein of Trypanosoma brucei is a nucleolar RNA-binding protein. J. Biol. Chem. 271:15675–15681 [DOI] [PubMed] [Google Scholar]
- 32. Parsons M, Ledbetter JA, Schieven GL, Nel AE, Kanner SB. 1994. Developmental regulation of pp44/46, tyrosine-phosphorylated proteins associated with tyrosine/serine kinase activity in Trypanosoma brucei. Mol. Biochem. Parasitol. 63:69–78 [DOI] [PubMed] [Google Scholar]
- 33. Callebaut I, Cossart P, Dehoux P. 1998. EVH1/WH1 domains of VASP and WASP proteins belong to a large family including Ran-binding domains of the RanBP1 family. FEBS Lett. 441:181–185 [DOI] [PubMed] [Google Scholar]
- 34. Woestenenk EA, Gongadze GM, Shcherbakov DV, Rak AV, Garber MB, Hard T, Berglund H. 2002. The solution structure of ribosomal protein L18 from Thermus thermophilus reveals a conserved RNA-binding fold. Biochem. J. 363:553–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Turner CF, Moore PB. 2004. The solution structure of ribosomal protein L18 from Bacillus stearothermophilus. J. Mol. Biol. 335:679–684 [DOI] [PubMed] [Google Scholar]
- 36. Xu RM, Jokhan L, Cheng X, Mayeda A, Krainer AR. 1997. Crystal structure of human UP1, the domain of hnRNP A1 that contains two RNA-recognition motifs. Structure 5:559–570 [DOI] [PubMed] [Google Scholar]
- 37. Shamoo Y, Krueger U, Rice LM, Williams KR, Steitz TA. 1997. Crystal structure of the two RNA binding domains of human hnRNP A1 at 1.75 A resolution. Nat. Struct. Biol. 4:215–222 [DOI] [PubMed] [Google Scholar]
- 38. Yonath A. 2005. Antibiotics targeting ribosomes: resistance, selectivity, synergism and cellular regulation. Annu. Rev. Biochem. 74:649–679 [DOI] [PubMed] [Google Scholar]
- 39. Harms JM, Schlunzen F, Fucini P, Bartels H, Yonath A. 2004. Alterations at the peptidyl transferase centre of the ribosome induced by the synergistic action of the streptogramins dalfopristin and quinupristin. BMC Biol. 2:4 doi:10.1186/1741-7007-2-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schlunzen F, Zarivach R, Harms J, Bashan A, Tocilj A, Albrecht R, Yonath A, Franceschi F. 2001. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature 413:814–821 [DOI] [PubMed] [Google Scholar]
- 41. Davis DR, Seth PP. 2011. Therapeutic targeting of HCV internal ribosomal entry site RNA. Antivir. Chem. Chemother. 21:117–128 [DOI] [PubMed] [Google Scholar]
- 42. Bhat P, Gnanasundram SV, Mani P, Ray PS, Sarkar DP, Das S. 2012. Targeting ribosome assembly on the HCV RNA using a small RNA molecule. RNA Biol. 9:1110–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gao H, Ayub MJ, Levin MJ, Frank J. 2005. The structure of the 80S ribosome from Trypanosoma cruzi reveals unique rRNA components. Proc. Natl. Acad. Sci. U. S. A. 102:10206–10211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xu Y, Shi J, Yamamoto N, Moss JA, Vogt PK, Janda KD. 2006. A credit-card library approach for disrupting protein-protein interactions. Bioorg. Med. Chem. 14:2660–2673 [DOI] [PubMed] [Google Scholar]
- 45. Yin H, Hamilton AD. 2005. Strategies for targeting protein-protein interactions with synthetic agents. Angew. Chem. Int. Ed. Engl. 44:4130–4163 [DOI] [PubMed] [Google Scholar]
- 46. Arkin MR, Wells JA. 2004. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat. Rev. Drug Discov. 3:301–317 [DOI] [PubMed] [Google Scholar]